
ABSTRACT
Malignant gliomas are resistant to natural killer (NK) cell immune surveillance. However, the mechanisms used by these cancers to suppress antitumor NK cell activity remain poorly understood. We have recently reported on a novel mechanism of innate immune evasion characterized by the overexpression of the carbohydrate-binding protein galectin-1 by both mouse and rat malignant glioma. Here, we investigate the cytokine profile of galectin-1-deficient GL26 cells and describe the process by which these tumors are targeted by the early innate immune system in RAG1−/− and C57BL/6J mice. Our data reveal that galectin-1 knockdown in GL26 cells heightens their inflammatory status leading to the rapid recruitment of Gr-1+/CD11b+ myeloid cells and NK1.1+ NK cells into the brain tumor microenvironment, culminating in tumor clearance. We show that immunodepletion of Gr-1+ myeloid cells in RAG1−/− mice permits the growth of galectin-1-deficient glioma despite the presence of NK cells, thus demonstrating an essential role for myeloid cells in the clearance of galectin-1-deficient glioma. Further characterization of tumor-infiltrating Gr-1+/CD11b+ cells reveals that these cells also express CCR2 and Ly-6C, markers consistent with inflammatory monocytes. Our results demonstrate that Gr-1+/CD11b+ myeloid cells, often referred to as myeloid-derived suppressor cells (MDSCs), are required for antitumor NK cell activity against galectin-1-deficient GL26 glioma. We conclude that glioma-derived galectin-1 represents an important factor in dictating the phenotypic behavior of monocytic Gr-1+/CD11b+ myeloid cells. Galectin-1 suppression may be a valuable treatment approach for clinical glioma by promoting their innate immune-mediated recognition and clearance through the concerted effort of innate myeloid and lymphoid cell lineages.
Introduction
Glioblastoma (GBM) is the most common and aggressive primary astrocytoma in adults carrying a median survival of 15–21 mo post-diagnosis despite the current standard-of-care.Citation1 GBM tumors are highly immunosuppressive due to secreted (i.e., TGF-β) and cell-surface bound factors (i.e., FasL, PD-L1, CD70, gangliosides, and certain HLAs) produced by their constituent cells.Citation2 Tumor-derived immunosuppressive factors are a major hurdle to the achievement of successful immunotherapeutic intervention.Citation3-5 Despite its immunosuppressive activities, GBM is highly infiltrated by immune cells of myeloid origin such as peripheral monocytes/macrophages and MDSCs.
MDSCs are a heterogeneous population of immature cells shown to accumulate in the blood and tumor microenvironment of humans and mice-bearing malignant tumors.Citation5-9 These cells have the capacity to suppress T cell proliferation and function through factors that include arginase I (Arg-I),Citation10 inducible nitric oxide synthase (iNOS),Citation6 reactive oxygen species (ROS),Citation11 indoleamine 2,3- dioxygenase (IDO) expression, accumulation of Foxp3+ T regulatory cells (Tregs),Citation12 and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX2).Citation6,13 MDSCs also influence innate immunity by altering macrophage cytokine production Citation14 and suppressing NK cell effector function.Citation15,16
Two major MDSC subsets are recognized, granulocytic (Ly-6Ghi/CD11b+/Ly-6Clow), and monocytic, (Ly-6Glow/CD11b+/Ly-6Chi).Citation6,17,18 Mounting evidence supports a role for glioma-derived factors in influencing the function of immature myeloid precursor cells;Citation19-25 however, the molecular mechanisms remain poorly understood.
Galectin-1 (gal-1) is a member of a family of β-galactoside-binding lectins characterized by the presence of one or more homologous carbohydrate recognition domains (CRDs) that mediate interactions with glycoproteins bearing the basic core disaccharide N-acetyllactosamine (LacNAc) found in N- and O-linked glycans.Citation26 Gal-1 induces apoptosis in activated antigen-specific T lymphocytes, a mechanism linked to tumor immune escape.Citation27-29 It has been further proposed that gal-1 dampens inflammatory (M1) monocyte/macrophage activities, in turn favoring those that are anti-inflammatory (M2) and pro-tumor.Citation30,31 Our own work has shown that mouse GL26 and rat CNS-1 glioma suppress NK-mediated tumor killing by expressing high-levels of this lectin.Citation32 Tumor-derived lactate dehydrogenase 5 (LDH5) represents an additional mechanism of innate immune suppression in human glioma causing the upregulation of NKG2D ligands on monocytes and leading to NK exhaustion and tumor progression.Citation33 Further work is required to fully elucidate the mechanistic details of innate immune escape in GBM.
Here, we demonstrate that intracranial GL26 glioma cells rendered gal-1-deficient through shRNA knockdown are marked by increased cytokine production and elimination through the concerted action of myeloid and lymphoid cells of the innate immune system. These tumors are rapidly infiltrated by a 7-fold higher number of Gr-1+/CD11b+ myeloid cells compared to gal-1-expressing GL26 glioma, which is followed by a 9-fold induction in the number of tumor-infiltrating NK1.1+ NK cells and ultimately tumor eradication. Further experiments reveal that the Gr-1+/CD11b+ myeloid cells shown to infiltrate early gal-1-deficient gliomas express CCR2 and Ly-6C, cell surface markers consistent with inflammatory monocytes.Citation34 Together our findings show that (1) glioma-derived gal-1 is an important mechanism of tumor-induced innate immunosuppression through its ability to cloak tumor cells from innate immune recognition and (2) that gal-1 favors the conversion of immature myeloid cells into anti-inflammatory MDSCs known to support glioma progression.Citation8
Results
Gal-1-deficient GL26 cells elicit innate immune rejection of co-implanted gal-1-expressing cells
We have previously demonstrated that GL26 cells grown in vitro secrete gal-1, an effect significantly diminished by shRNA-mediated gal-1 knockdown.Citation32 Based on this observation, we examined whether NK-resistant gal-1-expressing GL26 cells could, through a bystander effect, protect co-implanted NK-sensitive gal-1-deficient GL26 cells from innate immune-mediated rejection. To assess this, we performed Kaplan–Meier survival analysis on RAG1−/− mice bearing mixtures of orthotopically implanted GL26-Cit-NT cells (GL26 mouse glioma cells expressing mCitrine fluorescent protein for visualization purposes and a non-targeting control shRNACitation32) and GL26-Cit-gal1i cells (GL26 mouse glioma cells also expressing mCitrine fluorescent protein, but with a gal-1-specific shRNA.Citation32 These two cell lines will be referred to as GL26-NT and GL26-gal1i throughout the rest of this text.
A total of 2 × 104 glioma cells were implanted into the brain of each mouse at the following NT-to-gal1i ratios: 100:0, 80:20, 50:50, 20:80. Three reference groups were also included comprising 2 × 104, 1 × 104, and 4 × 103 GL26-NT cells alone. Our analysis revealed that gal-1-expressing cells did not protect gal-1-deficient cells from innate immune clearance. On the contrary, gal-1-deficient cells caused the rejection of the gal-1-expressing cells. This was evident by the fact that mouse median survival was extended in response to an increased percentage of GL26-gal1i cells in the co-implants. In fact, all mice receiving 80% gal-1-deficient glioma cells achieved long-term survival with no evidence of brain tumor burden 100-days post-implantation despite having also received 20% GL26-NT cells (). This result indicated that NK sensitive glioma cells are capable of eliciting an innate immune response, not only against themselves, but also against glioma cells that express normal levels of gal-1. The capacity of glioma cells to block innate immune killing therefore appears to be overcome under the right conditions of innate immune activation, as occurs when tumor-derived gal-1 is reduced.
Figure 1. Gal-1-deficient GL26 glioma cells are proinflammatory. (A) Kaplan–Meier survival analysis of RAG1−/− mice bearing GL26-NT cells alone (gray, blue and purple curves), or together with an increasing percentage of GL26-gal1i cells (green, orange and red curves). NT:gal1i ratios are indicated to the left of the three co-implant groups. Three alternative experimental outcomes are shown in the table below. The actual results are consistent with outcome number 3. Mantel–Cox log-rank test detected a significant survival difference between the indicated groups. *p <0.05; **p <0.005. (B) Percentage of circulating NK1.1+ NK cells in RAG1−/− mouse blood 5-d after intracranial engraftment of GL26-NT (n = 4), GL26-gal1i (n = 4), or injection with vehicle alone (n = 2). (C) Quantitative comparison of brain tumor size 5-days after implantation into RAG1−/− mice. GL26-NT (n = 4), GL26-gal1i (n = 4), and vehicle alone (n = 2) groups are shown. (D) Representative histology from the brain tumors represented in panel C showing tumor-derived mCitrine fluorescent protein (top micrographs) and granzyme B (GzmB) expression (bottom micrographs). (E) Circulating CD11blo/NK1.1+ NK cells from RAG1−/− mouse blood demonstrating the expression of GzmB (open black histogram) above isotype control (closed gray histogram). Experiment performed in triplicate. Data from a representative experiment is shown. (F) NK-mediated cytotoxicity assessed using an ATP-dependent luminescence assay showing the level of viable GL26-gal1i cells (RLU) alone, or in the presence of a 10:1 E:T ratio of circulating NK1.1+ NK cells from RAG1−/− mice after 4 h of co-culture (n = 4 technical replicates per group, experiment repeated × 3).
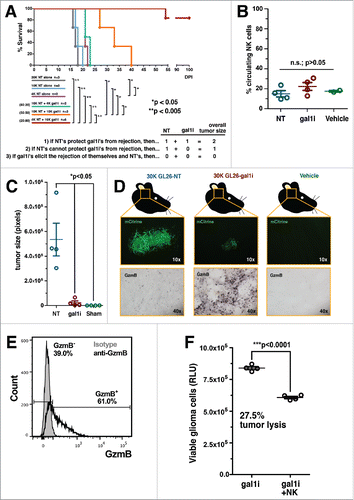
Orthotopically implanted gal-1-deficient glioma drives NK cells into the tumor microenvironment, but does not influence their abundance in the blood
We next asked if intracranial gal-1-deficient glioma cells would cause an increase in the number of circulating NK cells available to enter the tumor microenvironment, or whether these tumors would merely provoke the recruitment of existing numbers of these cells into the tumor microenvironment. To distinguish between these two alternatives, we engrafted 3 × 104 GL26-NT or GL26-gal1i cells into the striatum of RAG1−/− mice, and performed transcardial blood draws 5-days post-tumor implantation to assess the percentage of circulating NK cells in the blood stream. This time point corresponds both to tumors well vascularized by normal mouse brain blood vessels, and active tumor rejection as demonstrated by our previous work with GL26 cells.Citation32,35 A cohort of mice was included in the experiment that underwent intracranial injection with vehicle alone to control for potential inflammatory reactions due to the implantation procedure. The results of this experiment showed that the percentage of circulating NK cells in all three groups were similar (14.95 ± 3.16% NT vs. 22.25 ± 3.95% gal1i vs. 17.50 ± 0.80% vehicle alone; n.s.; p >0.05, one-way ANOVA followed by Tukey's post-test) (), suggesting that GL26-gal1i tumor rejection was not due to alterations in the profile of circulating NK cells, but rather due to a tropism of normal levels of NK cells toward the gal-1-deficient tumor microenvironment. Histologic analysis on the brains of these mice confirmed that GL26-gal1i tumors were indeed undergoing tumor rejection 5-days after tumor implantation, as the tumors were significantly smaller (5.35 × 105 ± 1.32 × 105 pixels NT vs. 2.27 × 104 ± 1.48 × 104 pixels gal1i; *p <0.05, one-way ANOVA followed by Tukey's post-test) and more highly infiltrated with granzyme B (GzmB) positive cells compared to GL26-NT tumors (). GzmB+ cells were completely absent from the brains of mice injected with vehicle alone, demonstrating the requirement of intracranial glioma cells to drive GzmB+ cells into the brain. Further experiments showed that 61.0% of circulating CD11blo/NK1.1+ NK cells in tumor-naive RAG1−/− mice expressed GzmB (53% of total NK cells) (), and that FACS-purified circulating NK1.1+ NK cells lyse GL26-gal1i cells by nearly 30% after 4 h of co-culture at a 10:1 effector:target (E:T) ratio without requiring ex vivo stimulation (8.39 × 105 ± 1.21 × 104 relative luminescence units (RLU) gal1i alone vs. 6.08 × 105 ± 9.51 × 103 RLU gal1i + NK cells; ***p <0.0001, unpaired, two-tailed student's t-test.) (). Our previous work had already demonstrated that gal-1-deficient glioma cells are more sensitive to NK-mediated lysis compared to gal-1-expressing cells.Citation32 These experimental results now indicated that circulating NK cells express cytotoxic granules and are active against gal-1-deficient glioma cells prior to tumor implantation.
Gal-1-deficient glioma cells exhibit enhanced chemokine production
Our results up to this point showed that (1) gal-1-deficient gliomas do not alter the percentage of circulating NK cells compared to gal-1-expressing gliomas within 5-days of tumor implantation, (2) circulating NK cells in tumor-naive mice express GzmB, and (3) GzmB+ NK cells lyse GL26 cells in vitro without requiring exogenous stimulation. The sum of these results suggested that proinflammatory cues within the gal-1-deficient tumor microenvironment were likely driving normal levels of cytotoxic NK cells from the circulation into the tumor microenvironment. We therefore speculated that GL26-gal1i cells might exhibit a proinflammatory cytokine signature compared to their gal-1-expressing counterparts. To test this hypothesis, we incubated whole cell lysate from GL26-NT and GL26-gal1i cells cultured in vitro with commercially available cytokine arrays capable of simultaneously detecting the relative abundances of 40 different cytokines. We found that gal-1-deficient GL26 cells exhibited a 27.8-fold induction in CXCL10/IP-10 (7.99 ± 0.82 mean pixel density (MPD) NT vs. 213.88 ± 14.93 MPD gal1i; **p = 0.0052), a 1.6-fold induction in CXCL12/SDF-1 (113.83 ± 1.72 MPD NT vs. 179.02 ± 6.00 MPD gal1i; **p = 0.0090), a 39.5-fold induction in CCL5/RANTES (0.21 ± 0.02 MPD NT vs. 8.29 ± 0.71 MPD gal1i; **p = 0.0076), an 8.5-fold reduction in CXCL1/KC (120.85 ± 5.49 MPD NT vs. 14.21 ± 3.68 MPD gal1i; **p = 0.0038), and a 6.7-fold reduction in IL-1ra (34.31 ± 1.13 MPD NT vs. 5.11 ± 0.57 MPD gal1i; **p = 0.0019) compared to GL26-NT cells as determined by unpaired, two-tailed student's t-tests (). Additional experiments in which GL26 conditioned media was used as cytokine array input corroborated our findings with whole cell lysate by showing that GL26-gal1i cells secrete 12.2-fold more CXCL10/IP-10 (17.30 ± 1.02 MPD NT vs. 211.61 ± 6.32 MPD gal1i; **p = 0.0011), 1.7-fold more CXCL12/SDF-1 (28.80 ± 0.27 MPD NT vs. 49.77 ± 4.29 MPD gal1i; *p = 0.0396), 188.2-fold more CCL5/RANTES (0.49 ± 0.02 MPD NT vs. 92.20 ± 3.47 MPD gal1i; **p = 0.0014), and 1.4-fold less CXCL1/KC (247.34 ± 1.817 MPD NT vs. 180.17 ± 4.08 MPD gal1i; **p = 0.0044) compared to GL26-NT cells as determined by unpaired, two-tailed student's t-tests ().
Figure 2. Gal-1-deficient GL26 glioma cells upregulate cytokine expression. (A–C) Relative expression values of detectable cytokines in GL26 whole cell lysate (A), GL26 conditioned media (B), and brain tissue homogenate containing GL26 gliomas 72 h post-engraftment (C). Red bars indicate GL26-gal1i cells, blue bars indicate GL26-NT cells. Numbers associated with each NT/gal1i bar graph pair correspond to the raw cytokine array data shown below. Error bars in panels A and B correspond to two technical replicates (n = 2). Error bars associated with the data in panel C correspond to two biological replicates (n = 2) of each tumor type. Positive control spots for each array are shown in the upper-left, upper-right, and lower-left corners. Negative control spots are at the lower-right corner of each array. The positive control spots in the arrays associated with panels A and B are overexposed and appear red. Statistical analysis was performed by unpaired, two-tailed student's t-tests. Associated p values are shown above each cytokine.
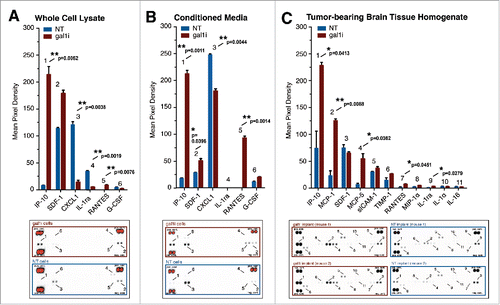
We next examined whether intracranially implanted gal-1-deficient GL26 cells would also express the cytokines we observed in vitro, and whether differences in additional cytokines might also be revealed between intracranial gal-1-deficient and gal-1-expressing gliomas. To test this, we used homogenized brain tissue from C57BL/6J mice inoculated 72 h earlier with 3 × 104 GL26-NT or GL26-gal1i glioma cells. These experiments confirmed the increased production of CXCL10/IP-10 and CCL5/RANTES associated with gal-1-deficient GL26-gal1i cells by revealing a 3.1-fold induction in CXCL10/IP-10 (73.98 ± 31.83 MPD NT vs. 228.28 ± 5.79 MPD gal1i; *p = 0.0413) and a 5.6-fold induction in CCL5/RANTES (1.25 ± 1.16 MPD NT vs. 7.01 ± 0.50 MPD gal1i; *p = 0.0451) in the gal-1-deficient tumor microenvironment compared to that of gal-1-expressing tumors using unpaired, two-tailed student's t-tests (). We also observed statistically significant differences in the levels of alternative cytokines not detected in GL26 cells grown in vitro. We found a 5.4-fold induction in CCL2/MCP-1 (22.99 ± 9.06 MPD NT vs. 125.29 ± 3.32 MPD gal1i; **p = 0.0088), a 9.3-fold induction in CCL12/MCP-5 (5.81 ± 2.43 MPD NT vs. 54.15 ± 9.14 MPD gal1i; *p = 0.0362), and a 5.9-fold-induction in IL-1ra (0.49 ± 0.31 MPD NT vs. 2.89 ± 0.26 MPD gal1i; *p = 0.0279) in the GL26-gal1i tumor microenvironment compared to that of GL26-NT tumors. The fact that we could detect increased levels of prototypical monocyte-derived chemokines such as CCL2/MCP-1 and CCL12/MCP-5 in the gal-1-deficient glioma microenvironment suggested that monocytes/macrophages might play a role in the eradication of gal-1-deficient glioma.
Gr-1+/CD11b+ myeloid cells accumulate in the gal-1-deficient tumor microenvironment prior to the recruitment of NK1.1+ NK cells
To comprehensively assess the different types of immune cells that penetrate the gal-1-deficient glioma microenvironment, we developed a protocol for the isolation and flow cytometric analysis of peripheral blood mononuclear cells (PBMCs) that infiltrate the early brain tumor microenvironment.Citation36 This procedure was used to characterize, and temporally resolve, the immune influx events associated with gal-1-deficient glioma rejection. C57BL/6J mice were engrafted with 3 × 104 GL26-NT or GL26-gal1i cells and euthanized 48 or 72 h post-tumor engraftment. GL26-gal1i tumors were infiltrated by 2.4-fold more CD45+ PBMCs at the 48-h time point (2,048 ± 212 NT vs. 4,941 ± 442 gal1i; **p = 0.0011, unpaired, two-tailed, Student's t- test). Of total CD45+ cells, Gr-1+/CD11b+ myeloid cells were the most disparate between the two tumor types, with 7-fold more of these cells in GL26-gal1i tumors compared to GL26-NT (235 ± 65 NT vs. 1,649 ± 275 gal1i; **p = 0.0024, unpaired, two-tailed, Student's t- test). We also detected a population of Gr-1int./CD11b+ myeloid cells that was 1.5-fold higher in gal-1-deficient tumors (1,222 ± 133 NT vs. 1,805 ± 136 gal1i; *p = 0.022, unpaired, two-tailed, Student's t- test) and a trend toward higher numbers of NK1.1+ NK cells in gal-1-deficient gliomas 48 h post-engraftment that failed to reach statistical significance (20 ± 7.2 NT vs. 141 ± 56 gal1i; n.s., p = 0.0759) as determined by unpaired, two-tailed, Student's t- tests ().
Figure 3. PBMCs preferentially infiltrate the early gal-1-deficient glioma microenvironment. (A) Gating strategy used to assess tumor-infiltrating PBMCs. Step 1: total cells extracted from a 37/70 density centrifugation media gradient interface, gated to exclude cellular debris (FSC-A less than ∼50 K). Steps 2 and 3: doublet discrimination gating. Step 4: CD45 gate to identify immune cells. Step 5: CD45+ cells stratified based on Gr-1 and CD11b expression. Distinct PBMC populations are color-coded. Step 6: color-coded PBMC populations stratified based on NK1.1 expression. Step 7: color-coded PBMC populations shown in Step 6, displayed in contour mode to better visualize the distribution of rare cell populations, backgated onto FSC-A versus SSC-A. NK1.1hi NK cells (orange population) are smaller on FSC-A compared to NK1.1lo myeloid cells (red and cyan populations), as expected, due to their smaller lymphoid size. (B and C) Comparison of the number of glioma-infiltrating PBMCs at 48- (B) and 72- (C) h post-intracranial engraftment. GL26-NT (n = 4) data points are shown in blue. GL26-gal1i (n = 4) data points are shown in red. Statistical analysis was performed using unpaired, two-tailed, Student's t-tests. p values are indicated above each PBMC type. (D) Representative fluorescence (top) and bright-field (bottom) micrographs of Gr-1 immunolabeled brain tissue sections bearing GL26-NT (left column) or GL26-gal1i (right column) tumors 5-d post-engraftment into the striatum of C57BL/6J mice. Bright-field micrographs show Gr-1 immunoreactivity corresponding to the same area shown in the respective fluorescence micrographs above. Insets show aspects of the respective bright-field micrographs at higher zoom for clarity.
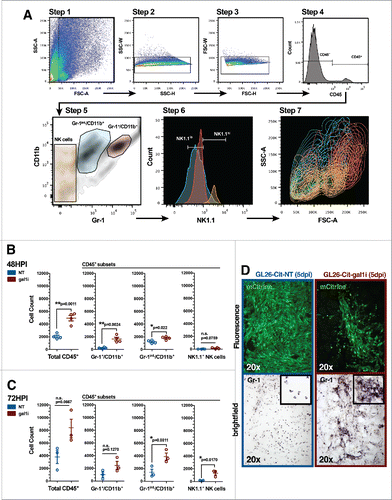
By the 72-h time point, the total number of CD45+ tumor-infiltrating PBMCs had increased in both groups, although the difference was now no longer statistically significant (3,802 ± 1,050 NT vs. 8,328 ± 1,474 gal1i; n.s., p = 0.0667). Of total CD45+ cells, the number of Gr-1+/CD11b+ myeloid cells also failed to reach statistical significance different between the two groups (1,008 ± 395 NT vs. 2,472 ± 651 gal1i; n.s., p = 0.1270). Conversely, statistical significance persisted in the Gr-1int./CD11b+ myeloid subset (1,411 ± 504 NT vs. 3,904 ± 576 gal1i, *p = 0.0311) and a significant 9.3-fold induction in the recruitment of NK1.1+ NK cells was now observed in the gal-1-deficient tumor microenvironment (142 ± 29 NT vs. 1,322 ± 298 gal1i; *p = 0.0170) as determined by unpaired, two-tailed, Student's t- tests ().
Although significantly more Gr-1+/CD11b+ myeloid cells accumulated in the tumor microenvironment of gal-1-deficient gliomas 48 h post-engraftment, the fact remained that these cells also accumulated in GL26-NT tumors. Immunohistochemical analysis of gliomas 5-days post-engraftment revealed that the Gr-1+ cells that infiltrate GL26-NT tumors are relatively spherical and evenly distributed throughout the tumor mass, while those infiltrating GL26-gal1i tumors display an amorphous shape, suggestive of cellular activation ().
Immunodepletion of Gr-1+ cells permits gal-1-deficient glioma growth in RAG1−/− mice
The enhancement of NK cell activity by collateral cells such as neutrophils,Citation37,38 monocytes,Citation39-41 macrophages,Citation42-44 and dendritic cellsCitation45-48 has been well documented. Many of these myeloid cells express Gr-1. We therefore examined whether the Gr-1+/CD11b+ myeloid cells shown to infiltrate the early gal-1-deficient glioma microenvironment played a significant role in NK-dependent tumor lysis. To test our hypothesis, we immunodepleted Gr-1+ cells in RAG1−/− mice using anti-Gr-1 monoclonal antibodies (clone: RB6-8C5) beginning one day prior to GL26-gal1i tumor implantation. GL26-gal1i tumors in mice treated with anti-Gr-1 antibodies were 9.4-times larger than those in mice treated with an equivalent regimen of control rat IgG antibodies after a 7-d growth period (1.94 × 105 ± 2.89 × 104 pixels rat IgG vs. 1.82 × 106 ± 1.91 × 105 pixels anti-Gr-1; **p = 0.0011, unpaired, two-tailed, Student's t-test) (). Further quantitative analysis revealed that GzmB expression in GL26-gal1i tumors treated with anti-Gr-1 antibodies was substantially lower on a per unit tumor area basis compared to GL26-gal1i tumors treated with rat IgG control antibodies (3.75 ± 0.36 rat IgG control vs. 0.63 ± 0.14 anti-Gr-1; **p = 0.0012; unpaired, two-tailed, Student's t-test) (). Control experiments showed that Gr-1+ myeloid cells are efficiently immunodepleted by 73 ± 1.3% (86 ± 2.06% rat IgG control vs. 12.85 ± 2.56% anti-Gr-1; ***p <0.0001, unpaired, two-tailed, Student's t-test) 48-h after a single 500 μg dose of anti-Gr-1 antibodies ().
Figure 4. Gr-1 immunodepletion permits GL26-gal1i tumor growth in RAG1−/− mice. (A) Representative fluorescence micrographs of GL26-gal1i gliomas 7-d post-engraftment into the striatum of RAG1−/− mice treated with rat IgG control antibodies (left; n = 3) or anti-Ly-6G/Ly-6C (i.e., Gr-1) antibodies (clone: RB6-8C5) (right; n = 3). Quantification of brain tumor size (in pixels) in each treatment group is shown to the right. (B) Representative fluorescence micrographs of GL26-gal1i gliomas 7-d post-engraftment into the striatum of RAG1−/− mice treated with rat IgG control antibodies (left; n = 3) or anti-Ly-6G/Ly-6C (i.e., Gr-1) antibodies (clone: RB6-8C5) (right; n = 3). Quantification of GzmB expression per unit tumor area (in pixels) in each treatment group is shown to the right. (C) Immunodepletion of Gr-1+ cells in RAG1−/− mouse blood in response to a single 500 μg dose of the RB6-8C5 clone. (D) Representative fluorescence micrographs of GL26-gal1i gliomas 7-d post-engraftment into the striatum of RAG1−/− mice treated with rat IgG control antibodies (left; n = 3) or anti-Ly-6G-specific antibodies (clone: 1A8) (right; n = 3). Quantification of brain tumor size (in pixels) in each treatment group is shown to the right. (E) Stacked bar graph showing the breakdown of total circulating leukocytes in RAG1−/− 24-h after a single 600 μg dose of the anti-Ly-6G 1A8 clone.
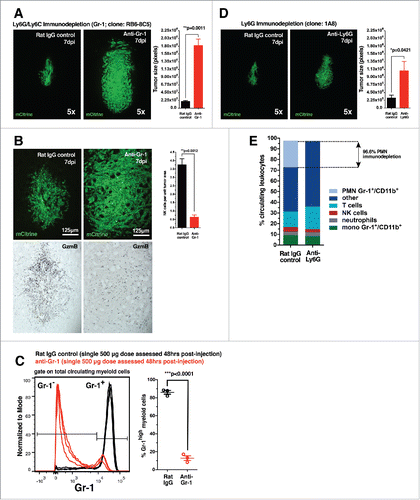
Two types of Gr-1+/CD11b+ myeloid cells exist, monocytic and polymorphonuclear. To determine which of these two cells was responsible for aiding NK-dependent gal-1-deficient glioma rejection, we specifically immunodepleted polymorphonuclear cells using anti-Ly-6G monoclonal antibodies (clone: 1A8). Treatment with anti-Ly-6G antibodies permitted a small, yet statistically significant, increase in the size of GL26-gal1i tumors compared to rat IgG control antibodies over a 7-d growth period (2.92 × 105 ± 8.33 × 104 pixels rat IgG vs. 1.08 × 106 ± 2.74 × 105 pixels anti-Ly-6G; *p = 0.0421, unpaired, two-tailed, Student's t-test) (). However, the average anti-Ly-6G treated tumor was still nearly twice as small as those which had grown in response to Gr-1 immunodepletion (1.08 × 106 ± 2.74 × 105 pixels anti-Ly-6G vs. 1.82 × 106 ± 1.91 × 105 pixels anti-Gr-1), which targets both Ly-6G+ and Ly-6C+ cells. Control experiments revealed that the 1A8 clone immunodepleted circulating polymorphonuclear Gr-1+/CD11b+ myeloid cells by 96.6% after 24-h in response to a single 600 μg dose, while only reducing the monocytic subtype by 8.5% (), thus demonstrating the specificity of anti-Ly-6G antibodies.
Gr-1+/CD11b+ myeloid cells that infiltrate early gal-1-deficient gliomas also express CCR2 and Ly-6C
Our data up to this point suggested that Ly-6C+ monocytes might play a more central role in the aiding of gal-1-deficient glioma rejection because (1) Gr-1 (Ly-6G/Ly-6C) immunodepletion had a greater effect on gal-1-deficient glioma growth compared to Ly-6G-specific immunodepletion and (2) our in vivo cytokine array analysis showed higher levels of monocyte chemoattractants in the gal-1-deficient glioma microenvironment. We therefore hypothesized that CCR2, the cognate receptor for CCL2/MCP-1 and CCL12/MCP-5, may be responsible for the chemoattraction of monocytic Gr-1+/CD11b+ myeloid cells into the brain tumor microenvironment. To test this hypothesis, we engrafted 3 × 104 GL26-gal1i cells into the striatum of wild-type C57BL/6J mice or B6.CCR2rfp/rfp knock-out mice, a model in which cells that would otherwise be CCR2+ express red fluorescent protein (RFP). Glioma growth in these two models was then compared. Quantitative histological analysis revealed that the tumors were equivalent in size 7-d post engraftment (4.76 × 104 ± 2.77 × 104 pixels C57 vs. 5.32 × 104 ± 1.76 × 104 pixels CCR2rfp/rfp; n.s., p = 0.8708, unpaired, two-tailed, Student's t-test) (). Scanning fluorescence confocal analysis further revealed that GL26-gal1i tumors in the B6.CCR2rfp/rfp mice were highly infiltrated by RFP+ cells (), thus demonstrating that the CCR2 signaling axis is not required for the trafficking of these cells into the tumor microenvironment. Flow cytometric analysis of circulating leukocytes from B6.CCR2rfp/rfp mice demonstrated that only monocytic Gr-1+/CD11b+ myeloid cells (not the polymorphonuclear subtype) are RFP+ (), implying that the RFP+ cells seen in confocal micrographs were likely monocytes. Immunohistochemical analysis with anti-Ly-6G (clone: 1A8) or anti-Ly-6C (clone: AL-21) antibodies on brain tissue sections from C57BL/6J mice bearing GL26-gal1i glioma revealed very few tumor-infiltrating Ly-6G+ cells, but numerous Ly-6C+ cells 7-d post-engraftment (). Flow cytometric analysis of PBMCs infiltrating the early gal-1-deficient tumor microenvironment in C57BL/6J mice confirmed that tumor infiltrating Gr-1+/CD11b+ myeloid cells co-express Ly-6C, and showed that the Gr-1int./CD11b+ cells are Ly-6C− ().
Figure 5. Gr-1+/CD11b+ myeloid cells that infiltrate early gal-1-deficient glioma express markers of inflammatory monocytes. (A) Quantification of GL26-gal1i tumor size 7-d after intracranial engraftment in wild-type C57BL/6J (n = 4) or B6.CCR2−/− (n = 4) mice. (B) Scanning fluorescence confocal analysis of GL26-gal1i glioma (green) 48-h post-engraftment into the brain of a B6.CCR2−/− mouse showing the presence of numerous RFP+ cells (red), a surrogate marker for cells that normally express CCR2. (C) Flow cytometric analysis of circulating leukocytes from tumor-naive B6.CCR2−/− mice reveals that only the monocytic subtype of Gr-1+/CD11b+ myeloid cell expresses RFP. (D) Fluorescence immunohistochemical analysis on brain tissue sections bearing GL26-gal1i 7-d post-engraftment into wild-type C57BL/6J mice (n = 4) using anti-Ly-6G (clone: 1A8) (left two panels) or anti-Ly-6C (clone: AL-21) (right two panels) antibodies, experiment repeated × 2. The aspects of the low-magnification micrographs outlined by the white boxes within the respective micrographs are shown at higher-magnification in the micrographs to the right, demonstrating the paucity of Ly-6G+ cells, but high degree of Ly-6C+ cells within the gal-1-deficient glioma microenvironment. Insets show examples of immunopositive cells whose nuclear morphology is consistent with Ly-6G+ polymorphonuclear cells (left panel) and Ly-6C+ monocytes (right panel). (E) Flow cytometric analysis of Gr-1int./CD11b+ (blue gate) and Gr-1+/CD11b+ myeloid cells (red gate) within the GL26-gal1i tumor microenvironment 6-d post-engraftment (left panel). Color-coded PBMC populations are further stratified based on Ly-6C expression in the histograms to the right, demonstrating that the Gr-1int./CD11b+ population is Ly-6C− while the Gr-1+/CD11b+ cells are Ly-6C+. Isotype control is shown (white histogram); experiment repeated × 2. (F) Flow cytometric analysis for CD11c (left) and F4/80 in FACS-purified Gr-1+/CD11b+ monocytic myeloid cells after 20-h of in vitro co-culture with GL26-NT or GL26-gal1i cells. The geometric mean of each color-coded histogram (bottom panels) is plotted as a bar graph above the respective histogram plots. The schematic between the two bar graphs shows the known fates of circulating monocytes toward either conventional myeloid dendritic cells (i.e., mDCs) or macrophages (MΦ) in response to different microenvironmental queues, experiment repeated × 2.
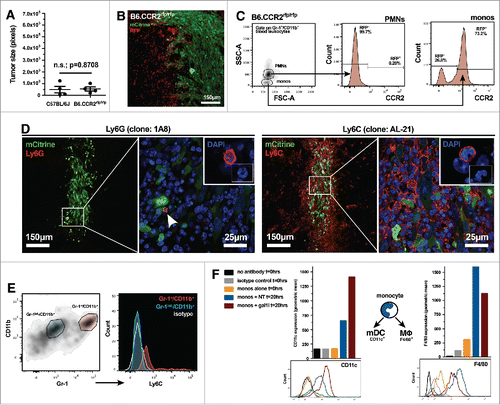
Specific microenvironmental cues cause circulating monocytes to differentiate into either macrophages or conventional dendritic cells upon extravasating from the blood into inflamed tissue.Citation49 We cultured FACS purified monocytic Gr-1+/CD11b+ cells with GL26-NT or GL26-gal1i glioma cells for 20-h, then assessed the resultant status of the myeloid cells. Our experiments revealed that monocytic Gr-1+/CD11b+ myeloid cells express more CD11c, a prototypical conventional dendritic cell marker, in the presence of gal-1-deficient GL26-gal1i cells (653 NT geometric mean vs. 1,410 gal1i geometric mean, experiment repeated × 2), and more F4/80, a prototypical macrophage marker, in the presence of gal-1-expressing GL26-NT cells (1,594 NT geometric mean vs. 1,127 gal1i geometric mean, experiment repeated × 2) showing that gal-1-deficient glioma cells favor the conversion of monocytic Gr-1+/CD11b+ myeloid cells toward the conventional dendritic cell phenotype in vitro (). A working model of gal-1-deficient glioma recognition and eradication through the concerted effort of monocytic Gr-1+/CD11b+ myeloid cells and NK cells is shown in ().
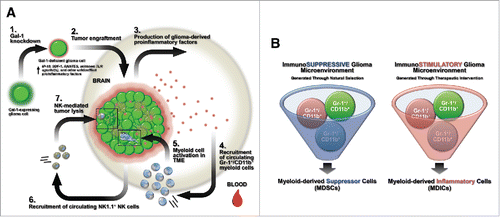
Figure 6. Summary models. (A) Schematic summary of innate immune-mediated gal-1-deficient GL26 glioma rejection. Step 1: gal-1-knockdown causes GL26 cells to increase production of the chemokines CXCL10/IP-10, CXCL12/SDF-1, and CCL5/RANTES. Step 2: gal-1-deficient GL26 cells are engrafted into the brain of RAG1−/− or C57BL/6 mice. Step 3: gal-1-deficient glioma cells produce proinflammatory factors in the brain. Step 4: circulating Gr-1+/CD11b+/Ly-6C+/CCR2+ monocytic myeloid cells are rapidly recruited to the brain tumor microenvironment. Step 5: once within the tumor microenvironment, these myeloid cells are influenced by tumor-derived proinflammatory factors, likely differentiating into conventional DCs. Step 6: circulating NK1.1+ NK cells then recruit to the brain tumor microenvironment. Step 7: NK cells lyse glioma cells leading to tumor eradication. (B) Myeloid-derived suppressor cells (MDSCs) versus myeloid-derived inflammatory cells (MDICs). Immunosuppressive malignant glioma generated through natural selective pressures influence the inherently plastic Gr-1+/CD11b+ myeloid precursor cell population to act as MDSCs with immunosuppressive or pro-tumor functionality. Gr-1 immunodepletion in these cancer systems is expected to extend survival (left panel). Experimental or therapeutic interventions that enhance the inflammatory state of the glioma microenvironment (i.e. tumor-derived gal-1 suppression) influence the same population of Gr-1+/CD11b+ myeloid precursor cells to act as MDICs with antitumor functionality. Gr-1 immunodepletion in these cancer systems is expected to reduce survival.
Discussion
Although immunotherapy for high-grade glioma is an active area of investigation, tumor localization within the immunospecialized CNS and the production of immunosuppressive factors act as major impediments to immune-mediated targeting of the disease. While the literature is well annotated with studies pertaining to mechanisms of glioma-induced adaptive immunosuppression, mechanisms of innate immunosuppression lack an equivalent depth of knowledge. We have recently contributed to a better understanding of glioma-induced innate immunosuppression by showing that orthotopically engrafted mouse and rat glioma cells rendered gal-1-deficient through shRNA knockdown are sensitized to NK-mediated recognition and clearance.Citation32 We now demonstrate that an unexpected population of Gr-1+/CD11b+ myeloid cells is central to the ability of NK cells to exert immunosurveillance activity against gal-1-deficient GL26 glioma.
Our data showing that monocytic Gr-1+/CD11b+ myeloid cells act as antitumor cells against gal-1-deficient glioma opposes the prevailing view of these cells as tolerogenic and immunosuppressive in murine cancer models.Citation5,50 Since we find that antitumor function in this myeloid cell subpopulation correlates with gal-1-deficiency in glioma cells, we propose that tumor-derived gal-1 may play an important role in the promotion of immunosuppressive MDSC expansion and activity. The work of others supports this hypothesis by demonstrating that gal-1 indeed favors the conversion of peripheral macrophages toward the M2 phenotype and deactivates M1 microglia within the CNS.Citation30,31
The view of tumor inflammatory status as a principle determinant of immature myeloid cell function helps explain why we and others investigating experimental glioma models with enhanced inflammatory characteristics ascribe antitumor activity to a population of myeloid cells conventionally thought to mediate immune regulatory effects in the context of cancer.Citation39,51,52 Our experiments with gal-1-deficient glioma reveal the capability of Gr-1+/CD11b+ cells to have immunostimulatory effects. We therefore suggest a definition of mouse MDSCs that goes beyond the cell surface markers Gr-1+/CD11b+ should be required to determine if such cells are indeed immunosuppressive (i.e., MDSCs), or immune-stimulating, as demonstrated here by us (). Such a functional definition may mitigate future controversies regarding disparate results obtained when Gr-1+ myeloid cells are immunodepleted in experimental murine cancer studies. A still outstanding question is whether immature human myeloid cells frequently defined as CD33+/CD11b+/HLA-DR−Citation53 are influenced in a similar manner by human-specific gal-1 which shares 88.1% identity with the rodent protein (HomoloGene database, NCBI), or if these cells also exhibit modularity in their phenotype in response to microenvironmental cues.
We suspect that immunotherapeutic strategies designed to suppress the expression or function of glioma-derived gal-1 may be of significant clinical value. Reductions of gal-1 protein by as little as 50% in mouse and rat glioma cells are sufficient to elicit robust recognition and eradication of both glioma models by the innate immune system of the respective species.Citation32 Despite this compelling observation, care must be taken in moving forward with early phase human clinical trials that implement anti-gal-1 immunotherapeutic strategies. We have yet to fully understand how glioma-derived gal-1 exerts its immunosuppressive activity. Simple application of extracellular gal-1 inhibitors may fail to provide significant clinical improvements in humans with malignant glioma. Aside from its extracellular functions, gal-1 also appears to play an important role in the suppression of intracellular inflammatory factors through a dampening of the uncoupled protein response (UPR) (i.e., ER stress response) as demonstrated by others.Citation54 Our own work supports this view by showing that gal-1-deficient GL26 cells exhibit increased inflammatory cytokine production, a potential indicator of heightened cell stress. The observation of elevated levels of proinflammatory cytokines produced by gal-1-deficient glioma cells may therefore be explained by an associated increase in intracellular stress. Assuming this to be a correct assumption, one would reasonably expect that further reductions in tumor-derived gal-1 (greater than the ∼50% decrease at the population level demonstrated by usCitation32) would lead to corresponding increases in intracellular stress and further production of proinflammatory cytokines. A situation such as this would be expected to lead to stronger influx of inflammatory Gr-1+/CD11b+ myeloid cells and NK cells into the tumor microenvironment. Investigations are now underway in our laboratory to identify the inflammatory factors responsible for heightened innate immune recognition and clearance of gal-1-deficient glioma, and to understand the molecular interactions necessary for CCR2+/Ly-6C+/Gr-1+/CD11b+ myeloid cells to license antitumor NK activity.
Materials and methods
Animal strains
Eight to 10 week-old female C57BL/6J, B6.129S7-Rag1tm1Mom/J (i.e., RAG1−/−) and B6.129(Cg)-Ccr2tm2.1Ifc/J (i.e., B6.CCR2rfp/rfp) mice were purchased from the Jackson Laboratory. RA/EGxdelCre mice were kindly provided by Angelika Bierhaus of the Department of Internal Medicine I, University of Heidelberg (Heidelberg, Germany). All animal experiments were conducted in accordance with procedures approved by the University Committee on Use and Care of Animals (UCUCA) and conformed to the policies and procedures of the Unit for Laboratory Animal Medicine (ULAM) at the University of Michigan.
Flow cytometry antibodies
The following fluorochrome-conjugated flow cytometric antibodies were used throughout this work (each used at a 1:100 dilution): Alexa Fluor 700-conjugated rat anti-mouse CD45 (clone: 30-F11), Cat#: 103128, Biolegend; PE-conjugated rat anti-mouse Gr-1 (clone:RB6-8C5), Cat#: 553128, BD Pharmigen; PerCP/Cy5.5-conjugated rat anti-mouse CD11b (clone: M1/70), Cat#: 101228, Biolegend; Pacific Blue-conjugated hamster anti-mouse CD3ε (clone: 500A2), Cat#: 558214, BD PharMingen; APC-conjugated mouse anti-mouse NK1.1 (clone: PK136), Cat#: 17–5941-82, eBioscience; Pacific Blue-conjugated mouse anti-mouse granzyme B (clone: GB11), Cat#: 515403, Biolegend; Pacific Blue-conjugated mouse IgG1, κ (clone: MOPC-21) isotype control, Cat#: 400151, Biolegend; APC-conjugated rat anti-mouse Ly-6C (Clone: AL-21), Cat#: 560595, BD Pharmingen; APC-conjugated rat IgM, κ isotype control (clone: RTK2118), Cat#: 400810, Biolegend; APC-conjugated Armenian Hamster anti-mouse CD11c (clone: N418), Cat#: 117310, Biolegend; APC-conjugated Armenian Hamster IgG isotype control (clone: eBio299Arm), Cat#: 17–4888-81, eBioscience; PE-conjugated rat anti-mouse F4/80 (clone: BM8), Cat#: 123109, Biolegend; PE-conjugated rat IgG2a, κ isotype control (clone: eBR2a), Cat#: 12–4321-80, eBioscience.
Immunohistochemistry antibodies
PE-conjugated rat anti-mouse Gr-1 (clone: RB6-8C5) (1:500), Cat#: 553128, BD Pharmigen, conjugated to polyclonal rabbit anti-rat immunoglobulins/biotinylated secondary antibodies (1:1,000), Cat#: E0468, Dako; Pacific Blue-conjugated mouse anti-human/mouse granzyme B (clone: GB11), Cat#: 515408, Biolegend, conjugated to polyclonal rabbit anti-mouse immunoglobulins/biotinylated secondary antibodies (1:1,000), Cat#: E0464, Dako; PerCP/Cy5.5-conjugated-rat anti-mouse Ly-6G (clone: 1A8) (1:500), Cat#: 127616, Biolegend, conjugated to Alexa Fluor® 594-conjugated goat anti-rat IgG (H+L) secondary antibodies (1:1,000), Cat#: A-11007, Life Technologies; APC-conjugated rat anti-mouse Ly-6C (clone: AL-21) (1:500), Cat#: 560595, BD Pharmigen, conjugated to Alexa Fluor® 594-conjugated goat anti-rat IgG (H+L) secondary antibodies (1:1,000). No surfactants or antigen retrieval steps were used in any immunohistochemical immunolabeling procedures.
Imaging modalities
Fluorescence and bright-field micrographs were taken with a Zeiss Axioplan-2 microscope equipped with a digital camera (Carl Zeiss MicroImaging, Inc.) and Axiovision Release 4.6 analysis software. Fluorescence scanning confocal micrographs were taken with a Leica DMIRE2 confocal microscope equipped with Leica Confocal Software version 2.61 (Leica Microsystems). Fluorescence channels were scanned sequentially to reduce inter-channel bleed.
Quantifying tumor size from PFA-fixed brain tissue sections
PFA fixed mouse brains were coronally sectioned 50 μm thick using a vibratome. Every sixth section was placed into the same well of a 12-well plate (only 6-wells of the plate were occupied per mouse brain). The contents of an entire well was then extracted and mounted on a glass microscope slide and coverslipped using Prolong Gold anti-fade reagent. Fluorescence images of all brain tissue sections containing an aspect of the mCitrine+ GL26 gliomas were taken using a 5× objective. Micrographs were then imported into ImageJ analytical software (National Institutes of Health, Bethesda, MD) and processed according to the following method: I. Image > Type > 8-bit. II. Image > Adjust > Threshold > Apply (an arbitrary threshold was chosen for each experiment; however, the threshold was not altered over the course of analyzing tissue from any one particular experiment). III. Process > Binary > Make Binary. IV. Analyze > Measure. The area output of each measurement was then summed to afford an estimate of the overall tumor size.
In vivo immunodepletion antibodies
The following antibodies were administered intraperitoneally for immunodepletion in RAG1−/− mice (per mouse): 500 μg of purified rat anti-mouse Gr-1 (Clone: RB6-8C5), Cat#: BE0075, Bio X Cell; 600 μg of rat anti-mouse Ly-6G (Clone:1A8), Cat#: BE0075-1, Bio X Cell. Both antibodies were diluted to a final volume of 200 μL in sterile DPBS and were administered one day before tumor implantation, then once every 4 d. Control mice received non-specific rat IgG immunoglobulins (Equitech-Bio Inc.) at an equivalent molar dose and volume.
Flow cytometric analysis
All analysis was performed using FlowJo analysis software v10.0.7 (Tree Star, Inc.).
Statistical analysis
Statistical analyses were performed using GraphPad Prism5 (GraphPad Software, Inc.). All data are reported as the mean ± SEM and were examined with the statistical tests specified throughout the results section or associated figure legend. Biological replicates of each experiment are reported in the associated figure legend. Values were considered significant at the p ≤0.05 level.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
590 G.J.B. and P.R.L. designed research; G.J.B., P.C. and D.Z. performed research; G.J.B., P.C., M.G.C. and P.R.L. analyzed data; G.J.B., M.G.C. and P.R.L. wrote the paper.
KONI_A_1163461_s02.docx
Download MS Word (48.8 KB)Funding
This work was supported by National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) grants R01-NS074387, R01-NS057711 and R21-NS091555 to M.G.C.; and NIH/NINDS grants R01-NS061107, R01-NS076991, R01-NS082311 and R21-NS084275 to P.R.L. The authors are thankful for the academic leadership and support received from Dr. Karin Muraszko and the Department of Neurosurgery; to M. Dahlgren, D. Tomford and S. Napolitan for superb administrative support; and to M. Dzaman for outstanding technical assistance.
References
- Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 2014; 16 Suppl 4:iv1-63; PMID:25304271; http://dx.doi.org/10.1093/neuonc/nou223
- Waziri A. Glioblastoma-derived mechanisms of systemic immunosuppression. Neurosurg Clin N Am 2010; 21:31-42; PMID:19944964; http://dx.doi.org/10.1016/j.nec.2009.08.005
- Hegde M, Bielamowicz KJ, Ahmed N. Novel approaches and mechanisms of immunotherapy for glioblastoma. Discov Med 2014; 17:145-54; PMID:24641957
- Weller M. Immunotherapy for glioblastoma: a long and winding road. Neuro Oncol 2010; 12:319; PMID:20308309; http://dx.doi.org/10.1093/neuonc/noq027
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499-506; PMID:19342621; http://dx.doi.org/10.4049/jimmunol.0802740
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, Garcia J, Vogelbaum MA, Finke J. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol 2011; 13:591-9; PMID:21636707; http://dx.doi.org/10.1093/neuonc/nor042
- Kohanbash G, Okada H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol Invest 2012; 41:658-79; PMID:23017140; http://dx.doi.org/10.3109/08820139.2012.689591
- Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother 2006; 55:237-45; PMID:16047143; http://dx.doi.org/10.1007/s00262-005-0048-z
- Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev 2008; 222:180-91; PMID:18364002; http://dx.doi.org/10.1111/j.1600-065X.2008.00608.x
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 2007; 13:828-35; PMID:17603493; http://dx.doi.org/10.1038/nm1609
- Yu J, Du W, Yan F, Wang Y, Li H, Cao S, Yu W, Shen C, Liu J, Ren X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol 2013; 190:3783-97; PMID:23440412; http://dx.doi.org/10.4049/jimmunol.1201449
- Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 2009; 182:5693-701; PMID:19380816; http://dx.doi.org/10.4049/jimmunol.0900092
- Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 2007; 179:977-83; PMID:17617589; http://dx.doi.org/10.4049/jimmunol.179.2.977
- Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol 2009; 182:240-9; PMID:19109155; http://dx.doi.org/10.4049/jimmunol.182.1.240
- Elkabets M, Ribeiro VS, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, Vosshenrich CA. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol 2010; 40:3347-57; PMID:21110318; http://dx.doi.org/10.1002/eji.201041037
- Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008; 111:4233-44; PMID:18272812; http://dx.doi.org/10.1182/blood-2007-07-099226
- Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 2008; 181:5791-802; PMID:18832739; http://dx.doi.org/10.4049/jimmunol.181.8.5791
- Rodrigues JC, Gonzalez GC, Zhang L, Ibrahim G, Kelly JJ, Gustafson MP, Lin Y, Dietz AB, Forsyth PA, Yong VW et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol 2010; 12:351-65; PMID:20308313; http://dx.doi.org/10.1093/neuonc/nop023
- Chae M, Peterson TE, Balgeman A, Chen S, Zhang L, Renner DN, Johnson AJ, Parney IF. Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro Oncol 2014; 17:978-91; PMID:25537019; http://dx.doi.org/10.1093/neuonc/nou343
- Kohanbash G, McKaveney K, Sakaki M, Ueda R, Mintz AH, Amankulor N, Fujita M, Ohlfest JR, Okada H. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res 2013; 73:6413-23; PMID:24030977; http://dx.doi.org/10.1158/0008-5472.CAN-12-4124
- Zou JP, Morford LA, Chougnet C, Dix AR, Brooks AG, Torres N, Shuman JD, Coligan JE, Brooks WH, Roszman TL et al. Human glioma-induced immunosuppression involves soluble factor(s) that alters monocyte cytokine profile and surface markers. J Immunol 1999; 162:4882-92; PMID:10202033
- Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 2013; 19:3165-75; PMID:23613317; http://dx.doi.org/10.1158/1078-0432.CCR-12-3314
- Gustafson MP, Lin Y, New KC, Bulur PA, O'Neill BP, Gastineau DA, Dietz AB. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol 2010; 12:631-44; PMID:20179016; http://dx.doi.org/10.1093/neuonc/noq001
- Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 2010; 12:1113-25; PMID:20667896; http://dx.doi.org/10.1093/neuonc/noq082
- Earl LA, Bi S, Baum LG. N- and O-glycans modulate galectin-1 binding, CD45 signaling, and T cell death. J Biol Chem 2010; 285:2232-44; PMID:19920154; http://dx.doi.org/10.1074/jbc.M109.066191
- Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature 1995; 378:736-9; PMID:7501023; http://dx.doi.org/10.1038/378736a0
- Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, Mordoh J, Fainboim L, Podhajcer OL, Rabinovich GA. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell 2004; 5:241-51; PMID:15050916; http://dx.doi.org/10.1016/S1535-6108(04)00024-8
- Banh A, Zhang J, Cao H, Bouley DM, Kwok S, Kong C, Le QT. Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res 2011; 71:4423-31; PMID:21546572; http://dx.doi.org/10.1158/0008-5472.CAN-10-4157
- Rostoker R, Yaseen H, Schif-Zuck S, Lichtenstein RG, Rabinovich GA, Ariel A. Galectin-1 induces 12/15-lipoxygenase expression in murine macrophages and favors their conversion toward a pro-resolving phenotype. Prostaglandins Other Lipid Mediat 2013; 107:85-94; PMID:23954858; http://dx.doi.org/10.1016/j.prostaglandins.2013.08.001
- Starossom SC, Mascanfroni ID, Imitola J, Cao L, Raddassi K, Hernandez SF, Bassil R, Croci DO, Cerliani JP, Delacour D et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity 2012; 37:249-63; PMID:22884314; http://dx.doi.org/10.1016/j.immuni.2012.05.023
- Baker GJ, Chockley P, Yadav VN, Doherty R, Ritt M, Sivaramakrishnan S, Castro MG, Lowenstein PR. Natural killer cells eradicate galectin-1-deficient glioma in the absence of adaptive immunity. Cancer Res 2014; 74:5079-90; PMID:25038230; http://dx.doi.org/10.1158/0008-5472.CAN-14-1203
- Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, Fong L, Campbell MJ, Cooper S, Oakes SA et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci U S A 2014; 111:12823-8; PMID:25136121; http://dx.doi.org/10.1073/pnas.1413933111
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011; 11:762-74; PMID:21984070; http://dx.doi.org/10.1038/nri3070
- Baker GJ, Yadav VN, Motsch S, Koschmann C, Calinescu AA, Mineharu Y, Camelo-Piragua SI, Orringer D, Bannykh S, Nichols WS et al. Mechanisms of glioma formation: iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 2014; 16:543-61; PMID:25117977; http://dx.doi.org/10.1016/j.neo.2014.06.003
- Baker GJ, Castro MG, Lowenstein PR. Isolation and Flow Cytometric Analysis of Glioma-infiltrating Peripheral Blood Mononuclear Cells. J Vis Exp. 2015 Nov 28; (105); PMID:26650233; http://dx.doi.org/10.3791/53676
- Jaeger BN, Donadieu J, Cognet C, Bernat C, Ordonez-Rueda D, Barlogis V, Mahlaoui N, Fenis A, Narni-Mancinelli E, Beaupain B et al. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J Exp Med 2012; 209:565-80; PMID:22393124; http://dx.doi.org/10.1084/jem.20111908
- Sporri R, Joller N, Hilbi H, Oxenius A. A novel role for neutrophils as critical activators of NK cells. J Immunol 2008; 181:7121-30; PMID:18981133; http://dx.doi.org/10.4049/jimmunol.181.10.7121
- Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood 2008; 112:4080-9; PMID:18753637; http://dx.doi.org/10.1182/blood-2008-03-143776
- Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 2012; 37:549-62; PMID:22940097; http://dx.doi.org/10.1016/j.immuni.2012.05.029
- Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol 2004; 173:6418-26; PMID:15528382; http://dx.doi.org/10.4049/jimmunol.173.10.6418
- Lapaque N, Walzer T, Meresse S, Vivier E, Trowsdale J. Interactions between human NK cells and macrophages in response to Salmonella infection. J Immunol 2009; 182:4339-48; PMID:19299734; http://dx.doi.org/10.4049/jimmunol.0803329
- Nedvetzki S, Sowinski S, Eagle RA, Harris J, Vely F, Pende D, Trowsdale J, Vivier E, Gordon S, Davis DM. Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood 2007; 109:3776-85; PMID:17218381; http://dx.doi.org/10.1182/blood-2006-10-052977
- Atochina O, Harn D. LNFPIII/LeX-stimulated macrophages activate natural killer cells via CD40-CD40L interaction. Clin Diagn Lab Immunol 2005; 12:1041-9; PMID:16148169; http://dx.doi.org/10.1128/CDLI.12.9.1041-1049.2005
- Chijioke O, Munz C. Interactions of human myeloid cells with natural killer cell subsets in vitro and in vivo. J Biomed Biotechnol 2011; 2011:251679; PMID:21541250; http://dx.doi.org/10.1155/2011/251679
- Kijima M, Yamaguchi T, Ishifune C, Maekawa Y, Koyanagi A, Yagita H, Chiba S, Kishihara K, Shimada M, Yasutomo K. Dendritic cell-mediated NK cell activation is controlled by Jagged2-Notch interaction. Proc Natl Acad Sci U S A 2008; 105:7010-5; PMID:18458347; http://dx.doi.org/10.1073/pnas.0709919105
- Schmitz M, Zhao S, Deuse Y, Schakel K, Wehner R, Wohner H, Hölig K, Wienforth F, Kiessling A, Bornhäuser M et al. Tumoricidal potential of native blood dendritic cells: direct tumor cell killing and activation of NK cell-mediated cytotoxicity. J Immunol 2005; 174:4127-34; PMID:15778372; http://dx.doi.org/10.4049/jimmunol.174.7.4127
- Valteau-Couanet D, Leboulaire C, Maincent K, Tournier M, Hartmann O, Benard J, Beaujean F, Boccaccio C, Zitvogel L, Angevin E. Dendritic cells for NK/LAK activation: rationale for multicellular immunotherapy in neuroblastoma patients. Blood 2002; 100:2554-61; PMID:12239169; http://dx.doi.org/10.1182/blood.V100.7.2554
- Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med 2012; 209:139-55; PMID:22231304; http://dx.doi.org/10.1084/jem.20101387
- Watanabe S, Deguchi K, Zheng R, Tamai H, Wang LX, Cohen PA, Shu S. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J Immunol 2008; 181:3291-300; PMID:18714001; http://dx.doi.org/10.4049/jimmunol.181.5.3291
- Shime H, Kojima A, Maruyama A, Saito Y, Oshiumi H, Matsumoto M, Seya T. Myeloid-derived suppressor cells confer tumor-suppressive functions on natural killer cells via polyinosinic:polycytidylic acid treatment in mouse tumor models. J Innate Immun 2014; 6:293-305; PMID:24192491; http://dx.doi.org/10.1159/000355126
- Thaci B, Ahmed AU, Ulasov IV, Wainwright DA, Nigam P, Auffinger B, Tobias AL, Han Y, Zhang L, Moon KS et al. Depletion of myeloid-derived suppressor cells during interleukin-12 immunogene therapy does not confer a survival advantage in experimental malignant glioma. Cancer Gene Ther 2014; 21:38-44; PMID:24434573; http://dx.doi.org/10.1038/cgt.2013.81
- Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer 2013; 13:739-52; PMID:24060865; http://dx.doi.org/10.1038/nrc3581
- Le Mercier M, Lefranc F, Mijatovic T, Debeir O, Haibe-Kains B, Bontempi G, Decaestecker C, Kiss R, Mathieu V. Evidence of galectin-1 involvement in glioma chemoresistance. Toxicol Appl Pharmacol 2008; 229:172-83; PMID:18313712; http://dx.doi.org/10.1016/j.taap.2008.01.009
- Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, Guenterberg K, Kondadasula SV, Chaudhury AR, La Perle KM et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res 2011; 71:5101-10; PMID:21680779; http://dx.doi.org/10.1158/0008-5472.CAN-10-2670