
ABSTRACT
The universal modular chimeric antigen receptor (UniCAR) platform redirects CAR-T cells using a separated, soluble targeting module with a short half-life. This segregation allows precise controllability and flexibility. Herein we show that the UniCAR platform can be used to efficiently target solid cancers in vitro and in vivo using a pre-clinical prostate cancer model which overexpresses prostate stem cell antigen (PSCA). Short-term administration of the targeting module to tumor bearing immunocompromised mice engrafted with human UniCAR-T cells significantly delayed tumor growth and prolonged survival of recipient mice both in a low and high tumor burden model. In addition, we analyzed phenotypic and functional changes of cancer cells and UniCAR-T cells in association with the administration of the targeting module to reveal potential immunoevasive mechanisms. Most notably, UniCAR-T cell activation induced upregulation of immune-inhibitory molecules such as programmed death ligands. In conclusion, this work illustrates that the UniCAR platform mediates potent anti-tumor activity in a relevant in vitro and in vivo solid tumor model.
Introduction
CARs are synthetic chimeric antigen receptors comprising an antigen recognition domain, mostly fused to the signaling domain of the CD3ζ chain with one or more co-stimulatory domain(s). CAR engineered immune cells can target surface antigens independently of MHC expression.Citation1-3 Despite impressive clinical responses observed in patients with hematological malignancies,Citation4-15 successful clinical utilization of CAR-T cells in solid tumors has not been achieved so far. Several obstacles prevent the broader application of the CAR technology in solid tumors. For instance, the microenvironment of solid neoplasms represents a mechanical barrier and provides an immunosuppressive milieu.Citation16,17 Almost all tumor associated antigens (TAAs) used as targets for CARs are not tumor specific but are also expressed to a varying extent on regular, non-malignant cells. CD19, the most successful target, is also expressed on regular B lymphocytes but sustained B cell aplasia in consequence of CD19-CAR therapy is clinically tolerable.Citation18 In contrast, TAAs of solid tumors are frequently shared between malignant and normal, more vital tissues.Citation19 Multiple strategies have been developed to increase the controllability of CAR-T cells to limit on-target/off-tumor toxicities and side effects such as the cytokine release syndrome.Citation20-24 The recently introduced UniCAR platform allows switching effector cells on and off in a controlled manner by splitting the antigen-binding and the signaling domain of conventional CARs.Citation25-28 Hence, the platform consists of 2 separate components: (i) the effector module, which is a genetically modified T cell that expresses the UniCAR with a binding moiety to a unique peptide epitope (5B9) derived from a human nuclear proteinCitation29 and (ii) a targeting module (TM). The TM is a fusion molecule of a binding domain which is directed against a tumor-associated antigen (TAA) and an epitope (5B9), which in turn is recognized by the UniCAR. Effector cells expressing the UniCAR remain inert until crosslinking to target cells in the presence of the TM. Reactivity of UniCAR-T cells is strictly related to presence of TM and the corresponding antigen. Moreover, formed UniCAR TM immune complexes can reversibly dissociate in a concentration dependent manner.Citation27 Therefore side effects associated with UniCAR-T cells can be encountered by an interruption of the administration of the TM. Furthermore, the extent of UniCAR-T cell activation correlates with the quantity of available TM. Hence, UniCAR-T cell activation can be controlled by dosing of the TM. Additionally, the UniCAR platform is more flexible than conventional CARs. Since the TM is solely responsible for the recognition of the TAA, UniCAR-T cells can be redirected to various TAAs simply by exchanging the TM.
Prostate stem cell antigen (PSCA) is a surface glycoprotein which is upregulated in prostate, urinary bladder and pancreatic cancer, while showing low expression in prostate epithelium, urinary bladder, kidney, esophagus, stomach, and placenta.Citation30-34 PSCA has been labeled an immunotherapeutic target and monoclonal antibodies (mAbs) recognizing PSCA have been tested in clinical trials including patients with metastatic prostate and pancreatic cancer.Citation35,36 However, more potent PSCA-directed targeting approaches may be required to achieve clinical responses.
Here we show that the UniCAR platform allows an efficient targeting of solid tumors both in vitro and in vivo by using PSCA over-expressing prostate cancer cells. Short-term application of the TMelicited significant in vivo anti-tumor activity in tumor-bearing mice engrafted with human UniCAR-T cells. To further elucidate potential immune resistance mechanisms, we characterized effector cells and target cells during therapy and found upregulation of targetable inhibitory immune checkpoint molecules. We conclude that a combination with immune checkpoint inhibitors might further potentiate the efficacy of UniCAR based therapies.
Results
Development and characterization of 7KATMPSCA
The TM consisting of an scFv fused to the epitope 5B9 has been described in the context of the “modular targeting system” and the “UniCAR platform.”Citation28,37,38 Here we used a modified anti-PSCA TM termed 7KATMPSCA. Schematic structure, biochemical characterization and verification of binding specificity are presented in the Fig. S1. In conclusion, this modified novel anti-PSCA TM shows a specific and concentration-dependent binding to PSCA expressing tumor cells. Most importantly, the accessibility of the 5B9-tag is maintained after cell surface binding which is a prerequisite for UniCAR functionality.
7KATMPSCA mediates efficient lysis of PSCA expressing tumor cells in presence of UniCAR-T cells in vitro
Chromium release assays were performed to quantify the killing efficacy in vitro and to demonstrate the functionality of the UniCAR system in dependence of its components (TM and UniCAR-T cell). Eradication of PC3PSCA occurred at nanomolar (nM) concentrations of 7KATMPSCA in the presence of UniCAR-T cells. At an e:t ratio of 1:1, a maximal lysis of 55% of PC3PSCA could be observed at a concentration of 1 nM 7KATMPSCA after 24 h of co-incubation. The half maximal effective concentration (EC50) was calculated as 0.4 ng/ml (Fig. S2A). After 48 h, the maximal killing efficacy increased to 67.5% (Data not shown). We observed no unspecific tumor cell lysis (Fig. S2B). Dependence of tumor cell lysis on functional UniCAR-T cells was further strengthened by the association of e:t ratio with lysis efficiency (Fig. S2B). We conclude that functionality of the UniCAR platform is strictly dependent on both TM, UniCAR-T cell and target cell expressing the corresponding TAA. Furthermore, the UniCAR platform achieves efficient killing of solid tumor cells at nanomolar concentrations of the TM.
Tumor biodistribution and plasma pharmacokinetics of 7KATMPSCA in vivo
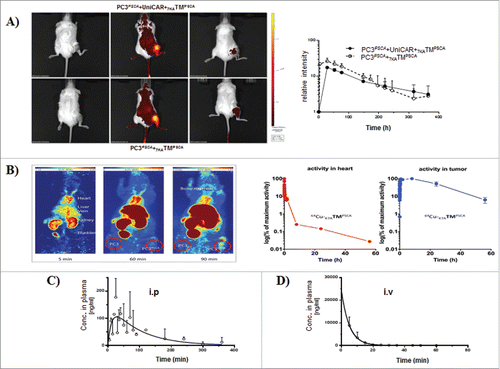
To characterize the behavior of 7KATMPSCA in vivo we analyzed the penetration capacity into the tumor tissue and the temporal persistence in the circulation after a single administration. Tumor-bearing NSG mice were iv injected with fluorochrome labeled 7KATMPSCA. Fluorescence intensity in the tumor region rapidly increased after administration and subsequently declined. However, even after 15 d residual fluorescence could still be detected in the tumor region irrespective of the presence of UniCAR-T cells (). Similar to fluorochrome labeled 7KATMPSCA, radioactivity rapidly increased in PC3PSCA-tumors of NMRI-nu mice after iv injection of 64Cu-NODAGA-7KATMPSCA. In contrast to the prolonged detection of fluorescence, radioactivity rapidly declined in the tumor tissue afterwards and the tumor half-life was calculated to be 20.8 h (). Comparable to the short-terminal half-life of 7KATMPSCA in the PB as detected by ELISA (terminal half-life of 98 min and 16 min after ip and iv injection of 250 ng 7KATMPSCA/g, respectively) (, ), also radioactivity in the heart as surrogate for the blood was rapidly cleared and the half-life of 7KATMPSCA was calculated to be7 min for the α phase and 2.6 h for the β phase (). Importantly, 7KATMPSCA could be detected in the PB of NSG mice even after ip injection. In conclusion, (i) 7KATMPSCA is rapidly cleared from PB, (ii) is enriched in the tumor tissue, and (iii) reaches the systemic circulation even after ip injection.
Figure 1. Tumor biodistribution and plasma pharmacokinetics of 7KATMPSCA in NSG mice (A) Tumor-bearing NSG mice ± UniCAR-T cells engraftment, were injected iv via tail vein with 50 µg fluorescence labeled 7KATMPSCA. Tumor uptake was measured longitudinally by in vivo fluorescence imaging. Left: Images of 2 representative mice before, 28 h and 15 d after injection are shown. Right: the mean relative fluorescence intensity in the region of interest over time. Error bars represent SD (n = 3 per group). (B) Tumor-bearing NMRI-nu mice were injected iv via tail vein with 3.8 MBq 64Cu-7KATMPSCA (NODAGA)1.5. Radioactivity was determined longitudinally in the region of interest. Left: Representative maximum intensity projections over 2 h presented as summed images with midframe times of 5, 60, and 90 min after a single iv injection. Right: PET-kinetics in tumor bearing NMRI-nu mice after a single iv injection. Data are presented as logarithm of maximum activity concentration in the heart (representative for the blood), and the PC3PSCA-tumor 2 h, 8 h, 24 h, and 56 h after injection. (C, D) NSG mice were injected either iv via tail vein (n = 3 for every time point) or ip (n = 3 for every time point) with 250 ng 7KATMPSCA/g bw. PB was obtained at different time points for evaluation of 7KATMPSCA plasma concentration. A terminal half-life time of 98 min and 16 min was calculated for 7KATMPSCA after ip and iv injection, respectively.
The UniCAR platform provides significant anti-tumor efficiency in vivo
Low tumor burden model
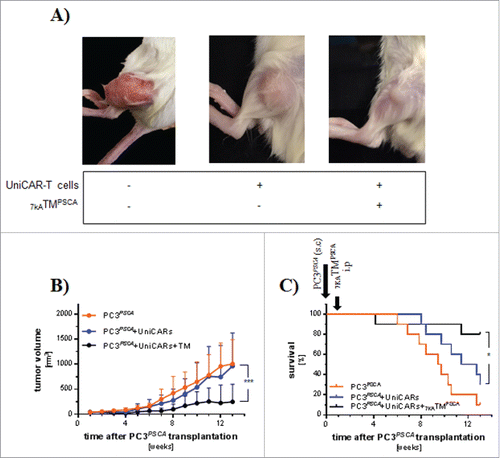
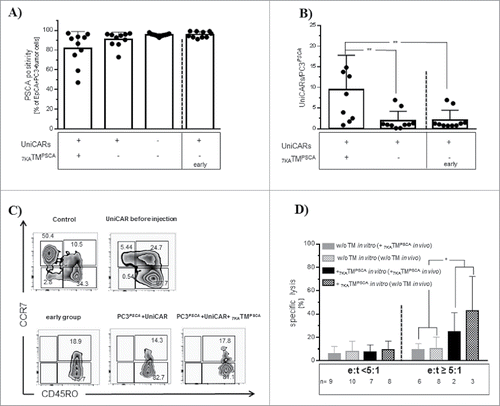
NSG mice ( ± UniCAR-T cell engraftment) were sc transplanted with PC3PSCA. After one week, treatment with 7KATMPSCA was started with ip injections bid for 7 consecutive days. The tumor volume at the start of treatment did not differ significantly between the groups () and mice engrafted with UniCAR-T cells showed comparable chimerism in the PB (Data not shown). Administration of 7KATMPSCA was associated with significant less tumor volume resulting in a significantly improved overall survival (). In the “PC3PSCA group,” all mice except one had to be killed before the scheduled end of the experiment due to the tumor size. In the “PC3PSCA + UniCAR + 7KATMPSCA group” 2 mice unexpectedly died at week 4 and 11.4 despite small tumors (21.4 mm3 and 865.17 mm3). The mere presence of UniCAR-T cells did not affect the tumor growth but numerically prolonged survival compared with mice in the “PC3PSCA group.” Taken together, these data demonstrate that the combination of 7KATMPSCA with the UniCAR platform can be used in vivo for efficient treatment of PSCA-positive solid cancers with low tumor burden.
Figure 2. The UniCAR platform mediates tumor growth inhibition and prolongs survival of small tumor bearing NSG mice. Three to 4 weeks after iv injection of 1 × 106 UniCAR-T cells, mice were sc transplanted with 1 × 106 PC3PSCA tumor cells. A separate group received only 1 × 106 PC3PSCA tumor cells. One week later 250 ng 7KATMPSCA/g bw was ip injected bid for 7 consecutive days in the study cohort. Tumors were measured 2−3 times weekly with a digital caliper. Mice were killed when predefined end points were met. (A) Representative pictures of tumors from all 3 groups at the end of the experiment. (B) Tumor volume ± SD during the observation period of 13 weeks (last observation carried forward method). The tumor volume at the start of treatment did not differ significantly among the groups (PC3PSCA: 39.2 ± 13.3 mm3, PC3PSCA + UniCAR: 33.7 ± 17.8 mm3were PC3PSCA + UniCAR + 7KATMPSCA: 45.9 ± 36.2 mm3) (C) Kaplan-Meier plot for the treatment groups associated with survival. Data are pooled from 2 independent experiments with individual donors, n = 10 (*p < 0.05, ***p ≤ 0.001).
High tumor burden model
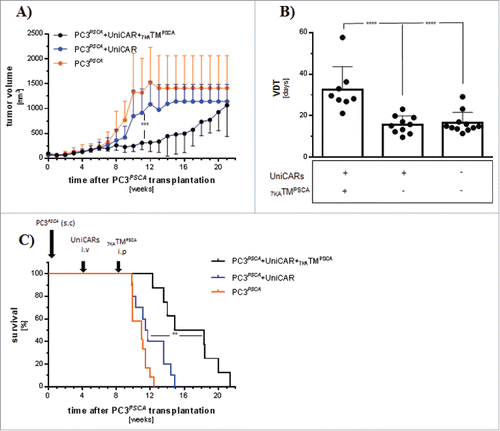
To increase the tumor load and to simulate the clinical situation more closely, NSG mice were first sc injected with PC3PSCA. After 4 weeks, mice had developed evident tumor burden and a subgroup was subsequently iv transplanted with UniCAR-T cells. Another 4 weeks later during the expected peak of UniCAR-T cell PB engraftment, tumor burden had further increased and mice were separated into 3 different treatment cohorts. The “PC3PSCA + UniCAR group” and the “PC3PSCA + UniCAR + 7KATMPSCA group” showed comparable tumor volumes. The tumor volume in the “PC3PSCA group” was slightly increased, although this difference did not reach statistical significance. There were no fundamental differences in effector cell chimerism between UniCAR-T cell engrafted cohorts (Data not shown). Treatment with 7KATMPSCA resulted in less tumor volume (, ) and significantly prolonged overall survival of tumor-bearing mice (). We conclude that the combination of 7KATMPSCA with the UniCAR platform might even provide potent anti-tumor activity in therapeutic situations with high tumor burden.
Figure 3. The UniCAR platform mediates potent anti-tumor activity in a high tumor burden model: Mice were transplanted sc with 1 × 106 PC3PSCA and 4 weeks later 1 × 106 UniCAR-T cells were iv injected. Eight weeks tumor post-inoculation, mice were treated with ip injections of 250 ng 7KATMPSCA/g bw bid for 7 consecutive days. Tumors were measured twice weekly. Mice were killed when predefined end points were met. (A) The tumor volume at the start of treatment was 362 ± 220 mm3, 265 ± 103.1 mm3 and 561.6 ± 386.5 mm3 in PC3PSCA + UniCAR, PC3PSCA + UniCAR + 7KATMPSCA and PC3PSCA, respectively. Tumor volume progress was significantly delayed by administration of 7KATMPSCA in tumor-bearing mice engrafted with UniCAR-T cells. (B) Tumor volume doubling time (VDT) was significantly increased in the study cohort compared with the controls. Bars represent mean ± SD. (C) Administration of 7KATMPSCA led to a significant improved overall survival of tumor-bearing, UniCAR-T cell engrafted NSG mice. Data are pooled from 2 independent experiments with individual donors, n = 8−12 mice per cohort (**p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
Response assessment
Additionally, we were interested in the monitoring of effector and target cell responses in dependence of 7KATMPSCA administration in the high tumor burden model. Therefore, we analyzed various factors including circulating effector cells in the PB immediately before and right after the treatment. Tumors were processed at the time point of euthanization (meaning at various time points when mice were killed because the end points were met) to investigate target and effector cells in situ. In separate experiments, we also analyzed tumors directly before and after the treatment period.
Peripheral blood
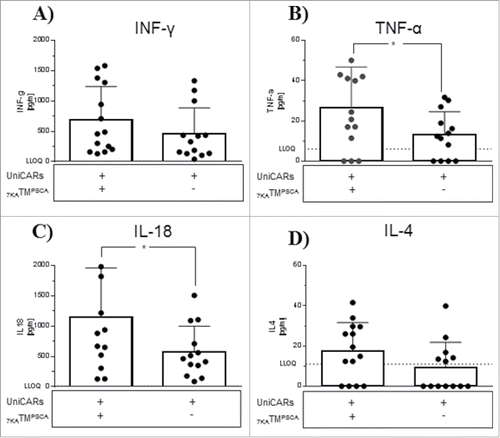
The percentage of circulating UniCAR-T cells remained unaffected by the administration of 7KATMPSCA. In general, PB chimerism post effector cell injection reached its maximum at 4−6 weeks and gradually dropped starting latest at 8 weeks. No relevant chimerism was detectable at the time point of euthanization (Data not shown). Right before the treatment, GFP as a surrogate for UniCAR expression had declined in circulating T cells compared with expression levels at injection. Application of 7KTMPSCA did not further affect GFP expression in PB circulating UniCAR-T cells (Fig. S3A). At the time point of injection, grafts consisted of equal proportions of CD4+ and CD8+ UniCAR-T cells. Four weeks later at treatment the majority of UniCAR-T cells in the PB were CD4+. However the CD4/CD8 ratio in PB was not affected further by the treatment (Fig. S3B and C). In addition to CD4 and CD8, the expression of activation markers such as CD25 and CD69 on circulating effector cells was also undistinguishable between both cohorts (Data not shown). Furthermore, plasma was analyzed for human Th1 and Th2 cytokines directly after the treatment period. We evaluated a wide range of Th1 cytokines, including INF-γ, TNF-α, IL-12, IL-2, IL-1β, GM-CSF, and IL-18 as well as Th2 cytokines, such as IL-13, IL-4, IL-5, and IL-6. Only INF-γ, TNF-α, IL-18, and IL-4 were mostly quantifiable. Addition of 7KATMPSCA induced a statistically significant elevation of TNF-α and IL-18, while INF-γ and IL-4 increased numerically (). In the therapy group, the expression level of the cytokines positively correlated with each other although not always necessary to a statistical significant level. There was no positive correlation observed between most cytokine levels in PB and the amount of intratumoral UniCAR-T cells (Fig. S4). In conclusion, circulating UniCAR-T cells were unsuitable for monitoring anti-tumor activity, while plasma cytokine measurement might be a helpful tool to characterize UniCAR activity in solid tumors.
Figure 4. Upregulation of Th1 and Th2 cytokine production in the plasma of UniCAR-T cell engrafted mice treated with 7KATMPSCA. The plasma of UniCAR-T cell engrafted, tumor-bearing mice was analyzed for human Th1/Th2 cytokines in dependence of 7KATMPSCA administration. INF-γ (A), TNF-α (B), IL-18 (C) and IL-4 (D) were the only detectable human cytokines. All of them were increased in mice receiving 7KATMPSCA, while only the increment of TNF-α and IL-18 was statistically significant. Bars represent mean ± SD. Data pooled from 3 independent experiments with individual donors (n = 13–14). (LLOQ = lower limit of quantification). Values below the LLOQ were set as zero for the statistical analysis (*p < 0.05).
Tumor tissue
Furthermore, we sought to monitor effector and target cells at the site of action. Hence, tumors were resected at the respective euthanization time point (earliest 1.9 week, latest 13.4 weeks after stop of 7KATMPSCA administration). In contrast to PB, substantial quantities of tumor-infiltrating UniCAR-T cells could be observed, which underlines the importance of investigating different compartments. Nevertheless, the extent of infiltrating UniCAR-T cells did not differ significantly between cohorts. Moreover, virtually all viable tumor cells expressed PSCA arguing against an outgrowth of antigen-loss variants as a potential mechanism of resistance. Most tumor-infiltrating UniCAR-T cells showed an effector memory phenotype (CCD7−CD45RO+) (Data not shown). In conclusion, although impressive anti-tumor response could be observed, these effects were not reflected by effector and target cells in situ. However, the interval between the treatment and the tissue analysis might have blurred the 7KATMPSCA-induced changes.
Therefore, we additionally analyzed tumors right before (“early group”) and immediately at the end of the treatment period. This time, we found a numerically but not statistically significant reduction in tumor cells expressing PSCA in the group of mice receiving 7KATMPSCA (). Unlike our previous observations, the proportion of tumor-infiltrating UniCAR-T cells significantly increased by adding of 7KATMPSCA to PC3PSCA tumor-bearing NSG mice (). The expression of activation markers such as CD25 and CD69 as well as the CD4/CD8 ratio was undistinguishable (Data not shown) and therefore not appropriate to monitor activity of the UniCAR system. The in vitro manufacturing process of UniCAR-T cells was associated with a more mature differentiation phenotype due to the polyclonal activation before transduction and most of CD4+ UniCAR-T cells used for transplantation displayed a central memory phenotype (CCR7+CD45RO+), while CD8+ UniCAR-T cells mostly showed an effector memory phenotype (CCR7−CD45RO+). In vivo, the proportion of effector memory phenotype UniCAR-T cells further increased in both subpopulations, irrespective of the injection of 7KATMPSCA (). To address the functionality of intratumoral UniCAR-T cells, we purified these effector cells and incubated them in vitro with 51Cr labeled PC3PSCA in the presence or in the absence of 7KATMPSCA. While at low e:t ratios (< 5:1) the addition of 7KATMPSCA did not result in induction of specific lysis, application of 7KATMPSCA at e:t ratios ≥ 5:1 induced specific lysis, which demonstrates that functionality of in vivo passaged UniCAR-T cells had been diminished irrespective of the preceding exposition toward 7KATMPSCA in vivo (). We conclude that in vivo passaged UniCAR-T cells remain functional and that 7KATMPSCA induced changes occur only in a tight timely correlation with 7KATMPSCA administration, which demonstrates again the strict dependence of the activity of UniCAR-T cells on the presence of the TM.
Figure 5. Phenotypic and functional characterization of effector and target cells in vivo. Tumors were excised right before start of the treatment and immediately after one week therapy with 7KATMPSCA and were analyzed for various factors associated with activity of the UniCAR platform. (A) The proportion of viable tumor cells (hCD45−hCD3−hEpCAM+), which did not express PSCA, was numerically increased in the “PC3PSCA + UniCAR + 7KATMPSCA group,” however the difference did not reach statistical significance. (B) Administration of 7KATMPSCA to UniCAR-T cell engrafted, PC3PSCA tumor-bearing NSG mice led to a significant increase in tumor-infiltrating viable UniCAR-T cells (hCD45+hCD3+GFP+) (PC3PSCA + UniCAR: 1.9 ± 2.3, PC3PSCA + UniCAR (early): 2,1 ± 2.4, PC3PSCA + UniCAR + 7KATMPSCA: 9.5 ± 8,3) (C) Representative dot blots of post-thymic differentiation of CD8+ T cells and CD8+ UniCAR-T cells. The addition of 7KATMPSCA in vivo did not further alter the differentiation phenotype. Most CD8+ effector cells were effector memory cells (CCR7−CD45RO+): graft 45.3 ± 19.7%, early group 83.5 ± 8.8%, PC3PSCA + UniCARs 87.5 ± 4.2%, PC3PSCA + UniCARs + 7KATMPSCA 90.3 ± 4.4%. (D) Tumor-infiltrating UniCAR-T cells were sorted from NSG mice immediately after the treatment period. Purified UniCAR-T cells were co-incubated for 24 h with 51Cr labeled PC3PSCA in vitro in the presence or in the absence of 7KATMPSCA. Most analyses were performed in triplicates. Data were pooled for e:t < 5:1 (range 1:1–4:1) and e:t ≥ 5:1 (range 5:1–24:1). At low e:t ratios, in both cohorts ( ± preceding exposition toward 7KATMPSCA in vivo) no specific lysis could be induced by the addition of 7KATMPSCA. At higher e:t ratios, addition of 7KATMPSCA still induced specific lysis, irrespective of the preceding exposition to 7KATMPSCA in vivo, which proves remaining functionality of the UniCAR platform after in vivo passage. Bars represent mean ± SD. Data pooled from 2 independent experiments with individual donors (*p < 0.05, **p ≤ 0.01).
Immunoevasive mechanisms induced by activity of the UniCAR platform in vitro and in vivo
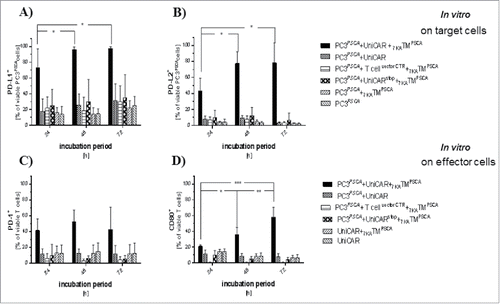
To explore counter-regulatory mechanisms with immunoevasive potential we analyzed the expression of several key immune checkpoint molecules, such as CD80, PD-L1, PD-L2, and PD-1 in vitro and in vivo.
In vitro, we found significant upregulation of PD-L1 and PD-L2 on target cells with a maximum after 48 h of co-incubation (, ) only in the presence of both UniCAR components. Especially PD-L1 was detected on virtually all tumor cells. On effector cells we identified an upregulation of PD-1 and CD80 (, ). PD-1 upregulation peaked at 24 h of co-incubation, while CD80 expression continually increased during the observation period. In contrast to the target cells, the upregulation was less homogeneous and restricted to subpopulations of UniCAR-T cells.
Figure 6. Upregulation of immune checkpoint molecules as a function of the activity of the UniCAR platform. PC3PSCA cells were (co)-incubated with/without UniCAR-T cells, non-signaling UniCAR-T cells (UniCARstop) and T cellvectorCTR in the presence or in the absence of 7KATMPSCA (EC90) at an e:t ratio of 1:1 for 24 h, 48 h and 72 h. The proportion of target (A, B) and effector cells (C, D), which expressed certain immune checkpoint molecules during co-incubation is shown. Upregulation of the mentioned immune checkpoint molecules was always restricted to the cohort encompassing all components necessary for activity of the UniCAR platform (target cell + TM + functional effector cell). (A) PD-L1 expression on tumor cells significantly increased in the presence of the active UniCAR platform. PD-L1 expression peaked at 48 h of co-incubation (13.9 ± 10.1% of PC3PSCA at baseline compared with 72.9 ± 24.1% at 24 hours, p ≤ 0.001). (B) PD-L2 expression on tumor cells was also significantly upregulated in the presence of the active UniCAR platform reaching its maximum at 48 h of co-incubation (4.1 ± 3.6% of PC3PSCA at baseline compared with 42.8 ± 16.1% at 24 hours, p ≤ 0.001). (C) PD-1 upregulation peaked after 24 h of co-incubation and did not further change during the observation period (12.8 ± 10.4% of UniCAR-T cells at baseline compared with 41.4 ± 14.8% at 24 hours, p < 0.05). (D) CD80 upregulation continually increased during the observation period (14.4 ± 3.5% of UniCAR-T cells at baseline compared with 21.2 ± 2.0% at 24 hours, p ≤ 0.01). Bars represent mean ± SD, data pooled from 4–5 independent experiments with individual donors (*p < 0.05, **p ≤ 0.01, ***p ≤ 0.0001).
In the high tumor burden model, tumors were excised right before and at the end of the treatment period and target cells as well as tumor-infiltrating effector cells were analyzed. In analogy to the in vitro findings, we observed a significant upregulation of PD-L2 on tumor cells only in the group of NSG mice engrafted with UniCAR-T cells and exposed to 7KATMPSCA.In contrast to the in vitro findings, a significant upregulation of PD-L1 was observed even in UniCAR-T cell engrafted tumor-bearing mice without application of 7KATMPSCA underlining the importance of the (micro-)environment on UniCAR-T cell/tumor cell interaction. Nonetheless, therapy with 7KATMPSCA further amplified expression of PD-L1 on tumor cells and ultimately both PD-L1 and PD-L2 expression was significantly elevated in UniCAR-T cell engrafted tumor-bearing mice receiving 7KATMPSCA compared with UniCAR-T cell engrafted tumor-bearing mice without administration of 7KATMPSCA at the end of the treatment period (, ). In contrast to our in vitro data, we did not observe upregulation of CD80 on tumor-infiltrating UniCAR-T cells in association with 7KATMPSCA administration. PD-1 instead was found to be expressed already on virtually all tumor-infiltrating UniCAR-T cells and the extent did not change under the influence of 7KATMPSCA (Data not shown). In tumors analyzed with time delay to the treatment period, PD-L2 expression on tumor cells remained significantly elevated in UniCAR-T cell engrafted tumor-bearing mice which had been treated with 7KATMPSCA compared with tumor-bearing mice without UniCAR-T cell injection. However, there was no significant difference anymore between both UniCAR-T cell engrafted cohorts (without 7KATMPSCA administration versus with). PD-L1 expression remained significantly elevated in both UniCAR-T cell engrafted tumor-bearing cohorts compared with tumor-bearing mice without UniCAR-T cell injection. The boosted expression of PD-L1 related to 7KATMPSCA treatment had declined and there was no significant difference observed anymore compared with those UniCAR-T cell engrafted mice which had never been exposed to 7KATMPSCA. After all, PD-L1 expression remained numerically elevated in both UniCAR-T cell engrafted cohorts compared with the time point before the treatment ( = early group,) (, ). These observations demonstrate the temporal association with 7KATMPSCA administration and its reversibility.
Figure 7. Activity of the UniCAR platform in solid tumors is associated with upregulation of PD-L1 and PD-L2 on tumor cells. PC3PSCA tumor-bearing NSG mice engrafted with UniCAR-T cells were treated for one week with 7KATMPSCA. Mice were either killed right before and after the treatment period (A,B) or at later time points due to the tumor size (C,D). Tumor cells were analyzed by flow cytometry. The mere presence of UniCAR-T-cells led to a significant upregulation of PD-L1 in vivo. PD-L1 expression was significantly further amplified after treatment with 7KATMPSCA (UniCAR-T cell engrafted mice without 7KATMPSCA administration killed after the treatment period compared with UniCAR-T cell engrafted mice receiving 7KATMPSCA, p ≤ 0.001) (A). An upregulation of PD-L2 on tumor cells was only found in association with the active UniCAR platform comprising target cells, effector cells and the TM (UniCAR-T cell engrafted mice without 7KATMPSCA administration compared with UniCAR-T cell engrafted mice receiving 7KATMPSCA, p < 0.05). (B). With time delay to the administration of 7KATMPSCA, PD-L1 expression declined comparing to the mice killed right after therapy but still remained significantly elevated compared with mice without UniCAR-T cell engraftment. The significant difference observed between both UniCAR-T cell engrafted cohorts right at the end of the treatment period had vanished. PD-L1 expression of both cohorts was still numerically elevated compared with UniCAR-T cell engrafted mice before the treatment period ( = early group) (C). PD-L2 expression did not decline and also remained significantly elevated in those mice which had been treated with 7KATMPSCA compared with mice without UniCAR-T cell engraftment. In contrast to earlier time points, there was no significant difference between both UniCAR-T cell engrafted cohorts anymore (D). Bars represent mean ± SD. Data are pooled from 2 independent experiments with individual donors (n = 10) (*p < 0.05, **p ≤ 0.001,***p ≤ 0.001, ****p ≤ 0.0001).
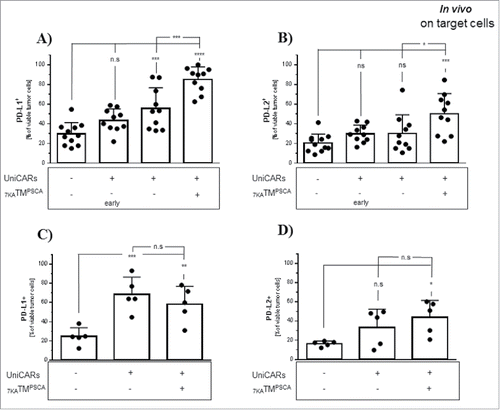
We conclude that activity of the combination of 7KATMPSCA with the UniCAR system in this solid tumor setting is associated with an upregulation of PD-L1 and PD-L2 on tumor cells in vitro and in vivo.
Discussion
The UniCAR platform provides a regulatable approach to CAR, thereby it aims to improve patient safety and the management of CAR-associated side effects. Furthermore, splitting the antigen binding moiety and the signaling domain allows a rapid change of the TAA to be targeted.Citation25-28 Both features are of special interest for a successful translation of CAR therapy to solid tumors. Here we demonstrate that the combination of a novel anti-PSCA TM with UniCAR T cells can efficiently target PSCA over-expressing solid neoplasms and mediate potent anti-tumor activity in vitro and in vivo. Specific anti-tumor cytotoxicity in vitro appeared at nanomolar concentrations. Moreover, we show that this UniCAR/TM combination induces phenotypic and functional changes in situ and is associated with counter-regulation of inhibitory immune checkpoint molecules providing a rationale for testing combinational therapeutic approaches to enhance anti-tumor efficacy.
PSCA is a representative example of a non-tumor-specific TAA with a need for optimized controllability to avoid excess of on-target/off-tumor toxicity. Conventional CAR-T cells targeting PSCA positive malignancies have been described previously.Citation39-43 First-generation CAR-T cells against PSCA were shown to efficiently kill PSCA-expressing pancreatic cancer cell lines in vitro.Citation39 However, using an in vivo model, the ip injection of these first-generation CAR-T cells only provided modest and temporarily restricted anti-tumor activity.Citation41 In addition to the well described limitations of first-generation CAR-T cells with regard to their in vivo activity, the same group also described downregulation and loss of TAAs as potential resistance mechanism. In our in vivo model, we also observed an increased proportion of tumor cells, which lacked PSCA expression, but the extent did not reach statistical significance and no outgrowth of PSCA negative tumors appeared. These in part contrary observations might be caused by utilization of cells endogenously expressing PSCA as compared with tumor cells that are genetically engineered to express PSCA. Interestingly, antigen loss was only significantly correlated with the amount of tumor-infiltrating effector cells in the cohort receiving the TM but not with cytokine levels in PB. Furthermore, one can speculate that the time-restricted selection pressure of the switchable CAR-T cells promotes less target escape variants compared with conventional CAR-T cells with continuous anti-tumor activity. This hypothesis assumes that CARs fail to completely eradicate TAA-expressing tumor cells (e.g. due to microenvironmental barriers) and that sustained CAR engraftment and activity induces a state of persisting minimal residual disease. In fact, long-term relapse-free survival for B cell malignancies has only been observed in patients with sustained engraftment of conventional CARs and therefore one cannot discriminate between cure and a chronic disease controlled by activity of the CARs.Citation7,13 In the case that complete eradication is achieved within a given continuous phase of TM administration, the selective pressure of the UniCAR platform would be certainly equal to conventional CARs. With regard to the UniCAR system, loss or downregulation of one TAA might be counteracted by application of another TM directed against an alternative TAA. Nevertheless, using conventional second-generation and third-generation CAR-T cells, several groups could show that PSCA-expressing malignancies can be successfully targeted in vivo.Citation22,40,42
A prerequisite for anti-tumor activity is the infiltration of the tumor tissue by functional CAR-T cells.Citation44 In contrast to conventional CARs, pharmacokinetic properties of the TM have to be considered in the context of the UniCAR platform. Here we showed that UniCAR-T cells were able to long-term infiltrate sc transplanted tumor tissues and that PB chimerism did not predict the presence of UniCAR-T cells within the tumors. Furthermore, the infiltration of the tumor tissue appeared regardless of TM availability, whereas the extent of infiltrating effector cells could be positively influenced by the administration of 7KATMPSCA. This observation suggested that 7KATMPSCA successfully penetrated into the tumor tissue. In addition, we observed a rapid increase of fluorescence intensity in the tumor after iv injection of fluorochrome and also of the 64Cu labeled 7KATMPSCA, which supports its intratumoral disposability. Interestingly, there were striking differences concerning the persistence of the TM in the tumor as suggested by detection of fluorescence and radioactivity. Fluorescence could be detected in the tumor region over a prolonged period, while radioactivity was cleared faster. In PB, radioactivity measurement and ELISA provided similar results and showed a short half-life of 7KATMPSCA in the circulation. Therefore, it should be taken into consideration that detection of fluorescence might have overestimated tumor-specific half-life of the TM. As both fluorescence and radioactivity are not necessarily equivalent to functional TM, it should be taken into account that different organs may vary in the tissue specific half-life and that the availability might also differ between various compartments of the organism.Citation27,45 In summary, we were able to demonstrate that the UniCAR platform can evoke potent anti-tumor activity in vivo despite temporally and spatially separated injection of both components.
In the era of immunotherapy, adaptive immunoevasion has emerged as a mechanism of resistance.Citation17 In the context of cancer, progressive T cell exhaustion has been associated with the expression of inhibitory receptors and specific cytokine profiles.Citation46 Tumor cells have been described to amplify the expression of co-inhibitory ligands under the influence of T cell derived cytokines promoting T cell exhaustion.Citation47 Correspondingly, we found a 7KATMPSCA-induced upregulation of PD-1 on effector cells in vitro. In contrast, tumor-infiltrating UniCAR-T cells inherently expressed PD-1 irrespective of TM application. It can be speculated that the tumor microenvironment mediated the upregulation of PD-1, as sustained xenogenic stimulation might have contributed to the PD-1 upregulation,Citation48 although no obvious signs of xenogeneic graft vs. host disease occurred.Citation49 Consistent with the expression of PD-1, we found a 7KATMPSCA-associated boost in the expression of the corresponding co-inhibitory ligands PD-L1 and PD-L2 on tumor cells in vitro and in vivo, which suggests that UniCAR-T cell efficacy might have been limited by stimulation of the PD-1 axis in the tumor microenvironment as shown for conventional CAR-T cells.Citation50 Importantly, PD-L1 expression on tumor cells was already upregulated in the mere presence of UniCAR-T cells irrespective of the TM administration, also hinting to a underlying activation of UniCAR-T cells related to xenoreactivity or alloreactivity in our model. Expression of CD80 on effector cells was inconsistent between in vitro and in vivo. Therefore, the role of CD80 in the modulation of UniCAR-T cell activity remains to be further elucidated, especially because CD80 can play controversial roles providing co-inhibitory signals e.g., by binding to PD-L1 as well as mediating co-stimulatory signals.Citation51-53 The effector memory phenotype of most tumor-infiltrating UniCAR-T cells was also compatible with exhaustion. Despite these phenotypic changes, UniCAR-T cells retained their functionality in vivo as 7KATMPSCA injection induced increased release of INF-γ, TNF-α, IL-18, and IL-4. In addition, intratumoral UniCAR-T cells still mediated specific cytotoxicity ex vivo. The absence of measurable IL-2, the only moderate increase in INF-γ secretion, as well as the reduced ex vivo killing capacity suggested that engrafted UniCAR-T cells had undergone exhaustion, which might be related to the tumor microenvironment or xenogeneic stimulation. However, the numerical increase of INF-γ and IL-4 secretion in vivo was well in line with the adaptive immunoevasion hypothesis, as INF-γ and IL-4 are potent inducers of PD-L1 and PD-L2 expression, respectively.Citation47,54 Analysis of cytokines in the treated cohort demonstrated a general correlation among all released cytokines. Most notably, INF-γ was significantly correlated only with IL-4 underscoring the importance of the PD-1 axis by simultaneous induction of PD-L1 and PD-L2 expression. Surprisingly, there was only a weak (non-significant) or even no correlation at all between cytokine levels and amount of infiltrating UniCAR-T cells. Cytokine release in the cohort of UniCAR-T cell engrafted tumor-bearing mice which did not receive 7KATMPSCA therapy was most likely due to subclinical xGvHDCitation55,56 or alloreactivity.Citation57 Importantly, we observed a decline in PD-L1 and PD-L2 expression on tumor cells after cessation of 7KATMPSCA treatment, which suggests that the induction of adaptive immunoevasion was restricted to the activity of the UniCAR platform. Therefore, breaks in TM application might be helpful to restore functionality of UniCAR-T cells. Vice versa, targeting of the PD-1 axis e.g., using immune checkpoint inhibitors might be helpful to restore functionality and activity of UniCAR-T cells.Citation43,50
In conclusion, we demonstrated that the combination of the novel 7KATMPSCA with UniCAR-T cells can be used to target specifically and efficiently a PSCA over-expressing solid neoplasm in vitro and in vivo. In a clinically relevant experimental setting, we showed that the spatially and temporally independent administration of both components of the UniCAR platform is feasible and associated with potent anti-tumor activity. For translation into the clinic, pharmacokinetic aspects of the respective TM will be of particular interest as both efficacy and toxicity largely depend on the presence of the TM in different tissues. Additionally, activity of the combination of 7KATMPSCA with the UniCAR platform was associated with an adaptive immunoevasive response. Further studies including different tumor models will have to reveal whether the combination with immune checkpoint inhibitors or the intermittent administration of the TM can revert immunosubversion and increase the effectivity of the UniCAR platform.
Materials and methods
Cell lines
Chinese Hamster ovarian (CHO), PC3wt (wild type) and PC3PSCA cells were cultured in RPMI 1640 medium (Biochrom, F 1215) supplemented with 10% FCS (Biochrom, S 0415), 100 U/ml penicillin and 100 μg/ml streptomycin (Biochrom A 2212), 2 mM N-acetyl-L-alanyl-L-glutamine (Biochrom, K 0302), 1% non-essential amino acids (Biochrom, K 0293) and 1 mM sodium pyruvate (Biochrom L 0473). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2. The cell lines were purchased from the American Type Culture Collection. PC3wt had been modified to express PSCA as described before resulting in the cell line PC3PSCA.Citation58
Generation of UniCAR vectors
The effector module consisted of a second-generation CAR-T cell recognizing the epitope 5B9 of the nuclear antigen La/SS-B. 25-28 The cloning of the anti-La/SS-B 5B9 scFv and generation of the hinge, transmembrane and signaling domain of the CAR has been described previously in detail.Citation38,59 The Stop variant of the UniCAR lacked intracellular sequences downstream of the CD28 transmembrane domain (UniCARstop).The open reading frame (orf) of the UniCAR constructs was fused upstream to a sequence encoding for eGFP separated by a Thosea Asigna virus 2A peptide..Citation59,60
Isolation and lentiviral transduction of T cells
The study was approved by the ethics committee of the medical faculty Carl Gustav Carus, TU Dresden (EK27022006) and the procedure has been described in detail before.Citation61 After successful genetic modification verified by concomitant expression of eGFP, T cells were purified by fluorescence-activated cell sorting (FACS) using a FACS Aria II (Becton Dickinson BD, Heidelberg, Germany).
Construction, purification and binding analysis of the targeting module
Cloning of the humanized anti-PSCA TM (TMPSCA) has been described in detail elsewhere.Citation28,38 In the present work, this TMPSCA was modified to contain a lysine-tag (7KA) instead of a myc-tag between the 5B9-tag and the His-tag, termed 7KATMPSCA. The lysine-tag was introduced to enable more efficient labeling with fluorochromes to facilitate pharmacokinetic experiments. A recombinant CHO cell line for permanent 7KATMPSCA production was generated and the 7KATMPSCA was purified from cell culture supernatants using Ni-NTA affinity chromatography (Qiagen, 31014). The methods used to quantify and characterize the purified protein were described previously.Citation58,62 Binding specificity of the purified 7KATMPSCA was verified using PC3wt and PC3PSCA cell lines. For saturation binding experiments, a total number of 1 × 106 PC3PSCA cells were labeled with the respective amount of 7KATMPSCA. Surface bound 7KATMPSCA was detected on viable cells (DAPI exclusion staining) using the His-tag. Binding characteristics were calculated using GraphPad Prism 6.0 (GraphPad Software, San Diego, California, USA). The extent of 7KATMPSCA binding was calculated as relative change in geometric mean fluorescence intensity (GeoMFI) compared with control.
Flow cytometry
Flow cytometry was performed on LSRII (BD) and MACSQuant (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). FACS was performed on Aria II (BD). All analyses were done using FlowJo vX.0.6 software (FlowJo, LLC, Ashland, Oregon, USA). All samples were prepared as single cell suspension and erythrocytes were removed using ACK lysing buffer (Thermo Fisher Scientific, A1049201). In tumor specimens, unspecific binding of mAb was reduced using human TruStain FcX (BioLegend, 422302) and purified anti-mouse CD16/CD32) (eBioscience, 14–0161–82). The following primary antibodies recognizing human antigens (clones and catalog numbers are given in parentheses) were used in various combinations. From BD: CD4 V500 (RPA-T4, 560769), CD45 V500 (HI30, 560777), and CD197 BV421 (150503, 562555). From eBioscience: CD3 APC eFluor780 (UCHT1, 147–0038–42), CD4 APC (RPA-T4, 17–0049–42), CD8a PE-Cy7 (SK1, 25–0087–42), CD25 PE (BC96, 12–0259–42), CD45 APC eFluor780 (HI30, 47–0459), CD45RO PerCP eFluor710 (UCHL1, 46–0457–41), CD80 PerCP eFluor710 (2D10.4, 46–0809–42), CD274 APC (MIH1, 17–5983–42), CD273 PE (MIH18, 12–5888–42) and CD326 PE-Cy7 (1B7, 25–9326). From BioLegend: CD3 Alexa Fluor 700 (HIT3a, 300242), CD45 Alexa Fluor647 (HI30, 304018), CD69 PerCP-Cy5.5 (FN50, 310926), CD86 BV421 (IT2.2, 305426), and CD279 PerCP eFluor710 (EH12.2H7, 329920). The anti-La/SS-B 5B9 (recognizing the epitope 5B9) was an in-house mAb and has been described before.Citation62,63 The following secondary/tertiary mAbs and derivatives were used: anti-His FITC (GG11–8F3.5.1, 130–092–675) and PE (GG11–8F3.5.1, 130–098–810) both from Miltenyi, and goat anti-mouse F(ab’)2 PE (polyclonal) from Beckman Coulter (IM0551). Murine leucocytes were stained using anti-CD45 Pacific Blue (30-F11, 103126) from BioLegend. Doublet discrimination was routinely performed and dead cells were excluded by DAPI and PI staining. In general, UniCAR-T cells were identified by their concurrent expression of CD45, CD3 and GFP, while PC3 cells were characterized by negativity for CD45, CD3 and positivity for CD326 (EpCAM).
Cytotoxicity assay
The cytotolytic potential of UniCAR-T cells was analyzed in vitro using a 51Cr release assay varying the e:t ratio and the concentration of 7KATMPSCA as described previously.Citation61 For the ex vivo cytotoxicity assay, UniCAR-T cells were isolated from PC3PSCA tumors of UniCAR-T cell engrafted NSG mice after one week treatment with 7KATMPSCA by FACS. These purified UniCAR-T cells were co-incubated with cell culture derived 51Cr labeled PC3PSCA cells for 24 h at different e:t ratios either in absence or at EC90 (as determined in the in vitro cytotoxicity assay) of 7KATMPSCA.
For the analysis of cell surface expression of various immune checkpoint molecules in the course of the in vitro cytotoxicity assay, UniCAR-T cells were incubated with PC3PSCA cells at an e:t ratio of 1:1 and with 7KATMPSCA at EC90. Non-signaling UniCAR-T cells (UniCARstop), and GFP-transduced T cells (T cellvectorCTR) were used as controls.
Animal experiments
All animal experiments were performed according institutional guidelines and the German animal protection law (Landesdirektion Sachsen, 24D-9168.11–4/2007–2, 24–9168.21–4/2004–1, TVV11/2014). Five- to 8-week-old male NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) and NMRI-nu (RjOrl:NMRI-Foxn1nu/Foxn1nu) mice were used. The NMRI-nu mouse PC3PSCA tumor model was established to evaluate the biodistribution and kinetics of radiolabeled 7KATMPSCA under non-sterile conditions in the positron-emission-tomography (PET).
A low and a high tumor burden xenotransplantation model were established in NSG mice. In the low tumor burden model, NSG mice were intravenously (iv) injected with 1 × 106 UniCAR-T cells. Three to 4 weeks after injection, mice were subcutaneously (sc) transplanted in the right flank with 1 × 106 PC3PSCA cells in 100 µl PBS:Matrigel® (1:1). One week later, mice were treated for 7 d by intraperitoneal (ip) injections twice daily (bid) of 250 ng 7KATMPSCA per gram body weight (bw). The elongated treatment period of 7 d was chosen, because extended exposition toward the TM enhances the cytolytic capacity in vitro and because in a previous preclinical AML model, a 2-day treatment period was associated with a numerical, but not statistical significant prolonged overall survival.Citation59 Survival and tumor size were monitored for up to 13 weeks after tumor implantation.
In the high tumor burden model, equal cell numbers of UniCAR-T cells and PC3PSCA were used. However, NSG mice were first sc injected with tumor cells, followed by iv application of UniCAR-T cells 4 weeks later. Another 3−4 weeks later, treatment with ip injections of 250 ng 7KATMPSCA/g bw bid for one week was initiated. In contrast to the low tumor burden model, follow up was not timely restricted. In both models 7KATMPSCA treatment was initiated around 4 weeks after UniCAR-T cell injection at the anticipated peak of PB engraftment and tumor-bearing mice were killed when deterioration of health occurred or when tumors exceeded 15 mm in diameter. All mice were weighed once weekly and tumor size was determined twice weekly using a caliper and expressed as volume [maximal length x (orthogonal width)2/2]. Additionally, in the high tumor burden model neoplasms were removed at the time point of euthanization for further analysis. In a separate cohort of the high tumor burden model, mice were killed immediately before (termed early group) and right after treatment with 7KATMPSCA.
For immuno-PET imaging and analysis, NMRI-nu mice were sc transplanted in the left hind leg with 1 × 106 PC3wt and in the right hind leg with 1 × 106 PC3PSCA cells in 100 µl PBS. Six to 8 weeks after cell inoculation, tumor bearing animals were selected for PET studies.
Preparation of single cell suspension from tumors
Before enzymatic digestion, xenograft tumors were cut up into small pieces and minced completely. To obtain single cell suspensions, the resulting tumor pieces were then mixed with 2 mg/ml collagenase Ia and DNAse (Sigma-Aldrich, C9891) in PBS containing 2% FCS and allowed to incubate (37°C, 40 min). Samples were mixed every 10 min. Finally, cells were filtered through a 40 µm cell strainer (BD) and washed with PBS containing 5% FCS.
Multiplex cytokine quantification assay
Mice were killed at the end of the treatment period in the high tumor burden model. PB was collected using micro-hematocrit capillaries (Brand GmbH, Wertheim, Germany) from mice before euthanization. Plasma was analyzed with a Human Th1/Th2 Extended 11 plex (ProcartaPlex, Affymetrix, eBioscience) according to the manufacturer's instructions on a Luminex® 200™ System.
Biodistribution of fluorochrome labeled 7KATMPSCA
7KATMPSCA was labeled with CF770SE according to the manufacturer's instructions (Biotium, Fremont, California, USA). All tumor bearing NSG mice ( ± UniCAR-T cell engraftment) received an iv bolus injection of 50 µg labeled 7KATMPSCA. In vivo optical imaging of near infrared labeled 7KATMPSCA was performed at different time points after injection (up to 15 days) using IVIS Spectrum (PerkinElmer, Walham, Massachusetts, USA). Data was analyzed using LivingImage® 4.3.1 (PerkinElmer). Relative fluorescence intensity was evaluated as background subtracted maximum radiant efficiency (REmax). Changes in fluorescence intensity were depicted as relative signal intensity calculated as REmax at a specific time point divided by REmax before 7KATMPSCA administration.
7KATMPSCA radiolabeling
The quality control of the conjugation with the chelator and the radio-labeling of 7KATMPSCA are summarized in Fig. S5.
NODAGA conjugation with 7KATMPSCA
7KATMPSCA was dialyzed against PBS and stored at a concentration of 1 mg/mL. For immuno-PET imaging studies 7KATMPSCA was conjugated with the chelator NODAGA. Prior to the conjugation, PBS buffer was exchanged by 0.1 M borate buffered saline (pH 8.5) using spin filtration (3 times at 7°C, 4000 rpm). Then 40 equivalents of 2,20-(7-(1-carboxy-4-((4-isothiocyanatobenzyl)amino)-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (p-SCN-Bz-NODAGA) ester were added to the TM solution. The mixture was left at 4°C for 20 h. Excess chelator was removed by spin filtration with PBS.
MALDI-TOF MS analysis
The average number of chelators per 7KATMPSCA molecule was determined using MALDI-TOF MS. A thin layer of a saturated sinapinic acid solution in ethanol was applied onto the target plate (Bruker Daltonik) and left to dry. On top of this layer, a mixture (50:50 v/v) of the sample and a saturated sinapinic acid solution in 0.1% TFA, 30% acetonitrile in water was added. The samples were left to dry before the analysis was performed (Autoflex, Bruker). To determine the number of attached chelator molecules the non-conjugated and the purified conjugate were mixed on the target plate and measured simultaneously. We estimated a ratio of 1.5 chelator per molecule of the TM. Therefore, the conjugated TM was termed as 7KATMPSCA(NODAGA)1.5.
Radiolabeling of 7KATMPSCA(NODAGA)1.5
The 64Cu solution was produced in house.Citation27,28,64,65 The radiolabeling yield and stability of the 64Cu-labeled antibody conjugate were monitored with radio-TLC (thin layer chromatography) and radio-HPLC (High performance liquid chromatography).
Immuno-PET imaging
General anesthesia was induced with 10% (v/v) and maintained with inhalation of 8% (v/v) desflurane (Suprane, Baxter) in 30/10% (v/v) oxygen/air. To evaluate tumor targeting of 64Cu-7KATMPSCA (NODAGA)1.5 and in vivo PSCA expression levels, immuno-PET imaging was performed in PC3PSCA- and PC3wt-tumor-bearing NMRI-nu mice. 64Cu-7KATMPSCA (NODAGA)1.5 (approximately 3.7 MBq) was iv injected into the lateral tail vein and dynamic scans were acquired over 2 h and static scans were done 24 and 48 h after injection using a small animal PET/CT scanner (NanoPET/CT, Mediso). The quantitative scales were expressed as SUV (standardized uptake value) using the activity concentration normalized to the body weight, which was calculated as tissue concentration (MBq/mL)/injected dose (MBq)/body weight (g) in (g/mL). Images were visualized using ROVER software (ABX). The time-activity curves were depicted as percent of maximum activity concentration.
Plasma pharmacokinetics
NSG mice were injected either iv or ip with 250 ng 7KATMPSCA/g bw and PB was obtained at the indicated time points. 7KATMPSCA plasma concentrations were determined with an in-house ELISA as recently described.Citation59 Pharmakokinetic parameters of the plasma were calculated using the non-compartmental analysis tool of PKSolver 2.0.Citation66
Statistics
For in vivo and in vitro experiments, we used unpaired student's t test to evaluate continuous variable of 2 groups and 1-way ANOVA either with post-test Fisher's least significance difference (LSD) to evaluate continuous variable of 3 groups, with Dunnet for evaluating more than 3 groups or with Tukey for evaluating multiple comparisons. Tumor-growth curves were illustrated using a last observation carried forward (LOCF) approach. The last available value of tumor-volume of killed mice was continually used to the scheduled end of the experiment. Tumor volume doubling time (VDT) was calculated using GraphPad Prism 6.0 based on the time-span after TM administration. Survival was analyzed by the Kaplan-Maier method and the log-rank (Mantel-Cox) test to compare pairs of groups. Statistics were computed using GraphPad Prism 6.0. Values of p < 0.05 were considered statistically significant.
Disclosure of potential conflicts of interest
Armin Ehninger, Gerhard Ehninger and Michael Bachmann are shareholders of the companies CPT and/or GEMoaB respectively, which hold licenses, patents and have filed patent applications related to the UniCAR platform. Marc Cartellieri, Armin Ehninger, Simon Loff and Johannes Spehr are used by CPT and GEMoaB, respectively.
Supplementary_materials.zip
Download Zip (450.4 KB)Acknowledgments
We thank Claudia Richter, Katrin Müller and Robert Kuhnert for technical assistance. The authors thank Martin Walther, Ulrike Gesche, and Christian Jentschel for the preparation of the 64Cu-isotope solutions, Andrea Suhr for her technical assistance with radiolabeling, and Regina Herrlich for assistance in the animal experiments.
Funding
This study was partly supported by grants provided to Ralf Bergmann by the Deutsche Forschungsgemeinschaft (BE 2607/1–2).
References
- Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 1989; 86:10024-28; PMID:2513569; https://doi.org/10.1073/pnas.86.24.10024
- Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol 2002; 20:70-5; PMID:11753365; https://doi.org/10.1038/nbt0102-70
- Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 2009; 21:215-23; PMID:19327974; https://doi.org/10.1016/j.coi.2009.02.009
- Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011; 118:4817-28; PMID:21849486; https://doi.org/10.1182/blood-2011-04-348540
- Brentjens RJ, Davila ML, Rivèire I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5:177ra38; PMID:23515080; https://doi.org/10.1126/scitranslmed.3005930
- Porter DL, Levine BL, Kalos M, Bagg A, June, CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Eng J Med 2011; 365:725-33; PMID:21830940; https://doi.org/10.1056/NEJMoa1103849
- Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7:303ra139; PMID:26333935; https://doi.org/10.1126/scitranslmed.aac5415
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3:95ra73; PMID:21832238; https://doi.org/10.1126/scitranslmed.3002842
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119:2709-20; PMID:22160384; https://doi.org/10.1182/blood-2011-10-384388
- Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015; 33:540-9; PMID:25154820; https://doi.org/10.1200/JCO.2014.56.2025
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Eng J Med 2013; 368:1509-18; PMID:23527958; https://doi.org/10.1056/NEJMoa1215134
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra25; PMID:24553386; https://doi.org/10.1126/scitranslmed.3008226
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Eng J Med 2014; 371:1507-17; PMID:25317870; https://doi.org/10.1056/NEJMoa1407222
- Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, Zheng Z, Vogl DT, Cohen AD, Weiss BM, et al. Chimeric Antigen Receptor T Cells against CD19 for multiple myeloma. N Eng J Med 2015; 373:1040-7; PMID:26352815; https://doi.org/10.1056/NEJMoa1504542
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015; 385:517-28; PMID:25319501; https://doi.org/10.1016/S0140-6736(14)61403-3
- Stewart TJ, Abrams SI. How tumours escape mass destruction. Oncogene 2008; 27:5894-903; PMID:18836470; https://doi.org/10.1038/onc.2008.268
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; https://doi.org/10.1038/nrc3239
- Brudno NJ, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016; 127:3321-30; PMID:27207799; https://doi.org/10.1182/blood-2016-04-703751
- Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: Current understanding and future directions. J Gene Med 2012; 14:405-15; PMID:22262649; https://doi.org/10.1002/jgm.2604
- Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma / leukemia effects and safety. Leukemia 2010; 24:1160-70; PMID:20428207; https://doi.org/10.1038/leu.2010.75
- Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 2013; 5:215ra172; PMID:24337479; https://doi.org/10.1126/scitranslmed.3006597
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 2013; 31:71-5; PMID:23242161; https://doi.org/10.1038/nbt.2459
- Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci USA 2016; 113:E450-8; PMID:26759368; https://doi.org/10.1073/pnas.1524193113
- Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci USA 2016; 113:E459-68; PMID:26759369; https://doi.org/10.1073/pnas.1524155113
- Koristka S, Cartellieri M, Feldmann A, Arndt C, Loff S, Michalk I, Aliperta R, von Bonin, M, Bornhäuser M, Ehninger E, et al. Flexible antigen-specific redirection of human regulatory T Cells Via a novel universal chimeric antigen receptor system. Blood 2014; 124, 3494; http://www.bloodjournal.org/content/124/21/3494. Paper presented at: 56th Annual Meeting and Exposition of the American Society of Hematology; 2014 Dec 6; San Francisco, USA
- Cartellieri M, Loff S, von Bonin M, Bejestani EP, Ehninger A, Feldmann A, Koristka A, Arndt A, Ehninger G, Bachmann MP. UniCAR: A novel modular retargeting platform technology for CAR T Cells. Blood 2015; 126, 5549; http://www.bloodjournal.org/content/126/23/5549. Paper presented at: 57th Annual Meeting and Exposition of the American Society of Hematology; 2015 Dec 8; Orlando, USA
- Albert S, Arndt C, Felmann A, Bergmann R, Bachmann D, Koristka S, Ludwig F, Ziller-Walter P, Kegler A, Gärtner S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunol 2017; 6(4):e1287246; PMID:28507794; https://doi.org/10.1080/2162402X.2017.1287246
- Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri C, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology ‘UniCAR’. Oncotarget 2017; 8(19):31368-85; PMID:28404896; https://doi.org/10.18632/oncotarget.15572
- Carmo-Fonseca M, Pfeifer K, Schröder HC, Vaz MF, Fonseca JE, Müller WE, Bachmann M. Identification of La ribonucleoproteins as a component of interchromatin granules. Exp Cell Res 1989; 185:73-85; PMID:2530103; https://doi.org/10.1016/0014-4827(89)90038-4
- Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, Wahl M, Nisitani S, Yamashiro J, Le Beau MM, et al. Prostate stem cell antigen: A cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci USA 1998; 95:1735-40; PMID:9465086; https://doi.org/10.1073/pnas.95.4.1735
- Saeki N, Gu J, Yoshida T, Wu X. Prostate stem cell antigen: A Jekyll and Hyde molecule? Clin Cancer Res 2010; 16:3533-8; PMID:20501618; https://doi.org/10.1158/1078-0432.CCR-09-3169
- Rodriguez JA, Li M, Yao Q, Chen C, Fisher WE. Gene overexpression in pancreatic adenocarcinoma : Diagnostic and therapeutic implications. World J Surg 2005; 29:297-305; PMID:15696394; https://doi.org/10.1007/s00268-004-7843-0
- Cunha, AC, Weigle B, Kiessling A, Bachmann M, Rieber EP. Tissue-specificity of prostate specific antigens : Comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett 2006; 236:229-38; PMID:16046056; https://doi.org/10.1016/j.canlet.2005.05.021
- Gu Z, Thomas G, Yamashiro J, Shintaku IP, Dorey F, Raitano A, Witte ON, Said JW, Loda M, Reiter RE. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000; 19:1288-96; PMID:10713670; https://doi.org/10.1038/sj.onc.1203426
- Morris MJ, Eisenberger MA, Pili R, Denmeade SR, Rathkopf D, Slovin SF, Farrelly J, Chudow JJ, Vincent M, Scher HI, et al. A phase I / IIA study of AGS-PSCA for castration-resistant prostate cancer. Ann Oncol 2012; 23:2714-9; PMID:22553195; https://doi.org/10.1093/annonc/mds078
- Wolpin BM, O'Reilly EM, Ko YJ, Blaszkowsky LS, Rarick M, Rocha-Lima CM, Ritch P, Chan E, Spratlin J, Macarulla T, et al. Global, multicenter, randomized, phase II trial of gemcitabine and gemcitabine plus AGS-1C4D4 in patients with previously untreated, metastatic pancreatic cancer. Ann Oncol 2013; 24:1792-801; PMID:23448807; https://doi.org/10.1093/annonc/mdt066
- Arndt C, Feldmann A, Koristka S, Cartellieri M, Dimmel M, Ehninger A, Ehninger G, Bachmann M. Simultaneous targeting of prostate stem cell antigen and prostate-specific membrane antigen improves the killing of prostate cancer cells using a novel modular T cell-retargeting system. Prostate 2014; 74:1335-46; PMID:25053443; https://doi.org/10.1002/pros.22850
- Arndt C, Feldmann A, Töpfer K, Koristka S, Cartellieri M, Temme A, Ehninger A, Ehninger G, Bachmann M. Redirection of CD4+ and CD8+ T lymphocytes via a novel antibody-based modular targeting system triggers efficient killing of PSCA+ prostate tumor cells. Prostate 2014; 74:1347-58; PMID:25053504; https://doi.org/10.1002/pros.22851
- Katari UL, Keirnan JM, Worth AC, Hodges SE, Leen AM, Fisher WE, Vera JF. Engineered T cells for pancreatic cancer treatment. HPB 2011; 13:643-50; PMID:21843265; https://doi.org/10.1111/j.1477-2574.2011.00344.x
- Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, Yu Z, Burns W, Miermont AM, Teper Y, Rudloff U, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic. cancer. Hum Gene Ther 2014; 25:1003-12; PMID:24694017; https://doi.org/10.1089/hum.2013.209
- Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, Fisher WE, Heslop HE, Rooney CM, Brenner MK, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther 2014; 22:623-33; PMID:24213558; https://doi.org/10.1038/mt.2013.262
- Hillerdal V, Ramachandran M, Leja J, Essand M. Systemic treatment with CAR-engineered T cells against PSCA delays subcutaneous tumor growth and prolongs survival of mice. BMC Cancer 14:30; PMID:24438073; https://doi.org/10.1186/1471-2407-14-30
- Liu X, Ranganathan R, Jiang S, Fan C, Sun J, Kim S, Newick K, Lo A, June C, Zhao Y, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res 2016; 76:1578-90; PMID:26979791; https://doi.org/10.1158/0008-5472.CAN-15-2524
- Zhang H, Ye ZL, Yuan ZG, Luo ZQ, Jin HJ, Qian QJ. New strategies for the treatment of solid tumors with CAR-T cells. Int J Biol Sci 2016; 12:718-29; PMID:27194949; https://doi.org/10.7150/ijbs.14405
- Mayer AT, Natarajan A, Gordon SR, Maute RL, McCracken MN, Ring AM, Weissman IL, Gambhir SS. Practical immuno-PET radiotracer design considerations for human immune checkpoint imaging. J Nucl Med 2017; 58:538-46; PMID:27980047; https://doi.org/10.2967/jnumed.116.177659
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15:486-99; PMID:26205583; https://doi.org/10.1038/nri3862
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha, TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013; 5:200ra116; PMID:23986400; https://doi.org/10.1126/scitranslmed.3006504
- Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, Foley JE, Massey PR, Felizardo TC, Riley JL, et al. The PDL1-PD1 axis converts human TH 1 cells into regulatory T cells. Sci Transl Med 2011; 3:111ra120; PMID:22133721; https://doi.org/10.1126/scitranslmed.3003130
- von Bonin M, Wermke M, Cosgun K, Thiede C, Bornhäuser M, Wagemaker G, Waskow C. In vivo expansion of co-transplanted T cells impacts on tumor re-initiating activity of human acute myeloid leukemia in NSG mice. PLoS One 2013; 8:e60680; PMID:23585844; https://doi.org/10.1371/journal.pone.0060680
- Cherkassky L, Morello A, Villena-vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 2016; 126:3130-44; PMID:27454297; https://doi.org/10.1172/JCI83092
- Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with B7-1 to inhibit T cell proliferation. Immunity 2007; 27:111-22; PMID:17629517; https://doi.org/10.1016/j.immuni.2007.05.016
- Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA4 Is a second receptor for the B Cell activation antigen B7. J Exp Med 1991; 174:561-9; PMID:1714933; https://doi.org/10.1084/jem.174.3.561
- Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci USA 1990; 87:5031-5; PMID:2164219; https://doi.org/10.1073/pnas.87.13.5031
- Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA 2003; 100:5336-41; PMID:12697896; https://doi.org/10.1073/pnas.0931259100
- Büchner SM, Sliva K, Bonig H, Völker I, Waibler Z, Kirberg J, Schnierle BS. Delayed onset of graft-versus-host disease in immunodeficent human leucocyte antigen-DQ8 transgenic, murine major histocompatibility complex class II-deficient mice repopulated by human peripheral blood mononuclear cells. Clin Exp Immunol 2013; 173:355-64; PMID:23607364; https://doi.org/10.1111/cei.12121
- Parmar S, Liu X, Tung SS, Robinson SN, Rodriguez G, Cooper LJ, Yang H, Shah, N, Yang H, Konopleva M, et al. Third-party umbilical cord blood-derived regulatory T cells prevent xenogenic graft-versus-host disease. Cytotherapy 2014; 16:90-100; PMID:24480547; https://doi.org/10.1016/j.jcyt.2013.07.009
- Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol 2012; 12:443-58; PMID:22576252; https://doi.org/10.1038/nri3212
- Feldmann A, Stamova S, Bippes CC, Bartsch H, Wehner R, Schmitz M, Temme A, Cartellieri M, Bachmann M. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: Comparison of different antibody formats. Prostate 2011; 71:998-1011; PMID:21541976; https://doi.org/10.1002/pros.21315
- Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J 2016; 6:e458; PMID:27518241; https://doi.org/10.1038/bcj.2016.61
- Szymczak AL, Workman CJ, Wang Y, Vignali K, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol 2004; 22:589-94; PMID:15064769; https://doi.org/10.1038/nbt957
- Cartellieri M, Koristka S, Arndt C, Feldmann A, Stamova S, von Bonin M, Töpfer K, Krüger T, Geib M, Michalk I, et al. A novel ex vivo isolation and expansion procedure for chimeric antigen receptor engrafted human T cells. PLoS One 2014; 9:e93745; PMID:24699869; https://doi.org/10.1371/journal.pone.0093745
- Feldmann A, Arndt C, Töpfer K, Stamova S, Krone F, Cartellieri M, Koristka S, Michalk I, Lindemann D, Schmitz M, et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T cells. J Immunol 2012; 189:3249-59; PMID:22875801; https://doi.org/10.4049/jimmunol.1200341
- Bippes CC, Feldmann A, Stamova S, Cartellieri M, Schwarzer A, Wehner R, Schmitz M, Rieber EP, Zhao S, Schäkel K, et al. A Novel modular antigen delivery System for immuno targeting of human 6-sulfo LacNAc-positive blood dendritic cells (SlanDCs). PLoS One 2011; 6:e16315; PMID:21283706; https://doi.org/10.1371/journal.pone.0016315
- Thieme S, Walther M, Pietzsch HJ, Henniger J, Preusche S, Mäding P, Steinbach J. Module-assisted preparation of 64Cu with high specific activity. Appl Radiat Isot 2012; 70:602-8; PMID:22326369; https://doi.org/10.1016/j.apradiso.2012.01.019
- Bergmann R, Splith K, Pietzsch J, Bachmann M, Neundorf I. Biological characterization of novel nitroimidazole-peptide conjugates in vitro and in vivo. J Pept Sci 2017; PMID:28332260; https://doi.org/10.1002/psc.2995
- Zhang Y, Huo M, Zhou J, Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed 2010; 99:306-14; PMID:20176408; https://doi.org/10.1016/j.cmpb.2010.01.007