
ABSTRACT
Tumor exome and RNA sequencing data provide a systematic and unbiased view on cancer-specific expression, over-expression, and mutations of genes, which can be mined for personalized cancer vaccines and other immunotherapies. Of key interest are tumor-specific mutations, because T cells recognizing neoepitopes have the potential to be highly tumoricidal. Here, we review recent developments and technical advances in identifying MHC class I and class II-restricted tumor antigens, especially neoantigen derived MHC ligands, including in silico predictions, immune-peptidome analysis by mass spectrometry, and MHC ligand validation by biochemical methods on T cells.
Introduction
T cells recognize tumor antigens (TA) as short peptides presented on MHC class I and II molecules (MHC I and II). TA can induce cancer-specific T cell immunity and comprise (1) Tumor-specific TA containing mutations that are specific for a given tumor.Citation1-3 (2) Shared TA, which are expressed on different tumors and can be divided in over-expressed and differentiation TA. (3) Cancer testis TA expressed on testis and cancer cells (http://cancerimmunity.org/peptide/). Comprehensive identification of cancer MHC ligands, especially immunogenic ones, is a challenging task due to the high polymorphism of MHC molecules, the vast diversity of TA, the difficulty to model antigen processing and presentation, and predict immunogenicity.
Many tumors are immunogenic, i.e. they induce adaptive immunity, and are infiltrated by T ells recognizing TA that can be used for adoptive T cell therapy (ACT).Citation5-7 In the tumor environment T cells tend to be suppressed, which is a major obstacle in cancer immunotherapy and can be reversed by checkpoint blockade, e.g. blocking of PD-1 and/or CTLA-4.Citation8 Among tumor infiltrating T cells, some recognize neoepitopes. Remarkably these cells expand upon checkpoint blockade and can be highly tumoricidal.Citation2,8-11 Neoepitope specific T cells usually have low frequencies due to cancer immunoediting.Citation2,12-14 However, they are attractive for ACT, because they are exquisitely tumor-specific and exhibit superior tumor control, presumably because they are not subject to central tolerance and hence may express high-affinity TCR.Citation11,12,Citation15,16 Even though tumor cells usually express no MHC II molecules, many tumor-infiltrated lymphocytes (TIL) are CD4+ T cells specific for neoepitopes and important players in immunotherapy.Citation7,10,Citation17
Immunogenic tumors are under pressure to evade immune destruction and some escape mechanisms involve aberrant TA presentation and processing, e.g., altered proteasomal degradation, RNA splicing, and/or post-translational modifications.Citation18-22 Most cancer cells express low levels of MHC I and no II molecules. However, they are prone to cell death and as a result can be taken up by antigen presenting cells (APC), namely DC and macrophages, which (cross)-present exogenous TA on their MHC molecules and induce TA-specific CD8+ and CD4+ T cells.Citation2,10,Citation23 Moreover, MHC restriction adds complexity to antigen presentation, e.g. by the polymorphism of MHC molecules. In humans, all cells express up to six HLA class I molecules and professional APC eight or more HLA II molecules, which in view of the >3,000 HLA alleles affords a vast diversity.Citation21,22
Whole exome and transcriptome NGS sequencing data provide detailed information on TA mutations, expression levels, aberrant mRNA splicing, and MHC types and can be mined for precise targeting of cancer immunotherapies, thus greatly increasing their efficacy.Citation2,4,Citation6,11 This calls for new high-throughput technologies that allow comprehensive identification of cancer-specific MHC ligands, T cell epitopes, especially neoepitopes and their MHC restrictions. Here, we review established technologies, present emerging new ones and discuss future perspectives.
Computational approaches to predict TA-derived MHC ligands
Detailed analysis of genomic and transcriptomic changes in tumors provides unprecedented opportunities to map differences between cancer and normal cells that are perceptible to the immune system. The recently observed correlation between the efficacy of checkpoint blockade and the mutational load of the patient's tumors triggered a broad interest in predicting and validating T cell neoepitopes.Citation1,2,Citation7-10,Citation17,24-26 Studies typically relied in a first part on computational approaches predicting mutation-containing MHC ligands.
Cancer exome and transcriptome analyses
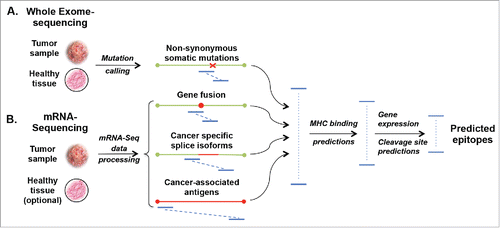
To identify genomic alterations resulting in amino acid changes, cancer exome sequencing is performed. For each patient, a tumor sample and a matched healthy tissue are sequenced and somatic variants are identified by comparing the differences between the two samples (). If coverage is sufficient, the majority of single nucleotide variants can be identified.Citation27 However, variants present in only a small fraction of cancer cells or in highly polymorphic regions may be missed and efforts to improve mutation-calling pipelines are still ongoing.Citation28,29 In addition, RNA sequencing allows for the identification of gene products selectively expressed by cancer cells like TA, cancer-specific splice variants, or gene fusions (). A major challenge in analyzing exome and RNA sequencing data consists in identifying cancer-specific changes relevant for T cell tumor immunity.
Figure 1. Computational approaches to identify cancer-specific MHC ligands. Comparing exome sequencing data (A) from tumor and healthy tissues allows identification of non-synonymous mutations. From mRNA sequencing (B) cancer-specific gene fusions, aberrant RNA specific, and expressed/overexpressed TA genes can be identified by comparison with databases of splice isoforms and healthy tissues. All possible peptides (blue lines) containing at least one cancer-specific amino acid mutation are then subjected to MHC-binding peptide predictions. For MHC I molecules 8- to 11-mer and for MHC II 14- to 20-mer peptides are considered for predictions. Predicted peptides are ranked based on their predicted binding affinity and possibly other variables, like immunogenicity, pMHC complex stability, predicted cleavage sites or level of expression of the corresponding genes.
Cancer MHC I ligand prediction
To elicit T cell immunity genetic variants must result in modified peptides that can be presented on MHC molecules. Different computational methods have been developed to predict peptide binding affinity to MHC molecules. MHC I molecules present usually 8–11 residue peptides to cytotoxic CD8+ T-lymphocytes (CTL) and the peptide's N- and C-termini are involved in MHC binding.Citation21 Therefore, MHC ligands of equal length can be naturally aligned and predictive models of binding affinity trained by identifying specific sequence motifs across known ligands for given MHC I alleles.Citation30 More modern approaches involve machine learning techniques, e.g., neural networks like NetMHC,Citation31,32 hidden Markov models or support vector machines that consider more intricate sequence patterns among peptide ligandsCitation33-35and peptides of different lengths.Citation36 Different in silico MHC ligand prediction algorithms can be combined to focus on peptides predicted by multiple strategies.Citation2,9,Citation37
These algorithms are trained on large datasets of peptide ligands such as those collected in IEDB database.Citation37 Therefore, they work best for HLA alleles with many known ligands. As the number of known HLA alleles expands, algorithms such as NetMHCpan have been developed to predict peptides binding to alleles for which no ligands are known.Citation38 These methods rely on HLA allele sequence homology and correlations between the HLA allele sequences and amino acid preferences in their ligands. Approaches integrating pMHC complex structures have been proposed.Citation39 However, including such information infers longer computation times and provides only modest improvement of prediction accuracy.Citation40
Most algorithms predict the affinity of peptide binding to MHC molecules, which may not correlate well with their immunogenicity, i.e., ability to elicit T cell immunity, which has been reported to correlate better with pMHC complex stability.Citation41 Moreover, cancer cells may not present predicted peptides, e.g. due to aberrant TA processing and/or presentation.Citation21 Different approaches have been employed to improve predictions, like filtering out low-expression genes, incorporating cleavage site, and peptide transport predictionsCitation42,43 or structural differences between mutant and wild-type peptides ().Citation25 However, it is challenging to adequately combine these parameters, which individually have only a poor predictive value for immunogenicity. Future developments may benefit from combining machine learning approaches with high-throughput validation and larger data sets of cancer-specific MHC ligands and T cell epitopes.
Cancer MHC II ligand prediction
The recognition that a substantial fraction of TIL are tumor-specific CD4+ T cells and that such T cells play important roles in tumor controlCitation7,10,Citation17 has motivated development of computational approaches for MHC II ligand predictions. This is challenging, because peptide binding to MHC II as compared to MHC I molecules is more promiscuous in terms of peptide length, binding sequence motifs, and binding registers; therefore, peptides need to be aligned first, which is often difficult.Citation44 In addition, except for HLA-DR molecules, the α and β chains are polymorphic, which dramatically increases the diversity of possible peptide binding specificity. To address these issues, different strategies have been proposed, like refining the alignment algorithms Citation33 or predicting peptide binding cores.Citation45 Several online servers are available for MHC II ligand predictions.Citation33,46,Citation47 In general in silico prediction of ligands for MHC II is less accurate than for MHC molecules.Citation45
The number of non-self MHC ligands predicted by in silico methods is typically vastly larger than the one for which T cell reactivity can be detected in cancer patients.Citation2-4, 26 Depending on the methods used for detection, some immunogenic ligands may escape detection, however, most of the predicted ligands are either not immunogenic or are not generated and presented.Citation1,17-21,Citation24,Citation48 In addition, inaccuracies of in silico predictions and lack of good correlations between pMHC complex stability, peptide binding affinity, and immunogenicity necessitate the use of generous cut-offs and calls for high-throughput in vitro validation.Citation41,48
MHC ligand identification and validation
MHC class I peptide binding validation
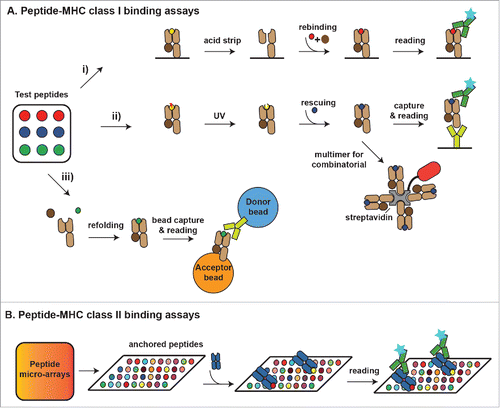
To reduce the number of in silico predicted MHC ligands, their MHC-binding affinities and complex stabilities are measured. Biochemical methods used to measure MHC peptide binding include: (1) A peptide-rebinding assay, referred to as iTopia, in which immobilized pMHC complexes containing an irrelevant peptide are acid stripped and upon re-incubation with test peptides and β2m newly formed pMHC complexes quantified by means of a conformational anti-HLA class I mAb, e.g.,W6/32 ().Citation49 (2) A peptide-rescuing assay in which pMHC complexes containing a photo-cleavable peptide are UV irradiated in the presence of test peptides, which depending on their MHC-binding strength can rescue the “empty” MHC molecule.Citation50,51 This method allows the generation of large numbers of different pMHC monomers and thus facilitates combinatorial multimer staining.Citation52,53 (3) A refolding assay in which denatured heavy and light chains are refolded in the presence of test peptides and pMHC complex formation is measured by means of the conformation-dependent anti-pan HLA class I antibody, W6/32, using a proximity-based immunoassay ().Citation41,54,Citation55 Data generated by this assay were used for training of prediction algorithms.Citation56, 57 It has been reported that pMHC complexes stability, as measured by a scintillation proximity assay, better correlates with immunogenicity than binding affinity.Citation41,58
Figure 2. MHC peptide binding assays. (A) Widely used MHC I peptide binding assays include the following: (i) Rebinding of test peptide (red dot) and β2m (brown dot) after acid strip of immobilized pMHC containing an irrelevant peptide (yellow dot). The newly formed complexes are quantified by means of a conformational mAb (green Y) containing a fluorescent label (blue star). (ii) Rescuing with test peptides (blue dot) of UV irradiated complexes containing a photocleavable peptide. The newly formed complexes can be captured and quantified or used for preparing combinatorial multimer libraries. (iii) Test peptide-driven refolding of β2m and biotinylated MHC I heavy chain is quantified by a luminescent oxygen channeling immunoassay reporting the proximity of an pMHC bearing acceptor bead and an acceptor bead coated with conformational pan-MHC I mAb. (B) Peptide-MHC class II binding assay using peptide microarray. Peptides (15 mer) (colored dots) synthesized on a microarray chip and incubated with receptive soluble MHC II molecules (blue dimers) and after washing peptide bound MHC II are quantified by means of a fluorescently labeled (blue star) conformational mAb (green Y) as described for A.
Peptide binding to MHC I molecules has also been assessed on cells by MHC stabilization assay on transporter associated with antigen presentation (TAP) deficient cells (e.g. T2 or RMA-S). However, such assays have limited throughput and limited accuracy due to overlay of peptide loading, competition, and MHC stabilization.Citation59
MHC II peptide binding validation
Like for MHC class I molecules, peptide binding to MHC II molecules can be assessed by peptide-driven refolding of α and β chains containing preformed intra-chain disulfide bonds.Citation60 Alternatively, using insect cell expression systems, soluble “empty” MHC II molecules can be produced and peptide binding assessed by means of biotin or fluorochrome labeled peptides.Citation61,62 MHC II-peptide complex stability can be measured via the disappearance of the tagged peptide over time. Moreover, because MHC II molecules have an open-ended peptide-binding site, they can bind to immobilized peptides, allowing the use of peptide microarrays for MHC class II peptide screening by incubating these with “empty” class II molecules and detection with anti-MHC II mAb ().Citation63 Peptide microarrays are comparatively inexpensive and having up to two millions peptides/chips, are attractive for high throughput screening.Citation64
Cancer immune-peptidome analysis
In silico MHC ligand prediction combined with biochemical validation is prone to identify peptides that are not expressed and not presented on cancer cells or fail to identify peptides containing post-translational modifications (e.g., phosphorylation) or sequences altered by proteasomal reverse splicing.Citation19-21 These caveats are circumvented by immunopeptidomics, a technology in which MHC molecules are isolated from cells, their peptide cargo isolated and analyzed by liquid chromatography and mass spectrometry (LC-MS).Citation65
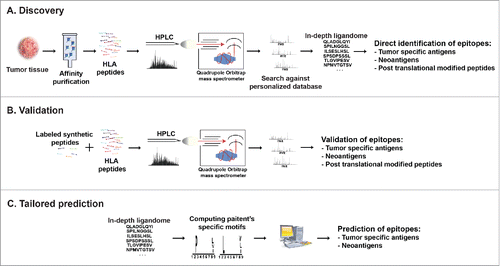
The best-established strategy for isolating HLA peptides is immunoaffinity purification of HLA-peptide complexes and recovery of their peptides (). Usually, pan anti-HLA I or II antibodies are used and peptides associated with all HLA molecules isolated and analyzed. Typically 1–5 × 108 cells from cell lines or one gram of tumor tissue are needed for in-depth immunopeptidome analysis, resulting in identification of thousands of peptides.Citation66-70 Remarkably detailed immunopeptidomes were determined on soluble HLA-peptide complexes isolated from plasma.Citation71 This methodology highly enriches HLA peptides, the vast majority (~95%) of which matches the binding motifs of the corresponding HLA molecules.Citation66,72
Figure 3. Cancer peptidome analysis. (A) Overview of immunopeptidome MHC ligand discovery. HLA molecules are isolated from cells, their peptide cargo isolated and analyzed by LC-MS. Matching the MS data to customized reference databases that include information of somatic mutations allows direct identification of MHC ligands derived from TA including those containing post-translational modifications. (B) For validation and quantification synthetic peptides labeled with heavy isotopes spiked into the eluted peptidome sample permit validation of in vivo presented MHC ligands. (C) Patient's-specific HLA peptide binding motifs can be resolved from in-depth ligandome data sets and may be used to predict private tumor-specific MHC ligands.
The remarkable sensitivity and accuracy of LC-MS analysis allows detailed analysis of immune-peptidomes. Three major MS data acquisition methods are available for HLA peptides: (1) targeted acquisitionCitation9,73-75; (2) data dependent acquisition, referred to as discovery or shotgun approach Citation66,67,Citation72,74,Citation76; and (3) data-independent acquisition, namely SWATH-MS-based acquisition.Citation65 In general, these methods allow identification of T cell (neo-) MHC ligands, including those containing post-translational modifications.Citation72,77
For identification of HLA eluted peptides MS/MS spectra are matched to theoretical spectra of peptide sequences in databases using search engines like MascotCitation78 or MaxQuant.Citation79 Generation of customized databases from genomic and transcriptomic information Citation70 allowed identification of private peptides that are not present in reference protein sequence databases. Recently, a targeted approach permitted identification and validation of two predicted neoepitopes from sarcoma cell lines.Citation10 Targeted MS is a highly reproducible and sensitive method, but is limited to hundreds of peptides. MS analysis of synthetic peptides guide selection of optimal transitions that can then be monitored in eluted peptide samples.Citation74 Conversely, using the discovery approach, seven MHC ligands were identified from a mouse cancer cell line, one of which was immunogenic and upon vaccination shown to control tumor growth.Citation25 It is expected that identification of mutation containing MHC ligands by the discovery approach from cancer tissues will become more efficient upon improvement of purification of HLA peptides, LC-MS technology, and bioinformatics algorithms.
The probability of identifying mutation-containing HLA ligands increases with the depth of the ligandome and the mutational content of the sample. Even in an in-depth analysis resulting in accurate identification of thousands of HLA peptides, only a few neoepitopes are expected. Stringent control over false discovery rate (FDR) and peptide spectrum match (PSM) scoring ensures accurate and reliable results; typically one to five percent FDR thresholds are applied to peptidomic datasets using the target-decoy approach.Citation80,81Inclusion of exhaustive lists of known mutations from repositories like the TCGA or COSMIC will increase the level of false positives and therefore better-personalized reference databases should be used. To quantify mutation containing HLA ligands, the sample can be spiked with synthetic peptides labeled with stable heavy isotopes ().Citation74 It is also recommended to resequence PCR amplicons of the mutated loci to confirm the mutation(s).
In depth interrogation of HLA II peptidomes by immune-peptidomic is straightforward, however, since HLA II molecules are typically expressed on immune cells infiltrating the tumor and only rarely on cancer cells, MS analysis may not be sensitive enough to detect peptides presented on APC in the tumor microenvironment. However, class II peptidomes can be obtained from patient's tissues or PBMC. From the repertoire of thousands detected MHC II peptides, specific binding motifs can be identified, similar as was shown for class I.Citation66,76,Citation80 As current tools for MHC II peptide binding predictions are relatively inaccurate, predictions based on these motifs may improve identification of patient-specific, mutation containing MHC II ligands ().
Conclusions and perspectives
Presently there exists no single method that allows comprehensive, truthful, and high throughput TA T cell epitope identification. Correlations between the immunogenicity of a peptide, its MHC-binding affinity and pMHC complex stability tend to be poor.Citation82-85 Each method has its advantages and shortcomings and depending on needs and means, different strategies for TA T cell epitope discovery are indicated. Bioinformatics sequencing data analysis, identification of mutated and overexpressed genes, and prediction of MHC ligands constitute a crucial initial part. For their validation three main strategies are used, each having its advantages and shortcomings: (1) Biochemical MHC ligand validation is tedious and costly in high throughput mode and is prone to identify peptides that are not presented by cancer cells or are not immunogenic. On the other hand, such data is valuable for training of prediction algorithms.Citation19-22,Citation52,85 (2) Immune-peptidome analysis has the big advantage to identify MHC ligands presented by cancer cells, including peptides containing post-translational modifications or altered sequences.Citation18-21 However, this analysis is prone to miss MHC ligands and provides no information on their immunogenicity. (3) T cell (neo)-epitope discovery using patient's T cells in functional assaysCitation1,10,Citation67,86-88 or combinatorial tetramer staining.Citation18,53,Citation89,90 These assays are reliable and provide information on patient's cancer-specific T cells, but they have limited throughput capabilities, e.g. because of the paucity of TILs, and especially neoepitope-specific T cells.Citation9,13 For personalized cancer vaccines based on TA peptides or corresponding RNA, in silico prediction and validation on patient's T cells is an established but cumbersome strategy.Citation1,7,Citation8,17,Citation86-88,Citation91 Alternatively, combination of in silico MHC ligand prediction and MS immune-peptidome analysis allowed identification of some cancer presented MHC ligands, but may miss poorly expressed ones.Citation25,67,Citation92,93
There is an urgent need to improve the accuracy of in silico TA MHC ligand predictions, especially for MHC II and for MHC I molecules for which little or no data exist. Moreover, poor correlations between binding affinity, pMHC complex stability, and immunogenicity motivate the development of machine-learning programs that integrate multiple parameters. The rapidly growing volume of data on validated TA presented on MHC molecules should support such efforts. Every progress in in silico MHC ligand prediction will save time and costs for TA epitope discovery. At the same time, high-content peptide validation assays need to be established that allow filtering out falsely predicted peptides and training of prediction algorithms. In view of recent progress in peptide microarray technology it may become possible to rapidly screen very high numbers of peptides binding to MHC II and perhaps also MHC I molecules.Citation63,64,Citation94 Alternatively, MS immune-peptidome analysis may be combined with UV irradiation induced exchange of conditional MHC ligands with libraries of predicted peptides.Citation49,50,Citation70 A powerful strategy of epitope discovery and T cell analysis consist in using peptide exchange combined with combinatorial multimer flow cytometry analysis of patient's TIL or PBMC.Citation5,52,Citation53,87 For more accurate selection of MHC ligands, this analysis can be combined with MS immune-peptidome analysis.Citation5, 50-53,Citation71,Citation87 By using mass cytometry the resolution of this technique can be further increased.Citation21 A promising perspective is pMHC multimer arrays allowing detection of hundreds of T cell specificities in one sample.Citation94 In view of the rapid progress in microfluidic multiplexing technologies the number of T cells that can be enumerated and analyzed is likely to increase dramatically in the near future.Citation95-97
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C et al. Exploiting the mutanome for tumor vaccination. Can Res 2012; 72:1081-91; PMID:22237626; http://dx.doi.org/10.1158/0008-5472.CAN-11-3722
- Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 2015; 125:3413-21; PMID:26258412; http://dx.doi.org/10.1172/JCI80008
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/10.1126/science.aaa4971
- van Buuren MM, Calis JJ, Schumacher TN. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. Oncoimmunology 2014; 3:e28836; PMID:25083320; http://dx.doi.org/10.4161/onci.28836
- Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, van Rooij N, Linnemann C, van Buuren MM, Urbanus JH et al. TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology 2012; 1:409-18; PMID:22754759; http://dx.doi.org/10.4161/onci.18851
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62-8; PMID:25838374; http://dx.doi.org/10.1126/science.aaa4967
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344:641-5; PMID:24812403; http://dx.doi.org/10.1126/science.1251102
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124-8; PMID:25765070; http://dx.doi.org/10.1126/science.aaa1348
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014; 515:577-81; PMID:25428507; http://dx.doi.org/10.1038/nature13988
- Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 2015; 21:81-5; PMID:25531942; http://dx.doi.org/10.1038/nm.3773
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/10.1126/science.aaa4971
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012; 482:400-4; PMID:22318521; http://dx.doi.org/10.1038/nature10755
- Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest 2015; 125:3981-91; PMID:26389673; http://dx.doi.org/10.1172/JCI82416
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015; 160:48-61; PMID:25594174; http://dx.doi.org/10.1016/j.cell.2014.12.033
- Tan MP, Gerry AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, Pumphrey NJ, Jakobsen BK, Price DA, Ladell K et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin Exp Immunol 2015; 180:255-70; PMID:25496365; http://dx.doi.org/10.1111/cei.12570
- Obenaus M, Leitão C, Leisegang M, Chen X, Gavvovidis I, van der Bruggen P, Uckert W, Schendel DJ, Blankenstein T. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat Biotechnol 2015; 33:402-7; PMID:25774714; http://dx.doi.org/10.1038/nbt.3147
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520:692-6; PMID:25901682; http://dx.doi.org/10.1038/nature14426
- Andersen RS, Andersen SR, Hjortsø MD, Lyngaa R, Idorn M, Køllgård TM, Met O, Thor Straten P, Hadrup SR. High frequency of T cells specific for cryptic epitopes in melanoma patients. Oncoimmunology 2013; 2:e25374; PMID:24073381; http://dx.doi.org/10.4161/onci.25374
- Berkers CR, de Jong A, Schuurman KG, Linnemann C, Meiring HD, Janssen L, Neefjes JJ, Schumacher TN, Rodenko B, Ovaa H. Definition of proteasomal peptide splicing rules for high-efficiency spliced peptide presentation by MHC class I molecules. J Immunol 2015; 195:4085-95; PMID:26401003; http://dx.doi.org/10.4049/jimmunol.1402455
- Dalet A, Robbins PF, Stroobant V, Vigneron N, Li YF, El-Gamil M, Hanada K, Yang JC, Rosenberg SA, Van den Eynde BJ. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc Natl Acad Sci U S A 2011; 108:E323-31; PMID:21670269; http://dx.doi.org/10.1073/pnas.1101892108
- Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst 2013; 105:1172-87; PMID:23852952; http://dx.doi.org/10.1093/jnci/djt184
- Newell EW. Higher throughput methods of identifying T cell epitopes for studying outcomes of altered antigen processing and presentation. Front Immunol 2013; 4:430; PMID:24367368; http://dx.doi.org/10.3389/fimmu.2013.00430
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015; 348:803-8; PMID:25837513; http://dx.doi.org/10.1126/science.aaa3828
- Kreiter S, Castle JC, Türeci O, Sahin U. Targeting the tumor mutanome for personalized vaccination therapy. Oncoimmunology 2012; 1:768-9; PMID:22934277; http://dx.doi.org/10.4161/onci.19727
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014; 515:572-6; PMID:25428506; http://dx.doi.org/10.1038/nature14001
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016; 35:1463-9; PMID:26940869; http://dx.doi.org/10.1126/science.aaf1490
- Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 2014; 15:121-32; PMID:24434847; http://dx.doi.org/10.1038/nrg3642
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015; 348:803-8; PMID:25837513; http://dx.doi.org/10.1126/science.aaa3828
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol 2015; 33:1152-58; PMID:26372948; http://dx.doi.org/10.1038/nbt.3344
- Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999; 50:213-9; PMID:10602881; http://dx.doi.org/10.1007/s002510050595
- Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 2003; 12:1007-17; PMID:12717023; http://dx.doi.org/10.1110/ps.0239403
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res 2008; 36:W509-12; PMID:18463140; http://dx.doi.org/10.1093/nar/gkn202
- Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics 2009; 10:296; PMID:19765293; http://dx.doi.org/10.1186/1471-2105-10-296
- Dönnes P, Elofsson, A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinformatics 2002; 3:25; PMID:12225620; http://dx.doi.org/10.1186/1471-2105-3-25
- Mamitsuka H. Predicting peptides that bind to MHC molecules using supervised learning of hidden Markov models. Proteins 1998; 33:460-74; PMID:9849933; http://dx.doi.org/10.1002/(SICI)1097-0134(19981201)33 :4<460::AID-PROT2>3.0.CO;2-M
- Andreatta M. Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 2015; PMID:26515819; http://dx.doi.org/10.1093/bioinformatics/btv639
- Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res 2015; 43:D405-12; PMID:25300482; http://dx.doi.org/10.1093/nar/gku938
- Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Røder G, Peters B, Sette A, Lund O et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2007; 2:e796; PMID:17726526; http://dx.doi.org/10.1371/journal.pone.0000796
- Antes I Siu SW, Lengauer T. DynaPred: a structure and sequence based method for the prediction of MHC class I binding peptide sequences and conformations. Bioinformatics 2006; 22, e16-24; PMID:16873467; http://dx.doi.org/10.1093/bioinformatics/btl216
- Yanover C, Bradley P. Large-scale characterization of peptide-MHC binding landscapes with structural simulations. Proc Natl Acad Sci U S A 2011; 108:6981-6; PMID:21478437; http://dx.doi.org/10.1073/pnas.1018165108
- Harndahl M, Rasmussen M, Roder G, Dalgaard Pedersen I, Sørensen M, Nielsen M, Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol 2012; 42:1405-16; PMID:22678897; http://dx.doi.org/10.1002/eji.201141774
- Nielsen M, Lundegaard C, Lund O, Keşmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005; 57:33-41; PMID:15744535; http://dx.doi.org/10.1007/s00251-005-0781-7
- Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, Nielsen M. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol 2005; 35:2295-303; PMID:15997466; http://dx.doi.org/10.1002/eji.200425811
- Nielsen M, Lund O, Buus S, Lundegaard C. MHC class II epitope predictive algorithms. Immunology 2010; 130:319-28; PMID:20408898; http://dx.doi.org/10.1111/j.1365-2567.2010.03268.x
- Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 2015; 67:641-50; PMID:26416257; http://dx.doi.org/10.1007/s00251-015-0873-y
- Singh, H, Raghava, GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics 2001; 17:1236-37; PMID:11751237; http://dx.doi.org/10.1093/bioinformatics/17.12.1236
- Reche PA, Glutting JP, Zhang H, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics 2004; 56:405-19; PMID:15349703; http://dx.doi.org/10.1007/s00251-004-0709-7
- Backert L, Kohlbacher O. Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med 2015; 119:1-12; PMID:26589500; http://dx.doi.org/10.1186/s13073-015-0245-0
- Fridman A, Finnefrock AC, Peruzzi D, Pak I, La Monica N, Bagchi A, Casimiro DR, Ciliberto G, Aurisicchio L. An efficient T-cell epitope discovery strategy using in silico prediction and the iTopia assay platform. Oncoimmunology 2012; 1:1258-270; PMID:23243589; http://dx.doi.org/10.4161/onci.21355
- Hadrup SR, Toebes M, Rodenko B, Bakker AH, Egan DA, Ovaa H, Schumacher TN. High-throughput T-cell epitope discovery through MHC peptide exchange. Methods Mol Biol. 2009; 524:383-405; PMID:19377960; http://dx.doi.org/10.1007/978-1-59745-450-6_28
- Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, Ovaa H. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc 2006; 1:1120-32; PMID:17406393; http://dx.doi.org/10.1038/nprot.2006.121
- Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebæk E, Svane IM, Schumacher TN, Thor Straten P, Hadrup SR. Dissection of T-cell antigen specificity in human melanoma. Cancer Res 2012; 72:1642-50; PMID:22311675; http://dx.doi.org/10.1158/0008-5472.CAN-11-2614
- Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw J, Hombrink P, Castermans E, Thor Straten P, Blank C, Haanen JB et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods 2009; 6:520-6; PMID:19543285; http://dx.doi.org/10.1038/nmeth.1345
- Hansen AM, Rasmussen M, Svitek N, Harndahl M, Golde WT, Barlow J, Nene V, Buus S, Nielsen M. Characterization of binding specificities of bovine leucocyte class I molecules: impacts for rational epitope discovery. Immunogenetics 2014; 66:705-18; PMID:25186069; http://dx.doi.org/10.1007/s00251-014-0802-5
- Harndahl M, Justesen S, Lamberth K, Røder G, Nielsen M, Buus S. Peptide binding to HLA class I molecules: homogenous, high-throughput screening, and affinity assays. J Biomol Screen 2009; 14:173-80; PMID:19196700; http://dx.doi.org/10.1177/1087057108329453
- Pedersen LE, Rasmussen M, Harndahl M, Nielsen M, Buus S, Jungersen G. A combined prediction strategy increases identification of peptides bound with high affinity and stability to porcine MHC class I molecules SLA-1*04:01, SLA-2*04:01, and SLA-3*04:01. Immunogenetics 2015; 68:157-65; PMID:26572135; http://dx.doi.org/10.1007/s00251-015-0883-9
- Lundegaard C, Lund O, Nielsen M. Prediction of epitopes using neural network based methods. J Immunol Methods 2011; 374:26-34; PMID:21047511; http://dx.doi.org/10.1016/j.jim.2010.10.011
- Harndahl M, Rasmussen M, Roder G, Buus S. Real-time, high-throughput measurements of peptide-MHC-I dissociation using a scintillation proximity assay. J Immunol Methods 2011; 374:5-12; PMID:21044632; http://dx.doi.org/10.1016/j.jim.2010.10.012
- Luft T, Rizkalla M, Tai TY, Chen Q, MacFarlan RI, Davis ID, Maraskovsky E, Cebon J. Exogenous peptides presented by transporter associated with antigen processing (TAP)-deficient and TAP-competent cells: intracellular loading and kinetics of presentation. J Immunol. 2001; 167:2529-37; PMID:11509592; http://dx.doi.org/10.4049/jimmunol.167.5.2529
- Justesen S, Harndahl M, Lamberth K, Nielsen LL, Buus S. Functional recombinant MHC class II molecules and high-throughput peptide-binding assays. Immunome Res 2009; 5:2; PMID:19416502; http://dx.doi.org/10.1186/1745-7580-5-2
- James EA, Moustakas AK, Bui J, Nouv R, Papadopoulos GK, Kwok WW. The binding of antigenic peptides to HLA-DR is influenced by interactions between pocket 6 and pocket 9. J Immunol 2009; 183:3249-58; PMID:19648278; http://dx.doi.org/10.4049/jimmunol.0802228
- Yin L, Stern LJ. Measurement of peptide binding to MHC class II molecules by fluorescence polarization. Curr Protoc Immunol 2014;10 6:5.10; PMID:25081912; http://dx.doi.org/10.1002/0471142735.im0510s106
- Gaseitsiwe S, Valentini D, Mahdavifar S, Reilly M, Ehrnst A, Maeurer M. Peptide microarray-based identification of Mycobacterium tuberculosis epitope binding to HLA-DRB1*0101, DRB1*1501, and DRB1*0401. Clin Vaccine Immunol 2010; 17:168-75; PMID:19864486; http://dx.doi.org/10.1128/CVI.00208-09
- Legutki JB, Zhao ZG, Greving M, Woodbury N, Johnston SA, Stafford P. Scalable high-density peptide arrays for comprehensive health monitoring. Nat Commun 2014; 5:4785; PMID:25183057; http://dx.doi.org/10.1038/ncomms5785
- Caron E, Kowalewski DJ, Chiek Koh C, Sturm T, Schuster H, Aebersold R. Analysis of major histocompatibility complex (MHC) Immunopeptidomes Using Mass Spectrometry. Mol Cell Proteomics 2015; 14:3105-17; PMID:26628741; http://dx.doi.org/10.1074/mcp.M115.052431
- Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics 2015; 14:658-73; PMID:25576301; http://dx.doi.org/10.1074/mcp.M114.042812
- Berlin C, Kowalewski DJ, Schuster H, Mirza N, Walz S, Handel M, Schmid-Horch B, Salih HR, Kanz L, Rammensee HG et al. Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy. Leukemia 2015; 29:647-59; PMID:25092142; http://dx.doi.org/10.1038/leu.2014.233
- Caron E, Vincent K, Fortier MH, Laverdure JP, Bramoulle A, Hardy MP, Voisin G, Roux PP, Lemieux S, Thibault P et al. The MHC I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Molecular systems biology 2011; 7:533; PMID:21952136; http://dx.doi.org/10.1038/msb.2011.68
- Granados DP, Sriranganadane D, Daouda T, Zieger A, Laumont CM, Caron-Lizott O, Boucher G, Hardy MP, Gendron P, Côté C et al. Impact of genomic polymorphisms on the repertoire of human MHC class I-associated peptides. Nat Commun 2014; 5:3600; PMID:24714562; http://dx.doi.org/10.1038/ncomms4600
- Granados DP, Yahyaoui W, Laumont CM, Daouda T, Muratore-Schroeder TL, Cote C, Laverdure JP, Lemieux S, Thibault P, Perreault C. MHC I-associated peptides preferentially derive from transcripts bearing miRNA response elements. Blood 2012; 119:e181-91; PMID:22438248; http://dx.doi.org/10.1182/blood-2012-02-412593
- Bassani-Sternberg M, Barnea E, Beer I, Avivi I, Katz T, Admon A. Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proc Natl Acad Sci USA 2010; 107:18769-76; PMID:20974924; http://dx.doi.org/10.1073/pnas.1008501107
- Mommen GP, Frese CK, Meiring HD, van Gaans-van den Brink J, de Jong AP, van Els CA, Heck AJ. Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc Nat Acad Sci USA 2014; 111:4507-12; PMID:24616531; http://dx.doi.org/10.1073/pnas.1321458111
- Tan CT, Croft NP, Dudek NL, Williamson NA, Purcell AW. Direct quantitation of MHC-bound peptide epitopes by selected reaction monitoring. Proteomics 2011; 11:2336-40; PMID:21598389; http://dx.doi.org/10.1002/pmic.201000531
- Hassan C, Kester MG, Oudgenoeg G, de Ru AH, Janssen GM, Drijfhout JW, Spaapen RM, Jiménez CR, Heemskerk MH, Falkenburg JH et al. Accurate quantitation of MHC-bound peptides by application of isotopically labeled peptide MHC complexes. Journal of Proteomics. 2014; 109:240-4; PMID:25050860; http://dx.doi.org/10.1016/j.jprot.2014.07.009
- Hogan KT, Sutton JN, Chu KU, Busby JA, Shabanowitz J, Hunt DF, Slingluff CL Jr Use of selected reaction monitoring mass spectrometry for the detection of specific MHC class I peptide antigens on A3 supertype family members. Cancer Immunol Immunother 2005; 54:359-71; PMID:15378283; http://dx.doi.org/10.1007/s00262-004-0592-y
- Hassan C, Kester MG, de Ru AH, Hombrink P, Drijfhout JW, Nijveen H, Leunissen JA, Heemskerk MH, Falkenburg JH, van Veelen PA. The human leukocyte antigen-presented ligandome of B lymphocytes. Mol Cell Proteomics. 2013; 12:1829-43; PMID:23481700; http://dx.doi.org/10.1074/mcp.M112.024810
- Abelin JG, Trantham PD, Penny SA, Patterson AM, Ward ST, Hildebrand WH, Cobbold M, Bai DL, Shabanowitz J, Hunt DF. Complementary IMAC enrichment methods for HLA-associated phosphopeptide identification by mass spectrometry. Nature Protocols 2015; 10:1308-18; PMID:26247297; http://dx.doi.org/10.1038/nprot.2015.086
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999; 20:3551-67; PMID:10612281; http://dx.doi.org/10.1002/(SICI)1522-2683(19991201)20 :18<3551::AID-ELPS3551>3.0.CO;2-2
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology 2008; 26:1367-72; PMID:19029910; http://dx.doi.org/10.1038/nbt.1511
- Mommen GP, Marino F, Meiring HD, Poelen MC, van Gaans-van den Brink JA, Mohammed S, Heck AJR, van Els ACM. Sampling from the proteome to the HLA-D ligandome proceeds via high specificity. Mol Cell Proteomics 2016; 15:1412-32; PMID:26764012; http://dx.doi.org/10.1074/mcp.M115.055780
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Method. 2007; 4:207-14; PMID:17327847; http://dx.doi.org/10.1038/nmeth1019
- Mazor R, Tai CH, Lee B, Pastan I. Poor correlation between T-cell activation assays and HLA-DR binding prediction algorithms in an immunogenic fragment of Pseudomonas exotoxin A. J Immunol Methods 2015; 425:10-20; PMID:26056938; http://dx.doi.org/10.1016/j.jim.2015.06.003
- Saethang T, Hirose O, Kimkong I, Tran VA, Dang XT, Nguyen LA, Le TK, Kubo M, Yamada Y, Satou K. PAAQD: Predicting immunogenicity of MHC class I binding peptides using amino acid pairwise contact potentials and quantum topological molecular similarity descriptors. J Immunol Methods 2013; 387:293-302; PMID:23058674; http://dx.doi.org/10.1016/j.jim.2012.09.016
- Trolle T, Nielsen M. NetTepi: an integrated method for the prediction of T cell epitopes. Immunogenetics 2014; 66:449-56; PMID:24863339; http://dx.doi.org/10.1007/s00251-014-0779-0
- Gilchuk P, Hill TM, Wilson JT, Joyce S. Discovering protective CD8 T cell epitopes-no single immunologic property predicts it! Curr Opin Immunol 2015; 34:43-51; PMID:25660347; http://dx.doi.org/10.1016/j.coi.2015.01.013
- Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014; 124:453-62; PMID:24891321; http://dx.doi.org/10.1182/blood-2014-04-567933
- Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y, Rosenberg SA, Robbins PF. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 2014; 20:3401-10; PMID:24987109; http://dx.doi.org/10.1158/1078-0432.CCR-14-0433
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y, Rosenberg SA. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013; 19:747-52; PMID:23644516; http://dx.doi.org/10.1038/nm.3161
- Frøsig TM, Lyngaa R, Met Ö, Larsen SK, Donia M, Svane IM, Thor Straten P, Hadrup SR. Broadening the repertoire of melanoma-associated T-cell epitopes. Cancer Immunol Immunother 2015; 64:609-20; PMID:25854582; http://dx.doi.org/10.1007/s00262-015-1664-x
- Newell EW, Sigal N, Nair N, Kidd BA, Greenberg HB, Davis MM. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat Biotechnol 2013; 31:623-9; PMID:23748502; http://dx.doi.org/10.1038/nbt.2593
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D et al. Tumor exome analysis reveal neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013; 31:e439-42; PMID:24043743; http://dx.doi.org/10.1200/JCO.2012.47.7521
- Walz S, Stickel JS, Kowalewski DJ, Schuster H, Weisel K, Backert L, Kahn S, Nelde A, Stroh T, Handel M et al. The antigenic landscape of multiple myeloma: mass spectrometry (re)defines targets for T-cell-based immunotherapy. Blood 2015; 126:1203-13; PMID:26138685; http://dx.doi.org/10.1182/blood-2015-04-640532
- Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L, Salih HR, Rammensee HG, Stevanovic S, Stickel JS. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci USA. 2015; 112:E166-75; PMID:25548167; http://dx.doi.org/10.1073/pnas.1416389112
- Carmona SJ, Nielsen M, Schafer-Nielsen C, Mucci J, Altcheh J, Balouz V, Tekiel V, Frasch AC, Campetella O, Buscaglia CA et al. Towards high-throughput immunomics for infectious diseases: use of next-generation peptide microarrays for rapid discovery and mapping of antigenic determinants. Mol Cell Proteomics 2015; 14:1871-84; PMID:25922409; http://dx.doi.org/10.1074/mcp.M114.045906
- Kwong GA, Radu CG, Hwang K, Shu CJ, Ma C, Koya RC, Comin-Anduix B, Hadrup SR, Bailey RC, Witte ON et al. Modular nucleic acid assembled p/MHC microarrays for multiplexed sorting of antigen-specific T cells. J Am Chem Soc 2009; 131:9695-703; PMID:19552409; http://dx.doi.org/10.1021/ja9006707
- Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, Koya RC, Liu CC, Kwong GA, Radu CG et al. A clinical microchip for evaluation of singleimmune cells reveals high functional heterogeneity in phenotypically similar Tcells. Nat Med 2011; 17:738-43; PMID:21602800; http://dx.doi.org/10.1038/nm.2375
- Volpetti F, Garcia-Cordero J, Maerkl SJ. A microfluidic platform for high-throughput multiplexed protein quantitation. PLoS One 2015;10:e0117744; PMID:25680117; http://dx.doi.org/10.1371/journal.pone.0117744