
ABSTRACT
Activation of the innate immune receptor retinoic acid-inducible gene I (RIG-I) by its specific ligand 5′-triphosphate-RNA (3pRNA) triggers antitumor immunity predominantly via NK cell activation and direct apoptosis induction in tumor cells. However, how NK cells are mobilized to attack the tumor cells remains elusive. Here, we show that RIG-I activation induced the secretion of extracellular vesicles (EVs) from melanoma cells, which by themselves revealed antitumor activity in vitro and in vivo. RIG-I-induced EVs from melanoma cells exhibited an increased expression of the NKp30-ligand (BAG6, BAT3) on their surface triggering NK cell-mediated lysis of melanoma cells via activation of the cytotoxicity NK cell-receptor NKp30. Moreover, systemic administration of RIG-I-induced melanoma-EVs showed a potent antitumor activity in a melanoma mouse model in vivo. In conclusion, our data establish a new RIG-I-dependent pathway leading to NK cell-mediated tumor cell killing.
Abbreviations
| 3pRNA | = | 5′-triphosphate-RNA |
| BAG6 | = | BCL2-Associated Athanogene 6 |
| DC | = | dendritic cells |
| EVs | = | extracellular vesicles |
| HCV | = | Hepatitis C virus |
| NK | = | Natural killer cells |
| NSCLC | = | non small cell lung cancer |
| PBMCs | = | peripheral blood mononuclear cells |
| RIG-I | = | retinoic acid inducible gene I |
| TLR | = | Toll-like receptor. |
Over the last years, communication by extracellular vesicles (EVs), e.g., exosomes, emerged as an important mechanism in regulating intercellular crosstalk. EVs are released by a multitude of malignant and non-malignant cells including melanoma cells. Depending on the EV releasing cell type, they can transfer several functional active molecules between cells inducing different effects in the recipient cell.Citation1-5 EVs derived from immune cells are described to enhance antitumor immunity. Dendritic cell (DC)-derived exosomes have the ability to activate NK cells against tumors leading to their evaluation in tumor therapy.Citation1,6 Clinical impact of NK cell activating DC-derived exosomes was recently documented in a Phase II trial in NSCLC stage IV patients.Citation7 In a fraction of patients presenting with defective NKp30 expression prolonged progression-free survival was associated with enhanced NKp30-dependent NK cell functions correlating with MHCII and BAG6 expression levels on exosomes.Citation7 BAG6 is an inducible surface ligand that binds to the activating natural cytotoxicity receptor NKp30 on NK cells.Citation8,9 Furthermore, BAG6-positive EVs have been identified as important regulators of NK cell activity.Citation9-12 In contrast to immune cell-derived EVs, it has been shown that tumor-derived EV (e.g., from melanoma) induce tolerance and thus strongly promote tumor growth and metastasis by a variety of different mechanisms, including the induction of an immunosuppressive environment.Citation1-4,13,14 Consequently, targeting of EVs has been suggested as a therapeutic strategy for the treatment of tumors.Citation15 However, there are only few experimental studies on the mechanisms how tumor-exosomes can be actively turned against the tumor.Citation16-19
There is emerging evidence that an antiviral immune response can induce changes in the composition and the function of EVs. For example, HCV-infected cells release exosomes containing viral components triggering an innate immune response.Citation20 Still, little is known about the impact of innate immune sensing receptors, initially responding to the viral infection, on the formation and function of EVs. Retinoic acid-inducible gene I (RIG-I) is a cytosolic immune sensing receptor detecting viral 5′-triphosphorylated RNA.Citation21,22 Unlike toll-like receptors (TLR), RIG-I is expressed in all nucleated cells, including tumor cells (e.g., melanoma cells).Citation23 Therapeutic stimulation of RIG-I with a synthetic 5′-triphosphate RNA oligonucleotide (3pRNA) induced a potent antitumor immune response mediated by NK cells and myeloid cells.Citation24-27 The mechanism by which NK cells are directed to specifically kill 3pRNA-treated tumor cells remains unknown.
Here, we demonstrate that in response to RIG-I activation tumor-derived EVs stimulated enhanced NK cell cytotoxicity via BAG6, an activating ligand of the NK cell-cytotoxicity receptor NKp30. Administration of RIG-I induced EVs led to potent antitumor activity in vivo. Thus, RIG-I activation turns tumor-derived EVs into vesicles with an immune-activating and tumor-suppressive phenotype.
Results
RIG-I stimulation triggers the release of extracellular vesicles (EVs)
To analyze the effect of RIG-I stimulation on formation and function of tumor-EVs, we used the human melanoma cell lines D04mel and Ma-Mel-86c.Citation28,29 In line with RIG-I as type I Interferon (IFN)-dependent gene, baseline expression of RIG-I in all used cell lines was strongly increased by type I IFN (). In response to activation of RIG-I with its ligand 3pRNA, D04mel and Ma-Mel-86c produced the IFN-dependent chemokine CXCL10 (), demonstrating a functional expression of RIG-I.
Figure 1. RIG-I stimulation triggers the release of extracellular vesicles (EVs). (A) Melanoma cells D04melCitation28 or Ma-Mel-86cCitation29 were analyzed for mRNA expression of RIG-I by quantitative real-time PCR in the presence or absence of type I Interferon (IFN) stimulation. Data are normalized to β-Actin. (n = 3) (B) Functionality of RIG-I signaling in D04mel and Ma-Mel-86c was determined by CXCL10 production 24 h after lipofection of cells with 1 µg/mL ctrl RNA or 3pRNA (n = 3) (C) Schematic overview of experimental procedures. Tumor cells (D04mel or Ma-Mel-86c) were transfected with 3pRNA (RIG-I ligand) or inert control RNA (non-RIG-I ligand) and EVs (RIG-I-EVs vs. ctrl-EVs) were purified using serial ultracentrifugation. Afterwards, EVs were analyzed regarding their effects on immune cells. (D) NTA analysis of purified vesicles (RIG-I-EVs vs. ctrl-EVs) derived from melanoma cells. (E) Cryo electron microscopy shows typical particles obtained by purification. One white or black scale bar indicates 100 nm. (F) Purified vesicles (RIG-I-EVs or ctrl-EVs) derived from D04mel cells were analyzed for expression of CD9, CD63, CD81 by western blot. (G) Purified vesicles (RIG-I-EVs vs. ctrl-EVs) derived from melanoma cells (D04mel, Ma-Mel- 86c) were analyzed for CD9 expression by flow cytometry (filled gray: isotype, dashed: ctrl-EVs, black line: RIG-I-EVs). (H) Amount of EVs after stimulation with 3pRNA (RIG-I-EVs) or ctrl RNA (ctrl-EVs) derived from melanoma cells (D04mel, Ma-Mel-86c) were estimated by quantification of proteins using Bradford Assay (n = 4–9). (I) Particle number of EVs after stimulation with 3pRNA (RIG-I EVs) or ctrl RNA (ctrl EVs) derived from melanoma cells (D04mel) was determined by NTA analysis (n = 6). All error bars reflect mean ± s.d. * indicates p < 0.05.
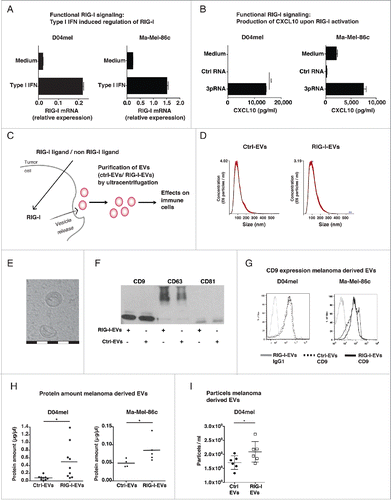
The experimental setting how EVs were analyzed is depicted in : Cells were transfected with the RIG-I ligand 3pRNA or an inert RNA (non-RIG-I-targeting) as control. Subsequently, the EV fractions were purified from cell culture supernatant as previously described and referred to as RIG-I-extracellular vesicles (RIG-I-EVs) or control RNA extracellular vesicles (ctrl-EVs).Citation11 Analysis of purified vesicles by nanoparticle tracking (NTA) as well as electron microscopy revealed vesicles around 100–140 nm with a cup-shaped structure (), as has been described for exosomes.Citation30,31 Western Blot () and flow cytometry () confirmed the expression of the exosomal markers CD9, CD63 and CD81. Stimulation of melanoma cells with 3pRNA increased the protein concentration in the EV fraction () in line with increased numbers of released EVs () in comparison to control RNA.
EVs derived from RIG-I-stimulated cells express enhanced levels of the NKp30-ligand BAG6
As RIG-I has been described to activate different cells of the immune system, we next analyzed whether RIG-I induced EVs are taken up by immune cells.Citation22,27,32 For this, we analyzed the association of CFSE-labeled D04mel-derived EVs with immune cells within peripheral blood mononuclear cells (PBMC) by flow cytometry. In comparison to ctrl-EVs, RIG-I-EVs demonstrated a significant higher association to NK cells arguing for enhanced binding or uptake of RIG-I-EV by this cell type (). Even if there was a slight increased association with CD3 positive cells as well, this was less pronounced than the association with NK cells.
Figure 2. Evs derived from RIG-I stimulated cells express enhanced levels of the NKp30-ligand BAG6. (A) CFSE labeled EVs (EV protein amount: 10 µg/mL) induced by 3pRNA (RIG-I-EVs) versus ctrl RNA (ctrl-EVs) were incubated with PBMCs and 24 h later CFSE staining of NK cells (CD3 negative, CD56 positive) or CD3 positive lymphocytes (CD3+ cells) were determined by flow cytometry (n = 3). (B) D04mel cells were transfected with 3pRNA (RIG-I-EVs) or ctrl RNA (ctrl-EVs) and the expression of MIC A/B, ULBP 1/2/3, Vimentin, B7-H6 and BAG6 on EVs was analyzed by flow cytometry (filled gray: isotype, dashed: ctrl-EVs, black line: RIG-I-EVs). One representative of four independent experiments is shown. (C) EVs induced by 3pRNA (RIG-I-EVs) vs. ctrl RNA (ctrl-EVs) were analyzed for binding of NKp30-fc by flow cytometry. Histogram shows one representative experiment (left, filled gray: isotype, dashed: ctrl-EVs, black line: RIG-I-EVs) and graph (right) shows quantification of x-fold induction of the geometric mean normalized to CD9 (n = 4). (D) Expression level of BAG6 on D04mel cells (left) or D04mel derived EVs (right) after transfection with 3pRNA or ctrl RNA was determined by flow cytometry (n = 5). (E) Purified EVs from melanoma (Ma-Mel-86c) cells induced by 3pRNA (RIG-I-EVs) versus ctrl RNA (ctrl-EVs) were analyzed for BAG6 expression on the surface by flow cytometry. Graphs show % induction of the geometric mean normalized to CD9 and s.e.m. of at least four independent experiments. (F) Exosomes from cells with siRNA mediated control knock down (ctrl kd) or RIG-I knock down (RIG-I kd) were analyzed for BAG6 expression on the surface by flow cytometry in response to 3pRNA (RIG-I-EVs) vs. ctrl RNA (ctrl-EVs). Graph shows geometric mean of BAG6 relative to CD9 (n = 4). All error bars reflect mean ± s.d. *, ** and *** indicates p < 0.05, p < 0.01 and p < 0.001.
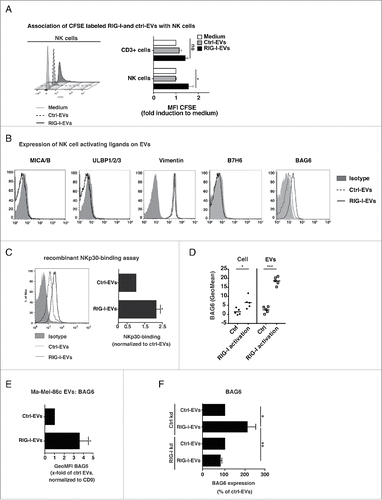
To unravel possible phenotypic differences between ctrl- and RIG-I-EVs, we analyzed the expression levels of ligands for activating NK cell receptors on tumor-EVs () and the cell surface of tumor cells (Fig. S1A) in response to 3pRNA. The expression of the NKG2D-ligands MICA, MICB, the UL binding proteins 1–3, the putative NKp46 ligand vimentin and the NKp30 ligand B7-H6 on tumor cells as well as EVs was not detectable or showed no difference irrespective of treatment ( and Fig. S1A). In contrast, the NKp30-ligand BAG6 was strongly induced on the surface of RIG-I-EVs but not on ctrl-EVs released from the melanoma cell line D04mel ( and Fig. S1B). In line with increased BAG6 expression on RIG-I-EVs, a recombinant NKp30-fc fusion protein bound stronger to RIG-I-EVs than ctrl-EVs (). Compared to the EV surface (), the expression of BAG6 on tumor cells was weak with only a slight increase in response to RIG-I activation (). Equal expression level of CD9 on EVs between RIG-I-EVs and ctrl-EVs suggest that equal amounts of EVs were analyzed (Fig. S1C) indicating that increased BAG6 expression on EVs does not simply reflect the cell surface but displays a specific EV composition. The increased BAG6 expression on RIG-I-EVs was also detectable on EVs from a different melanoma cell line (Ma-Mel-86c) (). Knock down of RIG-I (Fig. S1D) abolished the induction of BAG6 on RIG-I-EVs in response to 3pRNA treatment of the tumor cells confirming the specific role of RIG-I (). RIG-I activation did not lead to an upregulation of BAG6 mRNA expression in the D04mel cells (data not shown). This argues for a regulation of BAG6 on protein level and/or for enhanced transport of BAG6 in the EVs. Taken together, our results indicate that RIG-I activation leads to the release of BAG6-positive EVs.
BAG6-positive tumor-EVs derived from RIG-I-stimulated cells activate NK cells and promote NKp30-dependent cytotoxicity.
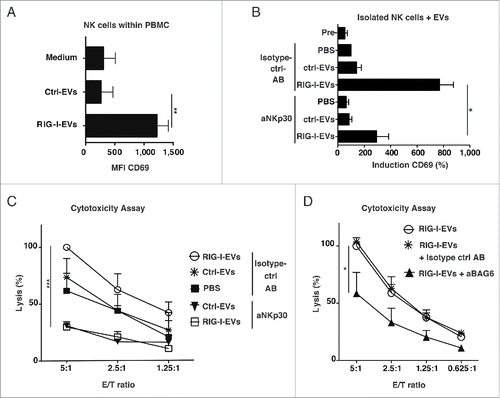
Since BAG6 is described to activate NK cells, we investigated the functional impact of the phenotypic differences between ctrl-EVs and RIG-I-EVs on the activation status of NK cells within PBMCs.Citation9,10 Incubation with RIG-I-EVs led to an enhanced expression of the activation marker CD69 on NK cells within PBMCs () and on primary naïve NK cells (). The observed NK cell activation was mainly NKp30-mediated, since antibody-mediated blockade of NKp30 strongly reduced the RIG-I-EV-dependent induction of CD69 on primary naïve NK cells. (). In addition, RIG-I-EVs derived from melanoma cells activated naïve NK cells to lyse untreated melanoma cells (D04mel) (). The inhibition of NKp30 function on NK cells () or BAG6 on melanoma-derived EVs () inhibited this effect. Thus, our data argue that EVs bind to and activate NK cells in a BAG6/NKp30 dependent manner, leading to an enhanced tumor cell lysis in vitro.
Figure 3. BAG6-positive RIG-I-EVs induce enhanced NK cell cytotoxicity in vitro. (A) PBMCs were incubated over night with RIG-I-EVs versus ctrl-EVs (EV protein amount: 10 µg/mL) and CD69 expression on NK cells (CD3 negative, CD56 positive) was determined by flow cytometry (n = 3). (B) Primary purified NK cells were left untreated (PBS) or incubated with 100 µg/mL (protein amount) RIG-I-EVs vs. ctrl-EVs for 36 h with (aNKp30) or without blocking (Isotype control antibody (AB)) of NKp30 (clone P30-15). Expression of CD69 (MFI) on NK cells after incubation with EVs was measured and normalized to PBS (n = 3). (C+D) Done as described in (B). (C) Cytotoxicity against untreated melanoma cells (D04mel) was assessed by europium release assay and results were normalized to 100% lysis by RIG-I-EVs in different E/T (n = 3). (D) Instead of NKp30 on NK cells, BAG6 was blocked on EVs with anti-BAG6 (aBAG6). Results were normalized to 100% lysis by RIG-I-EVs in an E/T-ratio of 5:1 (n = 3 for ctrl- and RIG-I-EVs + aBAG6, n = 2 for RIG-I-EVs + Isotype ctrl antibody). All error bars reflect mean ± s.d. *, ** and *** indicates p < 0.05, p < 0.01 and p < 0.001.
RIG-I induced tumor-EVs restrict tumor growth in vivo
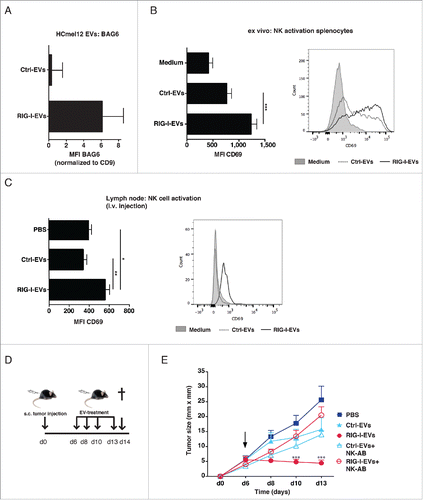
We next analyzed the antitumor activity of RIG-I-EVs from melanoma cells in vivo. EVs were prepared from the melanoma cell line HCmel12 which is derived from the spontaneous HGF-CDK4(R24C) melanoma mouse model and expressed vesicles marker CD81 and CD9 (Fig. S2A).Citation33 As shown for human EVs, RIG-I stimulation caused a upregulation of BAG6 on EVs derived from mouse melanoma cells (). Furthermore, like their human counterparts, HCmel12-derived RIG-I-EVs increased CD69 expression on mouse NK cells significant stronger than ctrl-EVs ex vivo (). RIG-I-EVs but not ctrl-EVs increased the expression of the activation marker CD69 on NK cells in draining lymph nodes () significantly, which is in line with the in vitro data. Next, HCmel12 cells were injected subcutaneously into the flank of C57BL/6 mice. After 6 d established melanomas were treated by four intratumoral injections of EVs derived from RIG-I-stimulated or control HCmel12 melanoma cells (). Treatment with RIG-I-EVs—but not with ctrl-EVs—effectively inhibited melanoma growth (). This effect was NK cell dependent, since depletion of NK cells (Fig. S2B) abrogated the antitumor effect mediated by RIG-I-EVs (). Thus, EVs derived from RIG-I stimulated tumor cells activate NK cells and suppress tumor growth of established tumors in vivo in a NK cell-dependent manner.
Figure 4. RIG-I-EVs lead to activation of NK cells and inhibition of tumor growth in vivo. HCmel12 cells were transfected with 3pRNA (RIG-I-EVs) or ctrl RNA (ctrl-EVs) and the expression of BAG6 on EVs was analyzed by flow cytometry (n = 3). (B) Activation of mouse splenocytes with 10 µg/mL (protein amount) EVs ex vivo. Graph (left) shows CD69 expression on NK cells (NK1.1+CD3−) was determined by flow cytometry (n = 5). Right histogram shows one representative experiment (left, filled gray: isotype, dashed: ctrl-EVs, black line: RIG-I-EVs) (C–E) Application of RIG-I- or ctrl-EVs in vivo. (C) HCmel12 mouse melanoma cell derived EVs (20 µg EV protein amount per mouse) were injected intravenously. Graph (left) shows expression of CD69 on NK cells (NK1.1+CD3−) purified from the lymph node was determined by flow cytometry (n = 5). Histogram (right) shows one representative experiment (left, filled gray: isotype, dashed: ctrl-EVs, black line: RIG-I-EVs) (D) Treatment schema of in vivo experiment. C57BL/6 mice were injected with HCmel12 mouse melanoma cells subcutaneously in the flank at day 0 and treated with melanoma-derived EVs at day 6, 8, 10, 13. Melanoma bearing mice were treated with PBS (ctrl), 20 µg protein amount of 3pRNA-induced EVs (RIG-I-EVs) or EVs induced by control RNA (ctrl-EVs), both derived from HCmel12 cells. Mice were sacrificed at day 14. (E) Tumor size was measured in treated and untreated mice with or without depletion of NK cells using antibody directed against NK1.1 (NK-AB). Mean tumor size and s.d. of 5–9 animals are shown. Arrow indicates begin of treatment, filled square: PBS, filled triangle: ctrl-EVs, filled circle: RIG-I-EVs, empty triangle: ctrl-EVs+NK-AB, empty circle: RIG-I-EVs+NK-AB. *, ** and *** indicates p < 0.05, p < 0.01 and p < 0.001.
Discussion
Spontaneously released tumor-EVs are mostly described to promote tumor growth and metastasis by diverse mechanisms, including immunosuppressionCitation1-4,34 such as: (i) inhibiting the NK cell response by decreasing their number and the expression of NKG2D,Citation35,36 (ii) carrying angiogenic and tumor supporting molecules like the EGFRvIII to neighboring cells,Citation37 (iii) supporting metastasis by enhanced homing to the sentinel lymph node,Citation38 (iv) by inducing a prometastatic niche,Citation14,39 v) shifting myeloid cells to a TGF-β expressing T cell suppressing differentiationCitation40 and (vi) inducing apoptosis in T cells via FAS ligand.Citation41 Thus, it can be assumed that EVs released by tumor cells serve the needs of the tumor rather than suppressing tumor growth. Therefore, the therapeutic reduction of the tumor-derived EV amount or manipulation of EVs has been suggested as a therapeutic strategy for the treatment of tumors.Citation15,42
In this work, we describe for the first time the novel and unexpected link between the immune sensing receptor RIG-I to the function of EVs as immune activating agents. This concept is supported by the findings that RIG-I stimulation (i) induced the increased release of EVs from tumor cells, (ii) induced a functional change in protein composition in EVs derived by malignant cells, (iii) stimulated the release of EVs that associate with NK cells and trigger NK cell activity and (iv) this was dependent on the upregulation of the ligand BAG6 engaging the cytotoxicity receptor NKp30 on NK cells.Citation9,43 The involvement of B7-H6, another NKp30 ligand is unlikely since we could not detect B7-H6 expression on tumor cells (data not shown) or EVs.Citation44 Moreover, both blocking of the receptor NKp30 or of the ligand BAG6 was sufficient to inhibit tumor cell killing by RIG-I-induced tumor-derived EVs. With NKp30 activated by BAG6, we identified one important factor critically involved in RIG-I-induced EV-mediated antitumor activity in the human system. However, since NKp30 is a pseudogene in the murine system and is not expressed as a functional receptor, the RIG-I-EVs mediated NK activating effects in vivo might be mediated by unknown receptors distinct from NKp30 that have been reported to exist in mice but which have not been identified yet.Citation45 Thus, further work is needed to analyze the role of RIG-I induced EVs and BAG6 in the mouse model.
BAG6 is a protein with multiple functions not only involved in immunological pathways such as the regulation of NK cell, macrophage or T cell responses.Citation43 A critical role of BAG6-positive EVs for tumor rejection and NKp30-dependent NK cell activation was previously reportedCitation7,11 and gene variations in the BAG6 gene are associated with lung cancerCitation46,47 and colon cancer.Citation48
Besides BAG6, additional molecular mechanisms may contribute to the potent antitumor activity of RIG-I-EVs. Different proteins, including other surface factors as well as cytokines, could be involved. Among these, heat shock proteinsCitation49,50 are strong candidates, as BAG6 binds directly to HSP70 via its BAG domain.Citation51 In line, expression of HSP70 on the surface of NK cell-stimulating exosomes was already described.Citation50,52 Moreover, the functional active transfer of nucleic acids (mRNA and miRNA) and also of RIG-I ligands has been demonstrated.Citation2,53,54 Thus, to fully understand the influence of RIG-I-EVs on NK cells, a comprehensive analysis of the proteins and nucleic acid content would be desirable. Activation of NK cells by activating ligands expressed on the surface of EVs poses the question how NK cells may retain their specificity against the damaged or infected cells. It is tempting to speculate that antigens of the target cell are transferred or presented to immune cells. HSP70 is known to be involved in the presentation of antigens and might thus confer specificity to EVs.Citation55 It was demonstrated that exosomes derived from different tumors contain tumor antigens, including melanomaCitation56 and that tumor-derived vesicles are in principle a potent source for vaccination.Citation57 Therefore, it remains to be analyzed whether, in addition to the effects on NK cells identified in this work, RIG-I stimulated tumor-derived EVs have more immune activating capabilities.
The here described effects of RIG-I activation on EV-function identifies a novel RIG-I-dependent defense pathway, which depends on the vesicle-mediated crosstalk between RIG-I-activated cells and immune cells.
Experimental procedure
Antibodies and reagents
Fluorophore conjugated antibodies against human CD3, CD9, CD56 and CD69 and murine CD3, NK1.1 and CD69 were obtained from BD (#563797, #555518, #557745, #553061, #561117, #562920) or BioLegend (#312105). For staining of EVs anti-human MICA/B. ULBP1, ULBP2, ULBP3 (all BamOMaB, #BAMO1, #AUMO2, #BUMO1, #CUMO3), CD9 and as secondary antibody goat-a-mouse-PE (BioLegend: #405307, #312103), monoclonal mouse-a-human-BAG6 (Pogge, unpublished, clone 3E4) were used. Binding of recombinant NKp30-fc protein (R&D Systems, #1849-NK-025) was detected by Cy3 anti-human fc from Dianova (#109-165-008). For blocking experiments, a human NKp30 (BioLegend, clone P30-15, #325202), monoclonal mouse-a-human BAG6 and IgG1-isotype control (BioLegend, #400101) were used. Recombinant human IFNα2a was purchased from Miltenyi (#130-093-874).
Immunostimulatory oligonucleotides
For generation of DNA-template-dependent in vitro-transcribed RNA (3pRNA), the oligonucleotide 1 (reverse) (5′-GGGACGCTGACCCAGAAGATCTACTATTTCTAGTAGATCTTCTGGGTCAGCGTCCCTATAGTGAGTCGTATTACAA-3′) was hybridized with oligonucleotide 2 (forward) (5′-TTGTAATACGACTCACTATAGGGACGCTGACCCAGAAGATCTACTAG AAATAGTAGATCTTCTGGGTCAGCGTCCC-3′, obtained from Biomers) in hybridization buffer (250 mM Tris-HCl, 250 mM NaCl, pH 7,4) for 5 min at 90°C. The hybridized product is directly used as a template for in vitro transcription reaction with a commercial in vitro T7 high-yield transcription kit (Thermofisher, #K0441) according to the manufactures protocol. Afterwards, the transcription product is digested with DNase I and purified with Mini Quick spin columns from Roche (Roche #11814419001). As negative control (ctrl RNA), a poly-A RNA obtained from Sigma (#P9403) was used.
Cell culture
The human melanoma cell line D04mel is available through the Australasian Biospecimen Network (Oncology) Cell Line Bank at the QIMR Berghofer Medical Research Institute and was a kind gift of C.W. Schmidt.Citation28 The human melanoma cell line Ma-Mel-86c was provided by A. Paschen.Citation29 The mouse melanoma cell line (HCmel12) was derived from a primary melanoma in HGF/SF—CDK4(R24C) mice by serial transplantation.Citation33 Cells were cultured in RPMI with penicillin (1%) and streptomycin (1%) and 10% FCS (Gibco). HEK Blue cells (InvivoGen, #hkb-ifnab) were maintained in DMEM containing 10% FCS and Pyruvate (1%). In case of EV isolation experiments cells were cultured with vesicle-reduced FCS. For culture of PBMCs, freshly prepared buffy coats from human healthy donors were obtained from the blood bank with the donors' written informed consent after approval by the responsible ethic committee. PBMCs were prepared by density gradient centrifugation using Biocoll (Biochrom, #L6113). Isolation of NK cells from PBMCs was performed by MACS using NK-Isolation Kit (Miltenyi, #130-092-657) according to the manufacturer's instruction. Purity of isolated NK cells was determined by FACS-Analysis to be ≥95%.
Isolation of splenocytes
Splenocytes were isolated from C57BL/6 mice. Spleens were mashed through a cell strainer and red blood cells were lysed. Per 96 well 400,000 splenocytes were used and incubated with 10 µg/mL EVs for 24 h followed by flow cytometric staining
Generation of RIG-I knockdown cells
For knockdown of RIG-I D04mel, cells were transfected with 20 pmol RIG-I siRNA or control siRNA (SantaCruz, USA, Texas, Dallas) with lipofection 24 h and 5 h prior transfection with RIG-I ligand or control RNA.
Transfection of melanoma cells
Melanoma cells were grown in 10 cm dishes and at a confluence of 70–80% (5 × 106 cells) cells were transfected with 3pRNA or poly-A RNA (ctrl RNA) as control. Therefor, 24 µg RNA were complexed with 60 µL Lipofectamin2000 according to the manual and cells were incubated for 3 h with the transfection complexes. Afterwards, cells were washed three times to remove lipofection complexes and cells were further cultured for 18 h in media supplemented with EV-reduced FCS for production of EVs.
Extracellular vesicle (EV) purification and labeling
Human melanoma cells (D04mel, Ma-Mel-86c) or mouse melanoma cells (HCmel12) were cultured in media with EV-reduced FCS (100,000 g for 90 min). Supernatant of cells for EV purification was centrifugated for 5 min at 400 g and twice for 15 min at 10,000 g. Vesicles were pelleted twice at 100,000 g for 90 min with intermediate resuspension in PBS (SW32Ti Rotor, Beckman Coulter). The amount of EV protein (approximatly 20–100 µg from 5 × 106 cells, dependent whether cells were activated with RIG-I ligand or not) was quantified by Bradford Assay (Carl Roth, #K015.2) or via Nanodrop (Peqlab, Erlangen, Germany) and equal amounts of EV protein were used in experiments (dependent on experiment between 10–100 µg/mL). To label EVs, melanoma cells were incubated with 5 µM carboxyfluorescein succinimidyl ester (CFSE) (eBioscience, #65-0850-84).
Nanoparticle tracking analysis
EVs were analyzed with NTA using the Nanosight NS300 (Malvern Instruments Ltd., Worcestershire, UK).
Stimulation of immune cells with extracellular vesicles (EVs)
NK cells or PBMCs were incubated with different amounts of EVs quantified by Bradford Assay or Nanodrop with incubation times between 24 h (for PBMC studies: 10 µg/mL EV amount) to 48 h (NK activation and cytotoxicity experiments: 100 µg/mL EV amount).
Quantitative real-time PCR
cDNA Synthesis was performed using VILO cDNA Synthesis Kit from Life Technologies (# 11754050) as described in the manual. For human, RIG-I cDNA was amplified in a total volume of 20 µL using LightCycler 480-System (Roche, Germany, Mannheim). Primer- and Probe-designs were performed using Universal Probe Library (Roche, Germany, Mannheim). Used Probes from Roche were #63 for human β-actin, #18 for human RIG-I. Following PCR conditions were used: 95°C for 10 min, followed by 50 cycles of 95°C for 10 s, 60°C for 30 s and 72°C for 1 min.
Flow cytometric analysis
For flow cytometric analysis, EVs were bound to carboxylated polysterene microbeads (4.5 µM, Polyscience Inc., #17140) and stained with antibodies. In addition, the expression of the tetraspanin CD9 was used for quantitation of EVs bound to the beads. Cells and EVs were measured using BD LSRII or FACS Calibur (Heidelberg, Germany) and analyzed using FlowJo (Tree Star, Olten, Switzerland) software. Activation of purified NK cells after 36 h incubation with 100 µg/mL EVs was analyzed by flow cytometry. NK cells were stained with CD69 and measured using FACS Calibur (Heidelberg, Germany).
Western blot
EVs or cells were either lysed (2 mM MgCl2, 50 mM Tris HCl pH 7.4, 150 mM NaCl, 1 mM DTT, 1% CHAPES, 1× Protease-Inhibitor) or loaded directly onto the gel. Equal amounts of total protein were separated by SDS gel electrophoresis and transferred onto a nitrocellulose membrane (GE Healthcare, Freiburg, Germany). For CD9, CD63, CD81 (all 1:200, all BioLegend, #312102, #353013, #349501,) detection membranes were incubated with the respective antibodies at 4°C overnight. HRP coupled secondary antibodies α-rat (1:5,000) or α-mouse (1:10,000) (both Jackson ImmunoResearch,) were incubated for 1 h at RT. Membranes were exposed to x-ray films after treatment with ECL western blotting substrate (Thermo Scientific, St. Leon-Rot, Germany).
Cytotoxicity assay
NK cell-mediated cytotoxicity was analyzed by a standard 3 h europium release assay in a 96-well microtiter plate as previously described (Strandmann et al. 2006). Briefly, NK effector cells were mixed with europium chloride (Sigma) labeled 5 × 103 target cells (D04mel) at different ratios. Supernatant was assayed for europium release after 3 h in a Wallac Victor 1420 multi-label counter. The percentage of specific lysis was calculated as 100 × [(experimental release − spontaneous release)/(maximal release − spontaneous release)]. NK cells were treated in the following way: Blocking of NKp30 was performed by pre-incubation of NK cells with 10 µg/mL of the blocking antibody clone P30-15 (BioLegend, #325202) or equivalent amount of an isotype control (ms IgG1, BioLegend, #400101) before addition of EVs (100 µg/mL) or PBS control. EVs were purified from 3pRNA or control RNA transfected D04mel cells. For BAG6-blocking experiments, EVs were pre-incubated with 10 µg/mL a-BAG6 antibody (clone 3E4) or corresponding isotype antibody for 30 min on ice. To prevent unspecific NK cell activation via CD16, NK cells were pre-incubated with 10 µg/mL human IgG antibody before addition of BAG6 or isotype ctrl pre-incubated EVs. NK cells were incubated for 40 h with EVs prior performance of the cytotoxicity assay.
Cryo electron microscopy
The vesicle pellet was suspended in 50 µL PBS. Approximately 3 μL were applied on a 400 × 100 mesh Quantifoil S7/2 holey carbon film on Cu grids (Quantifoil Micro Tools GmbH, Jena, Germany). After removal of excessive liquid, the grids were immediately shock-frozen by injection into liquid ethane. The grids were transferred into the transmission electron microscope (Leo 912 Ω-mega, Leo, Oberkochen, Germany) and analyzed under the atmosphere of liquid nitrogen (−183°C). The instrument was operated at 120 kV and pictures with a 6,300- to 12,500-fold magnification were taken.
In vivo experiments
Animal studies were approved by the local regulatory agency (Landesamt für Natur, Umwelt und Verbraucherschutz, NRW, Germany). For tumor-treatment experiments: 12 weeks old C57BL/6 mice were injected subcutaneously in the flank with 1.5 × 105 HCmel12 mouse melanoma cells. Treatment of mice was started at day 6 when all tumors were at least 2 × 2 mm in size. Tumor size (= length × width) was measured at days 6, 8, 10 and 13. HCmel12 cells were treated with 3pRNA (see above) or negative control RNA and EVs were purified from supernatant. EVs (20 µg EV protein per mouse) were injected into the tumor in 50 µL of PBS at day 6, 8, 10 and 13. Blood was taken 6 h before sacrificing mice at day 14. Mice were sacrificed when tumors reached 10 mm × 10 mm or tumor treatment day 14. NK cell depletion in mice was done using 100 µg NK1.1 antibody per mice (Bio X Cell, #BE-0036) by i.p. injection at day 4, 6, 8 and 13. To analyze NK cell activation within lymph nodes, HCmel12 derived EVs (20 µg EV protein per mouse) were injected intravenously. Lymph nodes were harvested after 18 h and CD69 expression on NK cells was measured.
Statistics
Graphs show mean and standard deviation if not stated differently. Statistical analysis was performed using non-parametric two-sided paired t-test. In case of multiple comparison, one way or two way ANOVA was used followed by Tukey test or Bonferroni to correct for multiple testing. * indicates p < 0.05, **p < 0.01 and ***p < 0.001.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1219827_s02.docx
Download MS Word (144.7 KB)Acknowledgments
We thank C.W. Schmidt for generation of D04mel cells, T. Wölfel for reciving D04mel from his lab and A. Paschen for Ma-Mel-86c. We thank A. Cerwenka for providing the anti-B7-H6 antibody (mAb 5.51.18).
Funding
This study was supported by grants of the Deutsche Forschungsgemeinschaft KFO286 (RP4 to E.P.v.S.), SFB832 (A12, A16, A19, A20), SFB670, SFB704 and KFO177 and BONFOR grants of the University Hospital Bonn to G.H., C.C., E.P.v.S., T.T. and M.S., G.H. and T.T. are members of the DFG-funded Excellence Cluster ImmunoSensation. This work contains parts of the PhD thesis of J.D-P. at the University of Bonn.
References
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 2009; 9:581-93; PMID:19498381; http://dx.doi.org/10.1038/nri2567
- van den Boorn JG, Daßler J, Coch C, Schlee M, Hartmann G. Exosomes as nucleic acid nanocarriers. Advanced Drug Delivery Reviews 2013; 65(3):331-5; PMID:22750807; http://dx.doi.org/10.1016/j.addr.2012.06.011
- Kharaziha P, Ceder S, Li Q, Panaretakis T. Tumor cell-derived exosomes: A message in a bottle. Biochimica et Biophysica Acta (BBA) - Rev Cancer 2012; 1826:103-11; PMID:22503823; http://dx.doi.org/10.1016/j.bbcan.2012.03.006
- Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res 2007; 67:2912-5; PMID:17409393; http://dx.doi.org/10.1158/0008-5472.CAN-07-0520
- Xiao D, Ohlendorf J, Chen Y, Taylor DD, Rai SN, Waigel S, Zacharias W, Hao H, McMasters KM. Identifying mRNA, MicroRNA and protein profiles of melanoma exosomes. PLoS ONE 2012; 7:e46874; PMID:23056502; http://dx.doi.org/10.1371/journal.pone.0046874
- Viaud S, Thery C, Ploix S, Tursz T, Lapierre V, Lantz O, Zitvogel L, Chaput N. Dendritic cell-derived exosomes for cancer immunotherapy: What's next? Cancer Res 2010; 70:1281-5; PMID:20145139; http://dx.doi.org/10.1158/0008-5472.CAN-09-3276
- Besse B, Charrier M, Lapierre V, Dansin E, Lantz O, Planchard D, Le Chevalier T, Livartoski A, Barlesi F, Laplanche A et al. Dendritic Cell-derived Exosomes as Maintenance Immunotherapy after First Line Chemotherapy in NSCLC. Oncoimmunology 2015; 5:e1071008; PMID:27141373; http://dx.doi.org/10.1080/2162402X.2015.1071008
- Binici J, Hartmann J, Herrmann J, Schreiber C, Beyer S, Güler G, Vogel V, Tumulka F, Abele R, Mäntele W et al. A soluble fragment of the tumor antigen BCL2-associated athanogene 6 (BAG-6) is essential and sufficient for inhibition of NKp30 receptor-dependent cytotoxicity of natural killer cells. J Biol Chem 2013; 288:34295-303; PMID:24133212; http://dx.doi.org/10.1074/jbc.M113.483602
- Pogge von Strandmann E, Simhadri VR, Tresckow von B, Sasse S, Reiners KS, Hansen HP, Rothe A, Böll B, Simhadri VL, Borchmann P et al. Human Leukocyte Antigen-B-Associated Transcript 3 Is Released from Tumor Cells and Engages the NKp30 Receptor on Natural Killer Cells. Immunity 2007; 27:965-74; PMID:18055229; http://dx.doi.org/10.1016/j.immuni.2007.10.010
- Simhadri VR, Reiners KS, Hansen HP, Topolar D, Simhadri VL, Nohroudi K, Kufer TA, Engert A, Pogge von Strandmann E. Dendritic Cells Release HLA-B-Associated Transcript-3 Positive Exosomes to Regulate Natural Killer Function. PLoS ONE 2008; 3:e3377; PMID:18852879; http://dx.doi.org/10.1371/journal.pone.0003377
- Reiners KS, Topolar D, Henke A, Simhadri VR, Kessler J, Sauer M, Bessler M, Hansen HP, Tawadros S, Herling M et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 2013; 121:3658-65; PMID:23509156; http://dx.doi.org/10.1182/blood-2013-01-476606
- Reiners KS, Daßler J, Coch C, Pogge von Strandmann E. Role of exosomes released by dendritic cells and/or by tumor targets: regulation of NK cell plasticity. Front Immunol 2014; 5:1074; PMID:24639679; http://dx.doi.org/10.3389/fimmu.2014.00091
- Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 2014; 14:195-208; PMID:24566916; http://dx.doi.org/10.1038/nri3622
- Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar CM et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 2012; 18:883-91; PMID:22635005; http://dx.doi.org/10.1038/nm.2753
- Iero M, Valenti R, Huber V, Filipazzi P, Parmiani G, Fais S, Rivoltini L. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ 2007; 15:80-8; PMID:17932500; http://dx.doi.org/10.1038/sj.cdd.4402237
- Chen T, Guo J, Yang M, Zhu X, Cao X. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J Immunol 2011; 186:2219-28; PMID:21242526; http://dx.doi.org/10.4049/jimmunol.1002991
- Lv LH, Wan YL, Lin Y, Zhang W, Yang M, Li GL, Lin HM, Shang CZ, Chen YJ, Min J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J Biol Chem 2012; 287:15874-85; PMID:22396543; http://dx.doi.org/10.1074/jbc.M112.340588
- Corrado C, Flugy AM, Taverna S, Raimondo S, Guggino G, Karmali R, De Leo G, Alessandro R. Carboxyamidotriazole-orotate inhibits the growth of imatinib-resistant chronic myeloid leukaemia cells and modulates exosomes-stimulated angiogenesis. PLoS ONE 2012; 7:e42310; PMID:22879938; http://dx.doi.org/10.1371/journal.pone.0042310
- Zhang Y, Luo C-L, He B-C, Zhang J-M, Cheng G, Wu X-H. Exosomes derived from IL-12-anchored renal cancer cells increase induction of specific antitumor response in vitro: a novel vaccine for renal cell carcinoma. Int J Oncol 2010; 36:133-40; PMID:19956842; http://dx.doi.org/10.3892/ijo_00000484
- Dreux M, Garaigorta U, Boyd B, Décembre É, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host & Microb 2012; 12:558-70; PMID:23084922; http://dx.doi.org/10.1016/j.chom.2012.08.010
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M et al. 5′-Triphosphate RNA Is the Ligand for RIG-I. Science 2006; 314:994-7; PMID:17038590; http://dx.doi.org/10.1126/science.1132505
- Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009; 31:25-34; PMID:19576794; http://dx.doi.org/10.1016/j.immuni.2009.05.008
- Barchet W, Wimmenauer V, Schlee M, Hartmann G. Accessing the therapeutic potential of immunostimulatory nucleic acids. Curr Opin Immunol 2008; 20:389-95; PMID:18652893; http://dx.doi.org/10.1016/j.coi.2008.07.007
- Besch R, Poeck H, Hohenauer T, Senft D, Häcker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest 2009; 119:2399-411; PMID:19620789; http://dx.doi.org/10.1172/JCI37155
- Glas M, Coch C, Trageser D, Daßler J, Simon M, Koch P, Mertens J, Quandel T, Gorris R, Reinartz R et al. Targeting the cytosolic innate immune receptors RIG-I and MDA5 effectively counteracts cancer cell heterogeneity in glioblastoma. Stem Cells 2013; 31:1064-74; PMID:23390110; http://dx.doi.org/10.1002/stem.1350
- Seya T, Kasamatsu J, Azuma M, Shime H, Matsumoto M. Natural killer cell activation secondary to innate pattern sensing. J Innate Immun 2011; 3:264-73; PMID:21454965; http://dx.doi.org/10.1159/000326891
- Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J et al. 5′-triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med 2008; 14:1256-63; PMID:18978796; http://dx.doi.org/10.1038/nm.1887
- O'Rourke MGE, Johnson M, Lanagan C, See J, Yang J, Bell JR, Slater GJ, Kerr BM, Crowe B, Purdie DM et al. Durable complete clinical responses in a phase I/II trial using an autologous melanoma cell/dendritic cell vaccine. Cancer Immunol Immunother 2003; 52:387-95; PMID:12682787; http://dx.doi.org/10.1007/s00262-003-0375-x
- Zhao F, Sucker A, Horn S, Heeke C, Bielefeld N, Schrörs B, Bicker A, Lindemann M, Roesch A, Gaudernack G et al. Melanoma lesions independently acquire T-cell resistance during metastatic latency. Cancer Research 2016; 76(15):4347-58; PMID:27261508; http://dx.doi.org/10.1158/0008-5472.CAN-16-0008
- György B, Szabó TG, Pásztói M, Pál Z, Misják P, Aradi B, László V, Pállinger É, Pap E, Kittel Á et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci 2011; 68:2667-88; PMID:21560073; http://dx.doi.org/10.1007/s00018-011-0689-3
- Crescitelli R, Lässer C, Szabó TG, Kittel Á, Eldh M, Dianzani I, Buzás EI, Lötvall J. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracel Vesicles 2013; 2:20677-; PMID:24223256; http://dx.doi.org/10.3402/jev.v2i0.20677
- Duewell P, Steger A, Lohr H, Bourhis H, Hoelz H, Kirchleitner SV, Stieg MR, Grassmann S, Kobold S, Siveke JT et al. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8+ T cells. Cell Death Differ 2014; 21:1825-37; PMID:25012502; http://dx.doi.org/10.1038/cdd.2014.96
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wölfel T, Hölzel M et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012; 490:412-6; PMID:23051752; http://dx.doi.org/10.1038/nature11538
- Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: Old sayings and new thoughts. Semin Cancer Biol 2011; 21:139-46; PMID:21251983; http://dx.doi.org/10.1016/j.semcancer.2011.01.002
- Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol 2008; 180:7249-58; PMID:18490724; http://dx.doi.org/10.4049/jimmunol.180.11.7249
- Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia Y, Kappes JC, Barnes S, Kimberly RP, Grizzle WE et al. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J Immunol 2006; 176:1375-85; PMID:16424164; http://dx.doi.org/10.4049/jimmunol.176.3.1375
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Curry WT, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008; 10:1470-6; PMID:19011622; http://dx.doi.org/10.1038/ncb1800
- Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res 2011; 71:3792-801; PMID:21478294; http://dx.doi.org/10.1158/0008-5472.CAN-10-4455
- Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015; 527:329-35; PMID:26524530; http://dx.doi.org/10.1038/nature15756
- Valenti R. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor- -mediated suppressive activity on T lymphocytes. Cancer Res 2006; 66:9290-8; PMID:16982774; http://dx.doi.org/10.1158/0008-5472.CAN-06-1819
- Abusamra AJ, Zhong Z, Zheng X, Li M, Ichim TE, Chin JL, Min W-P. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol Dis 2005; 35:169-73; PMID:16081306; http://dx.doi.org/10.1016/j.bcmd.2005.07.001
- Thuma F, Zöller M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin Cancer Biol 2014; 28:39-50; PMID:24631836; http://dx.doi.org/10.1016/j.semcancer.2014.02.011
- Binici J, Koch J. BAG-6, a jack of all trades in health and disease. Cell Mol Life Sci 2014; 71:1829-37; PMID:24305946; http://dx.doi.org/10.1007/s00018-013-1522-y
- Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med 2009; 206:1495-503; PMID:19528259; http://dx.doi.org/10.1084/jem.20090681
- Grover A, Izzo AA. BAT3 regulates mycobacterium tuberculosis protein ESAT-6-mediated apoptosis of macrophages. PLoS ONE 2012; 7:e40836; PMID:22808273; http://dx.doi.org/10.1371/journal.pone.0040836
- Etokebe GE, Zienolddiny S, Kupanovac Z, Enersen M, Balen S, Flego V, Bulat-Kardum L, Radojčić-Badovinac A, Skaug V, Bakke P et al. Association of the FAM46A gene VNTRs and BAG6 rs3117582 SNP with non small cell lung cancer (NSCLC) in croatian and norwegian populations. PLoS ONE 2015; 10:e0122651; PMID:25884493; http://dx.doi.org/10.1371/journal.pone.0122651
- Wang Y, Broderick P, Webb E, Wu X, Vijayakrishnan J, Matakidou A, Qureshi M, Dong Q, Gu X, Chen WV et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet 2008; 40:1407-9; PMID:18978787; http://dx.doi.org/10.1038/ng.273
- Ivanov I, Lo KC, Hawthorn L, Cowell JK, Ionov Y. Identifying candidate colon cancer tumor suppressor genes using inhibition of nonsense-mediated mRNA decay in colon cancer cells. Oncogene 2007; 26:2873-84; PMID:17086209; http://dx.doi.org/10.1038/sj.onc.1210098
- Elsner L, Muppala V, Gehrmann M, Lozano J, Malzahn D, Bickeböller H, Brunner E, Zientkowska M, Herrmann T, Walter L et al. The Heat Shock Protein HSP70 Promotes Mouse NK Cell Activity against Tumors That Express Inducible NKG2D Ligands. J Immunol 2007; 179:5523-33; PMID:17911639; http://dx.doi.org/10.4049/jimmunol.179.8.5523
- Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, Multhoff G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res 2005; 65:5238-47; PMID:15958569; http://dx.doi.org/10.1158/0008-5472.CAN-04-3804
- Thress K, Song J, Morimoto RI, Kornbluth S. Reversible inhibition of Hsp70 chaperone function by scythe and reaper. EMBO J 2001; 20:1033-41; PMID:11230127; http://dx.doi.org/10.1093/emboj/20.5.1033
- Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem 2005; 280:23349-55; PMID:15826944; http://dx.doi.org/10.1074/jbc.M502017200
- Xiao D, Ohlendorf J, Chen Y, Taylor DD, Rai SN, Waigel S, Zacharias W, Hao H, McMasters KM. Identifying mRNA, MicroRNA and Protein Profiles of Melanoma Exosomes. PLoS ONE 2012; 7:e46874; PMID:23056502; http://dx.doi.org/10.1371/journal.pone.0046874
- Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. - PubMed - NCBI. Cell 2014; 159:499-513; PMID:25417103; http://dx.doi.org/10.1016/j.cell.2014.09.051
- Chen T, Cao X. Stress for maintaining memory: HSP70 as a mobile messenger for innate and adaptive immunity. Eur J Immunol 2010; 40:1541-4; PMID:20468008; http://dx.doi.org/10.1002/eji.201040616
- Mears R, Craven RA, Hanrahan S, Totty N, Upton C, Young SL, Patel P, Selby PJ, Banks RE. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics 2004; 4:4019-31; PMID:15478216; http://dx.doi.org/10.1002/pmic.200400876
- Gu X, Erb U, Büchler MW, Zöller M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int J Cancer 2015; 136:E74-84; PMID:25066479; http://dx.doi.org/10.1002/ijc.29100