
ABSTRACT
Inflammation is a component of tumor progression mechanisms. Neutrophils are a common inflammatory infiltrate in many tumors, but their regulation and functions in neoplasia are not understood. Here, we showed, in detailed studies of c-Met molecule in 225 untreated patients with hepatocellular carcinoma (HCC), that high infiltration of neutrophils in HCC tissues determined malignant cell c-Met-associated clinical outcome of patients. High infiltration of neutrophils in HCCs determined malignant cell c-Met-associated clinical outcome of patients. Neutrophils were enriched predominantly in invading tumor edge of HCCs; the accumulated neutrophils were the major source of c-Met ligand HGF in HCCs. Exposure to HCC environments resulted in neutrophil activation and the following HGF production. Inhibiting the activities of Erk1/2, p38, and NF-κB, but not the phosphorylation of AKT or JNK, successfully attenuated the neutrophil HGF production induced by HCC environments. Further investigation revealed that GM-CSF was an important determinant in malignant cell-elicited neutrophil HGF production in vitro and in vivo. Moreover, we demonstrated that tumor neutrophils, via HGF/c-Met interaction, actively enhanced the metastasis of malignant cells in vitro and in vivo. These data provide direct evidence supporting the critical role of neutrophils in human tumor progression and reveal a fine-tuned collaborative action between cancer cells and immune cells in tumor milieu, which reroutes the immune activation into a tumor-promoting direction.
Abbreviations
| BCM | = | conditioned medium from blood neutrophils |
| CM | = | conditioned medium |
| DFS | = | disease-free survival |
| GM-CSF | = | granulocyte-macrophage colony stimulating factor |
| HCC | = | hepatocellular carcinoma |
| HGF | = | hepatocyte growth factor |
| MAPK | = | mitogen-activated protein kinase |
| OS | = | overall survival |
| PI3K/AKT | = | phosphoinositide 3-kinase/AKT |
| TCM | = | conditioned medium from tumor derived neutrophils |
| TSN | = | tumor culture supernatant |
Introduction
Tumor progression is now recognized as the product of evolving crosstalk between different cell types within tumors.Citation1-4 Hepatocellular carcinoma (HCC) is usually present in inflamed fibrotic and/or cirrhotic liver with extensive leukocyte infiltration. Thus, the immune status at a tumor site can largely influence the biologic behavior of HCC.Citation5-8 Recent studies have shown that activated monocytes/macrophages and IL-17-producing T cells in HCC can promote disease progression by stimulating cancer angiogenesis and B7-H1-associated immune privilege.Citation9-11 These observations suggest that local immune environments are important determinants for disease progression and cancer metastasis in humans.
Although less characterized than tumor macrophages and IL-17-producing T cellsCitation12,13, neutrophils are also emerging as important players in the pathophysiology of cancer.Citation14-16 Neutrophils are the most abundant leukocytes and serve as essential effector cells in the first line of host defense against invading microorganisms.Citation17,18 In addition to direct bactericidal activities, neutrophils can actively regulate angiogenesis and tissue re-modeling by releasing multiple proteases.Citation19,20 In a study of patients with HCC, it has been demonstrated that recruitment of neutrophils was regulated by IL-17+ T cell-induced epithelium-derived CXC chemokines, and those neutrophils were associated with the progression of cancer angiogenesis.Citation21 Notably, exposure of neutrophils to cancer environments also leads to autophagy-mediated survival of cells in vitro.Citation22 Thus, immune functional data and activated status of neutrophils in cancer environments are essential for understanding their roles and potential mechanisms in tumor immunopathogenesis.
Hepatocyte growth factor (HGF) is recognized as a potent mitogen for hepatocytes, and it plays important role in liver development and regeneration.Citation23,24 HGF receptor, the tyrosine kinase c-Met, is also highly expressed by malignant cells, and targeting HGF/c-Met axis in mice could significantly reduce the growth and metastasis of malignant cells.Citation25-27 However, such an approach is hampered in human cancers by the fact that the source and regulation of HGF are still unknown. The present study showed that high infiltration of neutrophils in HCC tissues determined malignant cell c-Met-associated clinical outcome of patients. Neutrophils were enriched predominantly in invading tumor edge of HCC tissues, and they were the major source of HGF in tissues. HCC environment-mediated activation was required for subsequent HGF production by neutrophils. Moreover, we also demonstrated that malignant cell-derived granulocyte-macrophage colony stimulating factor (GM-CSF) was an important determinant in neutrophil HGF production, which in turn enhanced the migration and invasion of malignant cells.
Results
Neutrophil infiltration determines malignant cell c-Met-associated clinical outcome of HCC patients
HGF/c-Met interaction activates a wide range of different cellular signaling pathways, including those involved in proliferation, motility, migration, and invasion of liver cells.Citation28 In individuals with untreated HCC (n = 60; ), we identified distinct expression patterns of c-Met in malignant cells by immunohistochemical staining in different paraffin-embedded samples (). In 58.3% of the samples analyzed, the c-Met+/high malignant cells were detected and present through the tumor tissues (). To evaluate the prognostic role of malignant c-Met in HCC pathology, 150 HCC patients () were divided into two groups according to the median value of tumor c-Met expression in transcriptional levels. However, as shown in , c-Met expression did not significantly affect the overall survival (OS) or disease-free survival (DFS) of patients, suggesting that c-Met alone could not effectively influence HCC progression.
Figure 1. Infiltration of neutrophils in HCC determines c-Met associated clinical outcome of patients. (A) Paraffin-embedded HCC samples (n = 60) were stained with anti-c-Met antibody. The representative micrographs of 60 HCC samples for c-Met staining are shown in down panel. (B) Expression of c-Met was unrelated to OS or DFS of HCC patients. 150 patients were divided into two groups according to the median value of transcriptional level of c-Met in tumor tissues. (C) 24-h production of HGF by HCC tumor-derived CD3+ T cells, CD14+ monocytes/macrophages, CD56+ NK cells, CD66b+ neutrophils, and fibroblasts (n = 4). (D) 24-h production of HGF by healthy or HCC blood-derived CD66b+ neutrophils (n = 4). (E) Paraffin-embedded HCC samples were stained with anti-CD15 antibody (n = 150). (F) Expression of c-Met was inversely correlated with OS or DFS in HCC patients with high peritumoral neutrophil infiltration. The patients were divided into four groups according to the median value of c-Met expression and peritumoral neutrophil infiltration in HCC tissues. Scale bar, 100 µm. Results represent mean ± SEM. *p < 0.01.
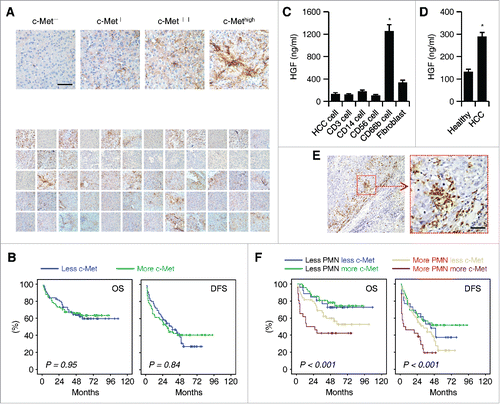
Table 1. Clinical characteristics of the 225 HCC patients.
We subsequently isolated primary HCC cells, fibroblasts, CD14+ myeloid cells, CD3+ T cells, CD56+ NK cells, and CD66b+ neutrophils form human HCC tissues and analyzed the production of c-Met ligand HGF. Primary HCC cells, CD14+ myeloid cells, CD3+ T cells, or CD56+ NK cells cultured for 24 h hardly produced HGF (). Strikingly, CD66b+ neutrophils derived from HCC tissues produced a large amount of HGF ex vivo, whereas fibroblasts also weakly secreted that factor (). Moreover, the neutrophils isolated from HCC blood, but not normal blood, also slightly produced HGF (). Thus, neutrophils might be the major source of HGF in HCCs, which prompted us to further investigate the role of neutrophils in c-Met-mediated tumor pathology.
The presence of neutrophils was visualized by staining of CD15 in paraffin-embedded HCC tissues (n = 150; ). CD15+ cells were often predominant in tumor-invading edge rather than in the cancer nest (). To further evaluate whether neutrophils contributed to c-Met-mediated tumor pathology, patients were stratified according to the median values of malignant c-Met expression and CD15 density in tumor-invading edge. As expected, in patients with high infiltration of CD15+ cells, there was a striking inverse association between malignant c-Met expression and both OS and DFS (p < 0.001 for both; ; ). By contrast, in patients with low infiltration of CD15+ cells, malignant c-Met expression was unrelated to the prognosis of either OS or DFS (; ). Together, neutrophils in tumor-invading edge determine the malignant c-Met-associated clinical outcome of HCC patients.
Table 2. Univariate and multivariate analyses of factors associated with survival and recurrence.
Exposure to HCC environments leads to neutrophil activation and subsequent HGF production
Having established the HGF production by neutrophils in HCC environments, we next set out to establish conditions under which this process can be reliably reproduced in vitro. Human neutrophils of ∼98% purity were left untreated or incubated with culture supernatants from primary HCC cells, three established hepatoma cell lines, and a normal liver cell line. Consistent with our in vivo findings, exposure of neutrophils to 30% tumor culture supernatants (TSNs) from both primary and established hepatoma cells, including HepG2, QGY-7703, and SK-Hep-1, resulted in a marked HGF production in a time-dependent manner (). By contrast, neutrophils cultured in medium alone or incubated with supernatant from normal liver cells (L02) marginally secreted those factors (). Such increased HGF production in neutrophils exposed to culture supernatants from primary and established hepatoma cells was further confirmed by real-time PCR ().
Figure 2. Activation of Erk1/2, p38, and NF-κB is essential for the induction of HGF in tumor associated neutrophils. (A–C) Purified neutrophils were left untreated or stimulated with culture supernatant from primary HCC cells, three heptatoma tumor cell lines or a normal liver cell line (L02) for 48 h. (A) The release of HGF in the culture supernatants at the indicated times were determined by ELISA. (B) The expression of HGF in neutrophils after 2-h treatment was detected by real-time PCR. (C) The activation of Erk1/2, p38, JNK, AKT, and IκBα at 0.5 h was measured by immunoblotting. (D) Inhibition of Erk1/2, p38, and IκBα impaired HGF expression in neutrophils exposed for 24 h to culture supernatant from primary HCC cells. The illustrated results represent mean ± SEM of four separate experiments. *p < 0.05, **p < 0.01, compared with medium; #p < 0.05, compared with DMSO.
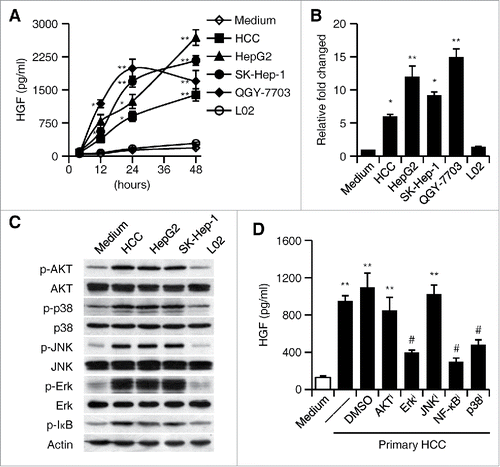
It has been demonstrated that phosphoinositide 3-kinase/AKT (PI3K/AKT), mitogen-activated protein kinase (MAPK), and NF-κB pathways are implicated in the regulation of neutrophil functions.Citation29-31 To further probe the mechanisms involved in the induction of neutrophil HGF production by cancer environment, we examined the activation of PI3K/AKT, MAPK, and NF-κB pathways in neutrophils. The activation patterns of the PI3K/AKT, MAPKs, JNK, Erk, and p38, and the NF-κB inhibitor IκBα in neutrophils left untreated or exposed to culture supernatants from hepatoma or liver cells coincided with the ability of the cells to produce HGF: Activation of these pathways was selectively enhanced in neutrophils stimulated with culture supernatants from both primary and established hepatoma cells (). Accordingly, using inhibitors to block the signal transduction of Erk1/2, p38, and NF-κB effectively impaired such TSN-induced neutrophil HGF production, whereas abolishing the phosphorylation of AKT and JNK had only a marginal effect (). These findings indicate that neutrophils are activated by HCC environments and subsequently acquire the ability to produce HGF.
GM-CSF is required for tumor neutrophil activation and HGF production
Our next endeavor was to determine the factor(s) involved in the induction of neutrophil HGF production by HCC environments. Recent studies have suggested that GM-CSF released by malignant cells contributes to the differentiation and protumorigenic functions of granulocytic MDSC in mice.Citation32 Indeed, we also observed a marked increase of GM-CSF in plasma from HCC blood, and that the level of GM-CSF positively correlated with the patients' TNM stage (). Analyzing the GM-CSF produced by primary and established hepatoma cells revealed a marked accumulation of GM-CSF in the culture supernatants within 24 h (). To investigate whether GM-CSF is also responsible for the generation of HGF-producing neutrophils in human HCC tumors, we initially tested the effect of recombinant human GM-CSF on HGF production by neutrophils. In support, GM-CSF, in a dose-dependent manner, did effectively induce HGF production (). Correspondingly, exposure of neutrophils to GM-CSF triggered rapid activation of PI3K/AKT, MAPKs, JNK, Erk, and p38, and the NF-κB inhibitor IκBα as those displayed by neutrophils treated with culture supernatants from primary and established hepatoma cells (Fig. S1; ); as expected, inhibiting the activities of Erk1/2, p38, and NF-κB, but not the phosphorylation of AKT or JNK, also successfully attenuated HGF production by GM-CSF-incubated neutrophils (). More importantly, using specific neutralizing antibody to abolish the effects of GM-CSF in culture supernatants from hepatoma cells and this treatment did efficiently inhibit HGF production by neutrophils ().
Figure 3. GM-CSF is responsible for the induction of HGF in tumor neutrophils. (A) Plasma concentration of GM-CSF in healthy donors (n = 22) and HCC patients (n = 39 for stage I+II and n = 36 for stage III+IV). Horizontal bars represent median values. (B) The concentration of GM-CSF in the culture supernatants from primary or established hepatoma cells. (C) Effect of GM-CSF on neutrophil HGF production after 24-h stimulation at indicated concentration (n = 4). (D) Inhibition of Erk1/2, p38, and IκBα impaired GM-CSF-mediated HGF production in neutrophils (n = 4). (E) Neutralization of GM-CSF attenuated the HGF production in neutrophils treated with culture supernatants from hepatoma cells (n = 4). (F) shGM-CSF in Hepa1-6 cells attenuated the GM-CSF expression in hepatoma tissues from mice and the HGF production in neutrophils derived from hepatoma tissues (n = 5 for each group). Results represent mean ± SEM. *p < 0.01, compared with those treated with DMSO, IgG, or shNC in corresponding group.
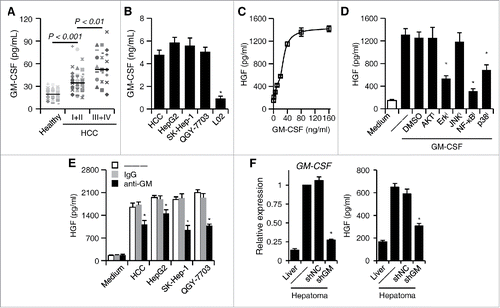
We afterward established a mouse hepatoma model to investigate the roles of malignant cell-derived GM-CSF in the induction of neutrophil HGF production. The shRNA retroviral vectors were applied to stably suppress the expression of GM-CSF in mouse hepatoma Hepa1-6 cells. The expression of GM-CSF in Hepa1-6 tissue was extremely higher than in normal mouse liver; stable transfection of shGM-CSF retroviral vectors in Hepa1-6 cells could markedly attenuate the expression of GM-CSF in hepatoma tissue (). Consistent with our hypothesis, the neutrophils derived from Hepa1-6 hepatoma that had been stably transfected with shGM-CSF retroviral vectors showed significant reduction of HGF production (). Thus, malignant cell-derived GM-CSF is an important determinant in tumor-elicited neutrophil HGF production.
Tumor neutrophils promote the metastasis of hepatoma cellsvia the HGF/c-Met axis
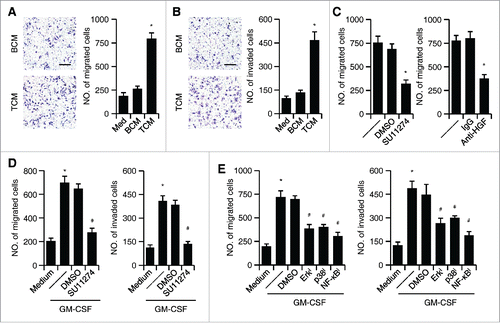
The results described above suggested that HCC environments trigger neutrophil HGF production, which in turn elicits protumorigenic effects of these cells. To test this hypothesis, we assessed the influence of tumor neutrophils on hepatoma cell proliferation, migration, and invasion. We collected the conditioned media (CM) from tumor neutrophils (TCM) and paired blood neutrophils (BCM). The TCM, but not BCM, significantly enhanced the migration and invasion of hepatoma QGY-7703 cells, whereas neither TCM nor BCM could affect the proliferation of cells (; Fig. S2). Supporting our hypothesis that tumor neutrophils promote hepatoma cell migration and invasion via the HGF/c-Met axis, using c-Met inhibitor SU11274 significantly impaired the increased hepatoma cell migration induced by TCM (). Analogously, neutralizing HGF in TCM also efficiently attenuated its ability to induce hepatoma cell migration (). In contrast, the control IgG did not affect that process (). Similar results were obtained when we used CM from neutrophils pre-incubated with GM-CSF to stimulate hepatoma QGY-7703 cells: The CM significantly increased QGY-7703 cell migration and invasion; these processes could be abolished by c-Met inhibitor SU11274 (). Notably, inhibiting the signals Erk1/2, p38, and NF-κB that regulated HGF production in GM-CSF-incubated neutrophils also effectively attenuated the ability of CM to induce hepatoma cell migration and invasion ().
Figure 4. The effect of HGF/c-Met axis on the migration and invasiveness of tumor cells. (A and B) Effects of TCM or BCM on hepatoma QGY-7703 cell migration (A) and invasion (B); untreated QGY-7703 cells, Med. (C) Suppression of c-Met activity by inhibitor SU11274 (left panel) or neutralization of HGF in TCM (right panel) effectively impaired hepatoma QGY-7703 cell migration. (D) Suppression of c-Met activity by inhibitor SU11274 effectively impaired hepatoma QGY-7703 cell migration (left panel) and invasion (right panel) stimulated by CM from GM-CSF-treated neutrophils. (E) Suppression of Erk1/2, p38, and IκBα activities in neutrophils treated with GM-CSF attenuated the effects of neutrophil CM on hepatoma QGY-7703 cell migration and invasion. All the data shown are representative of at least four separate experiments. Scale bar, 100 μm. *p < 0.01, compared with medium in D,E; #p < 0.05, compared with DMSO.
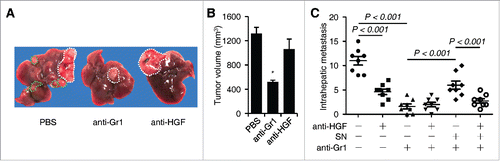
We finally used mouse hepatoma model to investigate the effect of neutrophil-derived HGF on intrahepatic metastasis. Depletion of tumor neutrophils by injecting anti-Gr1 Ab and suppression of HGF/c-Met axis by injecting anti-HGF Ab in mice both effectively impaired the incidence of intrahepatic metastasis, although anti-HGF Ab only weakly affected tumor sizes (). Supporting the hypothesis that neutrophils are important cellular source of HGF in hepatoma environments, the combined usage of Abs against Gr1 and HGF in mice exhibited similar capability in reducing the incidence of intrahepatic metastasis as those displayed by injecting anti-Gr1 Ab alone (). Consistently, peritoneal injection of supernatant derived from co-culture of neutrophils and Hepa1-6 cells partially restored the intrahepatic metastasis in mice treated with anti-Gr1 Ab, and this process could be abolished by adding anti-HGF Ab (). In contrast, the supernatant from the culture of neutrophils alone did not influence the intrahepatic metastasis (). These results together indicate that release of HGF by tumor neutrophils contribute to c-Met-mediated HCC metastasis.
Figure 5. Effects of neutrophils and HGF on hepatoma growth and intrahepatic metastasis in tumor-bearing mice (n = 8 for each group). (A–C) The Hepa1-6 hepatoma were treated with PBS or injected with anti-Gr1 and/or anti-HGF in the presence or absence of culture supernatant (SN) from coculture of blood neutrophils and Hepa1-6 cells as described in the methods. The representative images hepatoma with intrahepatic metastasis was shown in (A). The numbers of intrahepatic metastasis in different group were determined by immunohistochemical staining of hematoxylin and eosin (C). n = 8 for each group; *p< 0.01.
Discussion
Despite recent success in demonstrating the importance of HGF/c-Met axis in human cancers,Citation33,34 little is known about the source and regulation of HGF in cancer environments. The current study demonstrated that neutrophils were the major source of HGF in HCC tissues and the density of neutrophils in tumors determined malignant cell c-Met-associated clinical outcome of patients. More importantly, hepatoma cell-derived GM-CSF was an important determinant in neutrophil HGF production, which in turn enhanced the migration and invasion of malignant cells.
Tumor neutrophils exhibit distinct functional characteristics with impaired bactericidal activities, but increased potential in regulating angiogenesis and tissue remodeling.Citation35-37 In a recent study of human HCC, pro-inflammatory IL-17-producing cells have been found to recruit blood neutrophils into the peritumoral stroma of HCC by epithelium-derived CXC chemokines.Citation21 In the present study, we observed that, under the influence of local HCC environments, neutrophils were activated and acquired the ability to produce significant levels of c-Met ligand HGF, and that these cells, but not hepatoma cells, CD14+ myeloid cells, CD3+ T cells, or CD56+ NK cells, represented the major source of HGF in tissues. In fact, these activated neutrophils were unable to kill tumor cells and instead they promoted the in vitro migration and invasion of malignant cells via HGF/c-Met interaction, which suggests that such neutrophils can actually benefit tumor progression. This notion is supported by our current finding that, in patients with high infiltration of CD15+ cells, the malignant c-Met expression was inversely associated both OS and DFS. Consistent with our results, other investigators reported that cancer environments stimulated neutrophils to produce oncostatin M and MMP9, which in turn increased the metastatic potential of malignant cells.Citation21,38 PI3K/AKT, MAPK, and NF-κB pathways are all tightly associated with activation-induced neutrophil survival,Citation29 but the nature and regulation of these pathways in human tumor neutrophils remain largely unknown. The present study provided evidence that HCC environments could trigger rapid activation of PI3K/AKT, MAPKs JNK, Erk, and p38, and the NF-κB inhibitor IκBα in neutrophils, which in turn promoted the HGF-mediated hepatoma metastasis. Agents that prevent Erk1/2, p38, and NF-κB activation were found to block HGF production. In addition, in vitro migration assays showed that suppression of these pathways also effectively reduced tumor neutrophil-mediated increased motility of malignant cells. Such sequential neutrophil activation and enhanced malignant cell migration in tumors may reflect a novel immune-editing mechanism by which tumors render neutrophils able to perform a sustained protumorigenic function by stimulating neutrophil activation. This hypothesis is compatible with the studies showing that tumors educate macrophages to perform a suppressive role by inducing early activation of monocytes.Citation10,39,40
It is generally assumed that GM-CSF is efficient activator of M1 macrophage polarization, whereas M-CSF has been implicated for M2 macrophage polarization.Citation41,42 However, emerging evidence reveals that GM-CSF participate the differentiation of protumorigenic M2 macrophage.Citation43 Indeed, studies in mice also showed that GM-CSF released by malignant cells contributed to the differentiation and protumorigenic functions of granulocytic MDSC.Citation32 Supporting the protumorigenic roles of GM-CSF, our present study shows that induction of neutrophil HGF production by the hepatoma cell-derived GM-CSF plays a dominant role in maintaining prometastatic effect of neutrophils, as indicated by the results of four sets of experiments. First, significant GM-CSF was detected in plasma of HCC patients and in the culture supernatants of hepatoma cells, and that the levels of GM-CSF in plasma of patients were positively associated with their disease progression. Second, exposure of neutrophils to GM-CSF resulted in marked neutrophil activation and subsequent HGF production, and these neutrophils efficiently promoted migration and invasion of malignant cells via a HGF/c-Met-dependent manner. Third, Erk1/2, p38, and NF-κB were required for hepatoma cell-elicited neutrophil HGF production; these pathways were also responsible for GM-CSF-triggered neutrophil HGF production. Fourth, neutralizing GM-CSF effectively attenuated TSN-mediated neutrophil HGF production. These findings together suggest that increased GM-CSF in HCC environments may reroute neutrophils into a tumor-promoting direction by stimulating HGF production.
The biological effects of HGF are mediated by its interaction with its high-affinity tyrosine kinase receptor, c-Met, which is shown to be overexpressed and mutated in a variety of malignancies.Citation44 For HCC, there is a close relationship between overexpression of the c-Met and HCC metastasis, experimental studies and clinical investigations have shown that c-Met is a potential therapeutic target in HCC.Citation28 In the present study, elevated mRNA and protein expression of c-Met was observed in HCC tissues and hepatoma cell lines. In addition, our results showed that neutrophil-derived HGF induces significant migration of HepG2 and QGY hepatoma cells in vitro through interacting with c-Met receptor. However, the fact that anti-HGF antibodies did not completely abrogate such tumor cell migration suggests that other soluble motility factors are also involved. In accordance with our results, other investigators also reported that the neutrophil-derived oncostatin M and MMP9 promote the motility and invasiveness of mammary cancer cells.Citation21,38
Our results give important new insights into the production of HGF by tumor neutrophils. GM-CSF derived from hepatoma cells can trigger a rapid activation of Erk1/2, p38, and NF-κB in neutrophils, and thereby induce the production of HGF, which in turn leads to the migration and invasion of cancer cells in human HCC, reflecting a positive regulatory loop between tumors and their stroma. In support of this, the number of CD15+ neutrophils in primary HCC was inversely associated with the OS of the patients.Citation21 Therefore, it is possible that studies on the mechanisms that selectively modulate the activation of neutrophils will provide a novel strategy for anticancer therapy.
Materials and methods
Patients and specimens
HCC plasma and tissue samples were obtained from patients undergoing curative resection at the Cancer Center of Sun Yat-sen University (). Plasma samples (taken on day of surgery) were from 75 HCC patients who underwent surgical resections between December 2012 and June 2015 (Cohort 1; ). An additional 150 HCC patients who had undergone curative resection between 2005 and 2010 and had complete follow-up data (Cohort 2; ) were enrolled for analysis of OS and DFS. The inclusion criteria used in patient enrollment from the consecutive cohorts were absence of anticancer therapies or had distant metastasis prior to the operation, no concurrent autoimmune disease, HIV or syphilis, and availability of follow-up data. Of them, preoperative liver function was all classified as Child-Pugh class A. The clinical stage of tumors was determined according to the TNM classification system of International Union Against Cancer (edition 6). Tumor differentiation was graded by the Edmondson grading system. All samples were anonymously coded in accordance with local ethical guidelines (as stipulated by the Declaration of Helsinki). Written informed consent was obtained from the patients, and the protocol was approved by the Review Board of Sun Yat-sen University.
Preparation of culture supernatants from hepatoma cells and liver cells
Human hepatoma cell lines (HepG2 and SK-Hep-1) and mouse hepatoma Hepa1-6 cells were obtained from the American Type Culture Collection (Manassas, VA). Human normal liver cell line L02 and hepatoma cell line QGY-7703 were obtained from the Institute of Biochemistry and Cell Biology at the Chinese Academy of Sciences. All cells were proved to be mycoplasma free as routinely tested by a single-step PCR method,Citation45 and they were maintained in DMEM medium (Hyclone) supplemented with 10% FBS (Gibco). Culture supernatants from these cell lines were prepared as previously described.Citation39
Culture supernatant from primary HCC cells was acquired by culture of completely digested HCC tumor biopsy specimens. All of the samples were from patients without concurrent autoimmune disease, HIV, or syphilis. Ten Citation7 digested cells were resuspended in 10 mL of complete medium and cultured in 100-mm dishes. After 2 d, the supernatants were harvested, centrifuged, and stored at −80°C.
Immunohistochemistry
Paraffin-embedded formalin-fixed HCC samples were cut into 5-μm sections, which were processed for immunohistochemistry as previously described.Citation18 The sections were incubated with Abs against human CD15 (Lab Vision Corporation), or c-Met (Santa Cruz Biotechnology), and then stained in an Envision System (DakoCytomation). Evaluation of immunohistochemical variables was performed by two independent observers who were blinded to the clinical outcome.
Immunoblotting
The proteins were extracted as previously described.Citation46 Equal amounts of cellular proteins were separated by 10% SDS-PAGE, immunoblotted with Abs against AKT, p38, JNK, Erk1/2, phospho-AKT, phospho-p38, phospho-JNK, phospho-Erk1/2, phospho-IκB (Cell signaling Technology), and Actin (Boster), and then visualized with an ECL kit (Thermo Fisher Scientific).
Isolation and culture of neutrophils
Neutrophils were isolated from the peripheral blood of healthy donors or HCC patients.Citation46 Density-gradient separation on polymorphprep was performed at 340× g for 45 min at room temperature. The pale-red granulocyte layer was collected and the contaminated erythrocytes were lysed by a hypotonic buffer. Thereafter, the neutrophils were isolated using anti-CD66b Magnetic beads (Stem cell Technology, Canada).
Neutrophils of ∼98% purity in DMEM containing 10% FBS were plated at 5 × 106 per well in the presence or absence of culture supernatants from hepatoma or liver cells. In some experiments, the cells were pretreated with a c-Met inhibitor SU11274 (Pfizer, 5μM), a GM-CSF neutralization antibody (R&D Systems, 10 μg/mL), an HGF neutralization antibody (R&D Systems, 100 ng/mL), an Erk1/2 inhibitor U0126 (25 μM), a p38 inhibitor SB202190 (50 μM), a JNK inhibitor SP600125 (100 μM), an IκB inhibitor Bay11-7082 (20 μM), or an AKT inhibitor Triciribin Hydrate (100 μM), and subsequently exposed to indicated stimuli. In other experiments, the neutrophils were directly treated with recombinant GM-CSF (R&D Systems) with or without different inhibitors.
Isolation of leukocytes and fibroblasts from tissues
For the isolation of tumor-infiltrating leukocytes and fibroblasts, fresh HCC biopsy specimens were cut into small pieces and digested in RPMI 1640 supplemented with 0.05% collagenase IV (Sigma-Aldrich), 0.002% DNase I (Roche), and 20% FBS at 37°C for 30 min. Dissociated cells were filtered through a 150-μm mesh and separated by density gradient centrifugation. Thereafter, the leukocytes were harvested and the tumor-infiltrating neutrophils, monocytes, T cells, NK cells, and fibroblasts were isolated by anti-CD66b, anti-CD14, anti-CD3, anti-CD56, and anti-fibroblast magnetic beads (Miltenyi Biotec or Stem Cell Technology), respectively.
In vitro assays of QGY-7703 cell migration and invasion
The migration and invasion assay was performed in a 24-well Boyden chamber with an 8-μm pore size polycarbonate membrane (Corning). For migration assay, QGY-7703 cells were left untreated or stimulated for 24 h with supernatants from neutrophils with indicated treatments. Thereafter, the cells were removed from culture dishes by trypsinization, then washed, pelleted by centrifugation, and resuspended in serum-free medium at a density of 3 × 105/mL. 100 μL cell suspension was added to the upper chamber, whereas the lower compartment was filled with 600 μL of DMEM containing 10% FBS. After incubation at 37°C for 12 h, the cells remaining on the upper surface of the membrane were removed. The migrated tumor cells on the lower surface of the membrane were fixed and stained with crystal violet and subsequently counted under a light microscope. Invasion assay was done by the same procedure, except that the membrane was coated with 50 μg Matrigel to form a matrix barrier.
Construction of viral vectors
The candidate sequence for mouse shGM-CSF and a scrambled sequence for shNC were cloned into pSIF-H1-CopGFP-shRNA lentiviral vectors (System Biosciences). Thereafter, the lentiviral vectors were transfected into HEK293T cells together with their helper virus vectors pFIV-34N and pVSV-G (System Biosciences) using calcium phosphate. After 2 d, the viral particles were harvested and enriched by ultracentrifugation.
In vivo modulation of neutrophils and hepatoma cells
Mouse hepatoma Hepa1-6 cells were left untreated or stably transfected with shGM-CSF retroviral vectors. Hepa1-6-derived hepatomawas inoculated under the liver envelope as described.Citation47 After 25 d, tumor tissues were harvested and digested for isolation of tumor-infiltrating neutrophils. Thereafter, the production of HGF in tumor neutrophils was detected by ELISA. In another set of in vivo experiments, after 2-d inoculation of Hepa1-6-derived hepatoma, the animals were injected with Ab (s) against mouse Gr1 or HGF (10 mg/kg) or against Gr1 plus HGF in 100 μL buffered saline into the peritoneum every 3 d. In parallel, control animals were injected with 100 μL buffered saline. In some cases, the mice were first treated with an anti-Gr1 antibody (10 mg/kg) for 3 d. Thereafter, they were injected with anti-CD20 antibody plus 200 μL culture supernatant from co-culture of blood neutrophils and Hepa1-6 cells in the presence or absence of anti-HGF Ab every 3 d. After 21 d, tumors were harvested for subsequent immunohistochemical staining of hematoxylin and eosin. All mice were randomly grouped. Animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of Sun Yat-sen University.
Analysis of gene expression
Total RNA was extracted using the Trizol Reagent (Invitrogen) according to the manufacturer's recommendations. The mRNA level of HGF and c-Met was detected using THUNDERBIRD SYBR qPCR Mix (TOYOBO). GAPDH was used as the endogenous control. All reactions were run in triplicate and repeated in three independent experiments. The Data were analyzed by using the comparative Ct study. The specific primers used in this assay were as follows: human HGF, 5′-CTC ACA CCC GCT GGG AGT AC-3′ forward and 5′-TCC TTG ACC TTG GAT GCA TTC-3′ reverse; human MET, 5′-CCC CAC CCT TTG TTC AG-3′ forward and 5′-TCA GCC TTG TCC CTC CT-3′ reverse; human GAPDH, 5′-CAC CAT CTT CCA GGA GCG AG-3′ forward and 5′-GGG GCC ATC CAC AGT CTT C-3′ reverse; mouse GM-CSF, 5′-TGCCTGTCACGTTGA ATG AAG A-3′ forward and 5′-TGG TGA AAT TGC CCC GTA GA-3′ reverse; mouse GAPDH, 5′-GTATGACTCCACTCACGG-3′ forward and 5′-GGTCTGGCTCCTGGAAGA-3′ reverse.
ELISA
Concentrations of human HGF, mouse HGF, and human GM-CSF in the plasma or culture supernatants were determined using commercial ELISA kits (R&D system) according to the instructions provided by the manufacturer.
Statistical analysis
Statistical analyses were performed with the SPSS 17.0 (SPSS Inc., Chicago, IL) software. Results are expressed as means ± SEM. DFS was defined as the time from random assignment to recurrence, second primary cancer, or death without evidence of recurrence or second primary cancer. OS was defined as the time from random assignment to death as a result of all causes. Cumulative OS and DFS time were calculated using the Kaplan–Meier method and analyzed by the log-rank test. A multivariate Cox proportional hazards model was used to estimate adjusted hazard ratios and 95% confidence intervals and to identify independent prognostic factors. For categorical analysis, the median value was used as a cut point to dichotomize the series except serum AFP level and tumor size (for clinical applications). The χ2 test was used to test for relationships between categorical variables. The statistical significance of differences between groups was determined by Student's t-test. All data were analyzed using two-tailed tests unless otherwise specified, and p < 0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1219828_s02.doc
Download MS Word (1.4 MB)Funding
The study was supported by project grants from the National Natural Science Foundation of China (81503317, 81202319, 81201603), the Specialized Research Fund for the Doctoral Program of Higher Education (20120171120107), and the Youth Foundation of Guangzhou University of Chinese Medicine (2013GZYK10-6).
References
- Kuang DM, Xiao X, Zhao Q, Chen MM, Li XF, Liu RX, Wei Y, Ouyang FZ, Chen DP, Wu Y et al. B7-H1-expressing antigen-presenting cells mediate polarization of protumorigenic Th22 subsets. J Clin Invest 2014; 124:4657-67; PMID:25244097; http://dx.doi.org/10.1172/JCI74381
- Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, Vatan L, Szeliga W, Dou Y, Owens S et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014; 40:772-84; PMID:24816405; http://dx.doi.org/10.1016/j.immuni.2014.03.010
- Liu RX, Wei Y, Zeng QH, Chan KW, Xiao X, Zhao XY, Chen MM, Ouyang FZ, Chen DP, Zheng L et al. Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology 2015; 62:1779-90; PMID:26235097; http://dx.doi.org/10.1002/hep.28020
- Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, Chen DP, Zhao XY, Zhao Q, Li XF et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov 2016; 6:546-59; PMID:26928313; http://dx.doi.org/10.1158/2159-8290.CD-15-1408
- Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D, Li XF, Zheng L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013; 57:1107-16; PMID:23225218; http://dx.doi.org/10.1002/hep.26192
- Fu J, Zhang Z, Zhou L, Qi Z, Xing S, Lv J, Shi J, Fu B, Liu Z, Zhang JY et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology 2013; 58:139-49; PMID:22961630; http://dx.doi.org/10.1002/hep.26054
- Wang H, Gao B. MicroRNAs control hepatocarcinogenesis by regulating hepatocyte nuclear factor 4alpha-inflammatory signal feedback loops. Hepatology 2014; 60:1466-8; PMID:24996014; http://dx.doi.org/10.1002/hep.27287
- Su S, Zhao Q, He C, Huang D, Liu J, Chen F, Chen J, Liao JY, Cui X, Zeng Y et al. miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. Nat Commun 2015; 6:8523; PMID:26436920; http://dx.doi.org/10.1038/ncomms9523
- Zhao Q, Xiao X, Wu Y, Wei Y, Zhu LY, Zhou J, Kuang DM. Interleukin-17-educated monocytes suppress cytotoxic T-cell function through B7-H1 in hepatocellular carcinoma patients. Eur J Immunol 2011; 41:2314-22; PMID:21674477; http://dx.doi.org/10.1002/eji.201041282
- Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med 2009; 206:1327-37; PMID:19451266; http://dx.doi.org/10.1084/jem.20082173
- Kuang DM, Peng C, Zhao Q, Wu Y, Zhu LY, Wang J, Yin XY, Li L, Zheng L. Tumor-activated monocytes promote expansion of IL-17-producing CD8+ T cells in hepatocellular carcinoma patients. J Immunol 2010; 185:1544-9; PMID:20581151; http://dx.doi.org/10.4049/jimmunol.0904094
- Liou GY, Doppler H, Necela B, Edenfield B, Zhang L, Dawson DW, Storz P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov 2015; 5:52-63; PMID:25361845; http://dx.doi.org/10.1158/2159-8290.CD-14-0474
- Smith MP, Sanchez-Laorden B, O'Brien K, Brunton H, Ferguson J, Young H, Dhomen N, Flaherty KT, Frederick DT, Cooper ZA et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov 2014; 4:1214-29; PMID:25256614; http://dx.doi.org/10.1158/2159-8290.CD-13-1007
- Wu Y, Zhao Q, Peng C, Sun L, Li XF, Kuang DM. Neutrophils promote motility of cancer cells via a hyaluronan-mediated TLR4/PI3K activation loop. J Pathol 2011; 225:438-47; PMID:21826665; http://dx.doi.org/10.1002/path.2947
- Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011; 11:519-31; PMID:21785456; http://dx.doi.org/10.1038/nri3024
- Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006; 6:173-82; PMID:16498448; http://dx.doi.org/10.1038/nri1785
- Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol 2014; 14:302-14; PMID:24751955; http://dx.doi.org/10.1038/nri3660
- He M, Xu Z, Ding T, Kuang DM, Zheng L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol Immunol 2009; 6:343-52; PMID:19887047; http://dx.doi.org/10.1038/cmi.2009.45
- Tazzyman S, Niaz H, Murdoch C. Neutrophil-mediated tumour angiogenesis: subversion of immune responses to promote tumour growth. Semin Cancer Biol 2013; 23:149-58; PMID:23410638; http://dx.doi.org/10.1016/j.semcancer.2013.02.003
- Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP, Deryugina EI. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol 2011; 179:1455-70; PMID:21741942; http://dx.doi.org/10.1016/j.ajpath.2011.05.031
- Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, Yin XY, Zheng L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol 2011; 54:948-55; PMID:21145847; http://dx.doi.org/10.1016/j.jhep.2010.08.041
- Li XF, Chen DP, Ouyang FZ, Chen MM, Wu Y, Kuang DM, Zheng L. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J Hepatol 2015; 62:131-9; PMID:25152203; http://dx.doi.org/10.1016/j.jhep.2014.08.023
- Gui Y, Yeganeh M, Ramanathan S, Leblanc C, Pomerleau V, Ferbeyre G, Saucier C, Ilangumaran S. SOCS1 controls liver regeneration by regulating HGF signaling in hepatocytes. J Hepatol 2011; 55:1300-8; PMID:21703184; http://dx.doi.org/10.1016/j.jhep.2011.03.027
- Takeda S, Liu H, Sasagawa S, Dong Y, Trainor PA, Cheng EH, Hsieh JJ. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J Clin Invest 2013; 123:3154-65; PMID:23934123; http://dx.doi.org/10.1172/JCI65566
- Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer 2012; 12:89-103; PMID:22270953; http://dx.doi.org/10.1038/nrc3205
- Lordick F. Targeting the HGF/MET pathway in gastric cancer. Lancet Oncol 2014; 15:914-6; PMID:24965570; http://dx.doi.org/10.1016/S1470-2045(14)70273-6
- Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AA, Wauters E, Walmsley S, Prenen H, Granot Z et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015; 522:349-53; PMID:25985180; http://dx.doi.org/10.1038/nature14407
- You H, Ding W, Dang H, Jiang Y, Rountree CB. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011; 54:879-89; PMID:21618573; http://dx.doi.org/10.1002/hep.24450
- Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol 2012; 30:459-89; PMID:22224774; http://dx.doi.org/10.1146/annurev-immunol-020711-074942
- Hong CW, Kim TK, Ham HY, Nam JS, Kim YH, Zheng H, Pang B, Min TK, Jung JS, Lee SN et al. Lysophosphatidylcholine increases neutrophil bactericidal activity by enhancement of azurophil granule-phagosome fusion via glycine.GlyR alpha 2/TRPM2/p38 MAPK signaling. J Immunol 2010; 184:4401-13; PMID:20237295; http://dx.doi.org/10.4049/jimmunol.0902814
- Martinez D, Vermeulen M, Trevani A, Ceballos A, Sabatte J, Gamberale R, Alvarez ME, Salamone G, Tanos T, Coso OA et al. Extracellular acidosis induces neutrophil activation by a mechanism dependent on activation of phosphatidylinositol 3-kinase/Akt and ERK pathways. J Immunol 2006; 176:1163-71; PMID:16394005; http://dx.doi.org/10.4049/jimmunol.176.2.1163
- Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol 2010; 40:22-35; PMID:19941314; http://dx.doi.org/10.1002/eji.200939903
- Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 2010; 11:834-48; PMID:21102609; http://dx.doi.org/10.1038/nrm3012
- Blumenschein GR, Jr., Mills GB, Gonzalez-Angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol 2012; 30:3287-96; PMID:22869872; http://dx.doi.org/10.1200/JCO.2011.40.3774
- Mantovani A. The yin-yang of tumor-associated neutrophils. Cancer Cell 2009; 16:173-4; PMID:19732714; http://dx.doi.org/10.1016/j.ccr.2009.08.014
- Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008; 8:618-31; PMID:18633355; http://dx.doi.org/10.1038/nrc2444
- Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, Chen MB, Krall JA, DeCock J, Zervantonakis IK et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov 2016; 6:630-49; PMID:27072748; http://dx.doi.org/10.1158/2159-8290.CD-15-1157
- Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res 2005; 65:8896-904; PMID:16204061; http://dx.doi.org/10.1158/0008-5472.CAN-05-1734
- Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007; 110:587-95; PMID:17395778; http://dx.doi.org/10.1182/blood-2007-01-068031
- Zhao Q, Kuang DM, Wu Y, Xiao X, Li XF, Li TJ, Zheng L. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J Immunol 2012; 188:1117-24; PMID:22184722; http://dx.doi.org/10.4049/jimmunol.1100164
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011; 11:750-61; PMID:22025054; http://dx.doi.org/10.1038/nri3088
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010; 32:593-604; PMID:20510870; http://dx.doi.org/10.1016/j.immuni.2010.05.007
- Su S, Liu Q, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W, Zhang J et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014; 25:605-20; PMID:24823638; http://dx.doi.org/10.1016/j.ccr.2014.03.021
- Corso S, Giordano S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective. Cancer Discov 2013; 3:978-92; PMID:23901039; http://dx.doi.org/10.1158/2159-8290.CD-13-0040
- Uphoff CC, Drexler HG. Detection of mycoplasma in leukemia-lymphoma cell lines using polymerase chain reaction. Leukemia 2002; 16:289-93; PMID:11840297; http://dx.doi.org/10.1038/sj.leu.2402365
- Zheng L, He M, Long M, Blomgran R, Stendahl O. Pathogen-induced apoptotic neutrophils express heat shock proteins and elicit activation of human macrophages. J Immunol 2004; 173:6319-26; PMID:15528371; http://dx.doi.org/10.4049/jimmunol.173.10.6319
- Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology 2010; 51:154-64; PMID:19902483; http://dx.doi.org/10.1002/hep.23291