
ABSTRACT
Tumor antigen-targeting monoclonal antibodies (TA-targeting mAbs) are used as therapeutics in many malignancies and their capacity to mobilize the host immunity puts them at the forefront of anti-cancer immunotherapies. Both innate and adaptive immune cells have been associated with the therapeutic activity of such antibodies, but tumor escape from mAb-induced tumor immune surveillance remains one of the main clinical issues. In this preclinical study, we grafted immunocompetent and immunocompromised mice with the B16F10 mouse melanoma cell line and treated them with the TA99 TA-targeting mAb to analyze the immune mechanisms associated with the tumor response and resistance to TA99 monotherapy. In immunocompetent mice TA99 treatment strongly increased the fraction of CD8 and CD4 effector T cells in the tumor compared with isotype control, highlighting the specific immune modulation of the tumor microenvironment by TA99. However, in most mice, TA99 immunotherapy could not prevent immune effector exhaustion and the recruitment of regulatory CD4 T cells and consequently tumor escape from immune surveillance. Remarkably, anti-PD-1 treatment at the time of tumor emergence restored the Th1 effector functions of CD4 and CD8 T cells as well as of natural killer and γδT cells, which translated into a significant slow-down of tumor progression and extended survival. Our findings provide the first evidence that PD-1 blockade at the time of tumor emergence can efficiently boost the host anti-tumor immune response initiated several weeks before by the TA-targeting mAb. These results are promising for the design of combined therapies to sensitize non-responder or resistant patients.
Abbreviations
| ACK | = | ammonium-chloride-potassium |
| ADCC | = | antibody-dependent cell mediated cytotoxicity |
| ADCP | = | antibody-dependent cellular phagocytosis |
| Ag | = | antigen |
| BSA | = | bovine serum albumin |
| CD | = | cluster of differentiation |
| CFSE | = | carboxyfluorescein succinimidyl ester |
| CTL | = | cytotoxic T lymphocyte |
| CTLA4 | = | cytotoxic T-lymphocyte-associated protein 4 |
| DCs | = | dendritic cells |
| DMEM | = | Dulbecco's modified Eagle's medium |
| ELISA | = | enzyme-linked immunosorbent assay |
| EDTA | = | ethylenediaminetetraacetic acid |
| FACS | = | fluorescence activated cell sorting |
| FBS | = | fetal bovine serum |
| gp75 | = | glycoprotein 75 |
| gp100 | = | glycoprotein 100 |
| HEPES | = | 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid |
| HER2/ERB-B2 | = | Human Epidermal Growth Factor Receptor-2 |
| IC | = | isotype control |
| IFNγ | = | interferon gamma |
| Ig | = | immunoglobulin |
| IHC | = | immunohistochemistry |
| IL | = | interleukin |
| i.p. | = | intraperitoneal |
| i.v. | = | intravenous |
| mAb | = | monoclonal antibody |
| MDSCs | = | myeloid-derived suppressor cells |
| MHC | = | major histocompatibility complex |
| NK | = | natural killer |
| NSG | = | NOD-scid IL2Rgammanull |
| OVA | = | ovalbumin |
| PBMCs | = | peripheral blood mononuclear cells |
| PBS | = | phosphate buffered saline solution |
| PBST | = | PBS containing Tween |
| PD-1 | = | programmed cell death 1 |
| PFA | = | paraformaldehyde |
| PMA | = | phorbol 12-myristate 13-acetate |
| RPMI | = | Roswell Park Memorial Institute medium |
| s.c. | = | subcutaneous |
| SPECT/CT | = | single-photon emission computed tomography |
| TA | = | tumor antigen |
| TCR | = | T cell receptor |
| Th | = | T helper |
| TIL | = | tumor-infiltrating lymphocyte |
| TIM3 | = | mucin-domain containing-3 |
| Treg | = | regulatory T cells |
| TRP2 | = | tyrosinase-related protein 2 |
| TYRP-1/TRP-1 | = | tyrosinase-related protein 1 |
Introduction
Among the many cancer therapy approaches, the recent clinical success of inhibitors of immune checkpoints, such as CTLA-4 and PD-1, highlights the potential of treating cancer by immune modulation.Citation1 Moreover, preclinical and clinical studies have demonstrated that the long-term anti-tumor effects of conventional cancer treatments, such as chemotherapy and radiotherapy, rely on the host immunity.Citation2 Similarly, several studies have shown that the therapeutic activity of tumor antigen (TA)-targeting monoclonal antibodies (mAbs), such as trastuzumab (anti-HER-2/neu) and rituximab (anti-CD20), depends not only on direct effects on tumor cells, but also on immune cell-mediated effects through the activation of immunoglobulin G (IgG) Fc region gamma receptors (FcγR). Indeed, TA-targeting mAbs have the unique capacity to specifically target cancer cells and to kill them through FcγR-dependent mechanisms, such as antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). FcγR polymorphisms have been associated with differential clinical outcome in patients treated with trastuzumabCitation3,4 or rituximab,Citation5 supporting the importance of FcγR-mediated mechanisms. Moreover, the demonstration that rituximab immunotherapy elicits a lymphoma-specific T cell response also suggests that immunotherapy with TA-targeting mAbs can result in the induction of a specific cellular immune response against tumor-associated antigens.Citation6
Using a mouse model of virus-induced leukemia, we provided the first evidence of mAb-induced vaccine-like effects associated with an adaptive memory immune response and long-lasting protection.Citation7-9 We demonstrated that immune complexes formed between infected cells and an anti-viral mAb improve dendritic cell (DC) maturation, antigen presentation and cross-priming, leading to immunological memory.Citation10 Moreover, Stagg et al. demonstrated, using immunocompetent BALB/c mice with established TUBO breast tumors and treated with anti-TRAIL-R2 and anti-ERBB-2 mAbs, that the adaptive anti-tumor immunity is essential for complete tumor regression after treatment with TA-targeting mAbs.Citation11 The dependence of TA-targeting mAbs therapeutic effect on the adaptive immune response was confirmed using immunocompetent mice with CD20+ or HER2+ tumors and treated with anti-CD20Citation12,13 or anti-HER2Citation14,15 mAbs, respectively. In agreement with these preclinical results, increased levels of tumor-infiltrating lymphocytes (TILs) at diagnosis have been associated with higher response rates to adjuvant trastuzumab in patients with early breast cancer.Citation16 Similarly, higher expression of immune markers, such interferon gamma (IFNγ) and STAT1, and of metagenes linked to the adaptive immune system in pre-treatment biopsies from patients with breast cancer who received trastuzumab- or pertuzumab-based therapies has been associated with higher rate of pathological complete response.Citation17
However, tumor escape from immune surveillance remains one of the main clinical issues. Indeed, malignant cells acquire the ability to exploit the host immunosuppressive mechanisms to avoid recognition and elimination by the host immune system and/or to drive the impairment of anti-tumor effector immune cells.Citation18,19 The precise phenotypic and functional analysis of the tumor immune microenvironment at the time of tumor escape and a better understanding of its changes during tumor progression should bring key information for the development of new combinatorial strategies to delay or prevent tumor relapse and increase the overall survival of patients treated with TA-targeting mAbs.
Therefore, we decided to characterize the tumor escape mechanisms during immunotherapy with a TA-targeting mAb in mice grafted with B16F10 melanoma cells and treated with the TA-targeting mAb TA99, as monotherapy. In this preclinical model, tumor progression is associated with a strong immunosuppressive tumor microenvironment and the response to TA99 therapy is heterogeneous. Indeed, in some mice TA99 leads to a long tumor-free survival, whereas in others it cannot efficiently control tumor progression. Using this preclinical model, we first characterized the cellular mechanisms of TA99-induced anti-tumor immunity in long-term protected mice. Then, we described the tumor immune microenvironment at the time of tumor escape in TA99-treated mice and proposed a combined therapeutic approach based on the synergistic effects of the TA-targeting mAb and an anti-immune checkpoint that resulted in an increased anti-tumor immunity and prolonged mice survival.
Materials and methods
Materials
The tyrosinase-related protein 2 (TRP2) (180–188; Ser-Val-Tyr-Asp-Phe-Phe-Val-Trp-Leu), gp100 (25–33; Glu-Gly-Ser-Arg-Asn-Gln-Asp-Trp-Leu), and ovalbumin (OVA) (257–264; Ser-Ile-Ile-Asn-Phe-Glu-Lys-Leu) peptides were purchased from Anaspec.
For flow cytometry, the following antibodies were used: anti-mouse CD45-PerCPVio700 (clone 30F11) and anti-mouse MHC2-FITC (clone M5 114.15.2) from Miltenyi Biotech; anti-mouse CD4-BV650 (clone RM4–5), anti-mouse TCRγδ−Βς605 (clone GL3), anti-mouse CD25-PECy5 (clone B173002), anti-mouse IFNγ-BV421 (clone XMG1.3), anti-mouse NKp46-FITC (clone 29A1.4) and anti-human TIM3-BV711 (clone F38 2E2) from Biolegend; anti-mouse CD8-PECy5.5 (clone 53–6.7), anti-mouse PD-1-FITC (clone J43), anti-mouse TIM3-PE (clone RMT 3–23), anti-mouse F4/80-APCeF780 (clone BM8), anti-mouse FOXP3-APC (clone FJK-16S) and anti-human CD45-PECy5.5 (2D1) from eBioscience; anti-mouse CD3-BV711 (clone 17A2), anti-mouse CD11c-PE (clone HL3), anti-mouse CD11b-APC (clone M1/70), anti-mouse CD107a-BV786 (clone 1D4B), anti-mouse IL2-APC-Cy7 (clone JES6–5H4), anti-mouse IL12-V450 (clone C15.6), anti-human CD3-PerCPCy5.5 (clone SK7), anti-human CD4-BV650 (Clone SK3), anti-human CD8-BV605 (clone SK1), anti-human PD1-BV786 (clone EH12.1), anti-human IL2-PE (clone MQ1 17H12) and anti-human IFNγ-PECy7 (clone 4SB3) from BD PharMingen. The LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit for 405 nm excitation was purchased from Invitrogen.
The anti-mouse tyrosinase-related protein 1 (TYRP-1/ TRP-1) TA99 and anti-mouse PD-1 (CD279) mAbs and their respective mouse IgG2a C1.18.4 and rat IgG2A 2A3 isotype controls were purchased from BioXcell. All antibodies were certified murine pathogen free.
Phorbol 12-myristate 13-acetate (PMA) and ionomycin were purchased from Sigma Aldrich; BD GolgiPlug™ Protein Transport inhibitor (containing brefeldin A) and BD GolgiStop™ Protein Transport inhibitor (containing monensin) were from BD Bioscience, fixation and permeabilization buffers from eBioscience, and ammonium-chloride-potassium (ACK) lysing buffer from BioWhittaker. The C57BL/6 murine melanoma B16F10 and colon adenocarcinoma MC38 cell lines were from ATCC.
Animals
Eight-week-old female NOD-scid IL2Rgammanull (NSG) mice were purchased from Charles River and 8-week-old female C57Bl/6N or athymic nude Foxn1nu mice from Envigo. Mice were maintained according to protocols approved by the French national committee of animal care.
Cell culture
B16F10 and B16F1 cells were maintained in complete Dulbecco's modified Eagle's medium (DMEM, GE Healthcare Life Sciences) supplemented with 10% (vol/vol) fetal bovine serum (FBS), and 2 mg/ml gentamycin (Life Technology 15710–049). MC38 cells were grown in complete RPMI 1640 medium supplemented with 10% (vol/vol) FBS, 16 mM HEPES (Gibco by Life Technologies 15630–056) and 2 mg/ml gentamycin. All cell lines were cultured at 37°C and 5% CO2 and tested regularly for mycoplasma contamination and for rodent pathogens.
Tumor models and treatments
For primary tumor graft, 5.104 B16F10 or B16F1 cells in 100 µl of phosphate buffered saline solution (PBS) were inoculated subcutaneously (s.c.). At day 2 post-graft, treatment by intraperitoneal (i.p.) injection of TA99 mAb, IgG2a isotype control (IC) or PBS was started (400 µg for the first injection followed by 5 injections of 200 μg at day 3, 4, 7, 9 and 11). Tumors were measured 3 times per week with a caliper and mice were killed when the tumor reached a volume of 1500 mm3. Tumor volume was calculated using the following formula: length × width × width/2. For the challenge, 5.104 B16F10 or 5.105 MC38 cells in 100 µl PBS were inoculated s.c. in the opposite flank.
For testing the effect of immune checkpoint inhibition, 200 µg of anti-PD-1 mAb in 100 µl PBS was administered i.p. twice a week starting at the time of tumor emergence (30–50 mm3 tumor volume). For the lung metastasis model, 105 B16F10 cells were inoculated intravenously (i.v.) in tumor-free TA99-treated or naive (i.e., ungrafted and never treated) mice that were killed 14 d after to analyze lung invasion. The total number and number of large foci (> 3 mm) were assessed by using a binocular microscope.
Quantification of anti-B16F10 immunoglobulins by enzyme-linked immunosorbent assay (ELISA)
After retro-orbital sinus blood sampling at different time points or intra-cardiac blood sampling after euthanasia, serum was recovered and stored at −20°C. For ELISA assays, B16F10 cells were plated overnight at 37°C in 96-well plates. The day after, cells were rinsed twice with PBS and fixed with 4% paraformaldehyde (PFA). Cells were washed twice with PBS and incubated at room temperature (RT) with PBS/0.1%Tween 20 (PBST) and 1% bovine serum albumin (PBST-BSA) for 1 h. Serum samples were diluted in PBST-BSA and incubated at RT for 2 h. After 3 washes in PBST, a peroxidase-conjugated anti-mouse IgG (γ chain specific) (Sigma), diluted 1/1000 in PBST-BSA, was added at RT for 1 h. After 2 washes in PBST and one wash in PBS, 50 µl of TMB solution (TMB Substrate Reagent Set BD OptEIA™ Cat: 55514) was added at RT in the dark for 15 min. The reaction was stopped by addition of 50 µl of H2SO4 and absorbance was read at 450 nm. The total serum IgG concentrations were calculated using the standard curve obtained with purified TA99.
Serum transfer experiments
Serum samples from untreated (naive serum) or challenged tumor-free TA99 treated-mice (immune serum) were collected and pooled. Mice were grafted with 5.104 B16F10 cells and received 6 i.p. injections of 100 µl of immune or naive serum at day 2, 3, 4, 7, 9 and 11 post-graft.
Isolation of immune cells
Spleen, lymph nodes and tumors were recovered in ice-cold PBS containing 0.5% BSA and 2 mM EDTA (dissociation buffer) and were mechanically dissociated. For spleen, red blood cells were eliminated by adding 2 ml ACK lysing buffer. Then white blood cells were recovered by centrifugation, washed with PBS and re-suspended in RPMI 1640 plus 10% FBS for functional assays or in FACS buffer (2% fetal calf serum, 1 ml 0.5 M EDTA, 1 µg/ml sodium azide in PBS) for staining and flow cytometry analysis. For tumors and lymph nodes, after dissociation, cells were filtered through 70 µm filters (Falcon Cell Strainer Nylon 352340) and centrifuged. Pellets were re-suspended in RPMI 1640 plus 10% FBS for functional assays or in FACS buffer for staining and flow cytometry analysis.
In vivo cytotoxicity assays
Splenocytes from naive mice were suspended in RPMI 1640 plus 10% FBS, and loaded at 37°C with a mix of gp100 and TRP2 peptides or OVA (all at 1 µg/ml) for 2 h. After washes with PBS to eliminate the peptide excess, gp100 and TRP2-loaded splenocytes were labeled with 5 µM carboxyfluorescein succinimidyl ester (CFSE) (CFSEhigh) and OVA-loaded splenocytes with 0.5 µM CFSE (CFSElow) at 37°C for 10 min. Cells were washed again in ice-cold PBS, re-suspended in PBS, counted and mixed in a 1:1 ratio. Then, 1.5 107 splenocytes were injected i.v. in recipient mice 12 d after challenge. Five days later, mice were killed for flow cytometry quantification of splenic CFSElow and CFSEhigh cells. The CTL activity against gp100/TRP2-loaded splenocytes was calculated according to the CFSEhigh/CFSElow cell ratio normalized to the CFSEhigh/CFSElow cell ratio assayed before splenocyte injection in recipient mice.
Flow cytometry analysis
When tumors reached a volume of 300–400 mm3, mice were killed. Spleen, lymph nodes and tumors were recovered and dissociated as described above. Cells were suspended in RPMI 1640/10% FBS (unstimulated conditions), or RPMI 1640/10% FBS with 20 ng/ml PMA and 1 µg/ml ionomycin (stimulated conditions) in 96-well plates at 37°C for 5 hours. The anti-mouse CD107a-BV786 antibody (BD Horizon) was added simultaneously to evaluate degranulation. After 1 hour, GolgiPlug™ and GolgiStop™ were added in each well to block cytokine secretion (according to the manufacturer's protocol; BD Biosciences). Then, at the end of the 5 hours, cells were washed once with PBS and antibodies for surface markers were added at 4°C for 1 hour. After extracellular staining, cells were fixed and permeabilized, according to the eBioscience fixation and permeabilization procedures, and intracellular staining was performed overnight at 4°C. Then, samples were washed, fixed in 1% PFA, and processed for data acquisition using a FORTESSA flow cytometer (Beckton Dickinson). Data were analyzed using the Flowjo 10 software.
Histology
For the evaluation of TRP-1 expression on human melanoma biopsies, immunohistochemical staining of TRP-1 was performed on 6 of the 15 metastatic patients included in our study and 4 primary patients selected from the database of the tumor library from ICM-Val d'Aurelle. Four-µm thin sections of formalin-fixed paraffin-embedded tissues were mounted on Flex microscope slides (Dako) and allowed to dry overnight at room temperature before immunohistochemistry processing. PT-Link® system (Dako) was used for pre-treatment, allowing simultaneous de-paraffinization and antigen retrieval. Heat-induced antigen retrieval was executed for 15 minutes in High pH Buffer (Dako) at 95°C. Immunohistochemistry procedure was performed using the Dako Autostainer Link48 platform. Briefly, endogeneous peroxidase was quenched using Flex Peroxidase Block (Dako) for 5 min at room temperature. Slides were then incubated with the anti-TYRP1 mouse monoclonal antibody (Abcam, Clone TA99; 13.8 µg/ml) for 30 min at room temperature. After 2 rinses in buffer, the slides were incubated with a horseradish peroxidase-labeled polymer coupled to secondary anti-mouse and anti-rabbit antibodies for 20 min, followed by appliance of 3,3′-Diaminobenzidine for 10 min as substrate. Counterstaining was performed using Flex Hematoxylin (Dako) followed by washing the slides under tap water for 5 min. Finally, slides were mounted with a coverslip after dehydratation. All samples were also processed with irrelevant IgG2A isotypic control at the same concentration as TRP-1 antibody, and used as negative control. Normal skin was used as positive control.
Patient samples
Tumor biopsies and blood samples were recovered from patients with metastatic melanoma before any treatment. Informed consent was obtained from all patients. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation, as described by the manufacturer (Eurobio). Tumor biopsies were cut in small pieces of 0.5 cm3 in RPMI 1640 medium and dissociated by combining mechanical disruption (GentleMACS™ dissociator, Miltenyi Biotec) with enzymatic digestion using 500 µg/ml collagenase type IV-S (Sigma). After dissociation, samples were filtered (40 µM nylon filter) to remove any remaining larger particles from the cell suspension and the recovered cells were used immediately.
Primary melanomas were selected from the database of the tumor library from ICM-Val d'Aurelle. Tumor samples were collected following French laws under the supervision of an investigator and declared to the French Ministry of Higher Education and Research (declaration number DC-2008–695). All patients were informed about the use of their tissue samples for biologic research and the study was approved by the local translational research committee.
Ethics Statement
All procedures for animal handling and experiments were approved by the local animal facility “ComEth” Institutional Review Board under the supervision of the French LR Regional CEEA ethics committee on animal experimentation (Chairman: Pr M Michel, Montpellier).
Statistical analysis
All results were expressed as the mean ± SEM of at least 2 independent experiments. Statistical analyses were performed with the non-parametric Mann-Whitney test when comparing 2 groups, the Kruskal-Wallis test when comparing multiple groups and the Kaplan-Meir test for survival analyses. Statistical analyses were performed using the Prism 6 software (GraphPad, San Diego, CA, USA). Differences between groups with a p value p < 0.05 were considered as statistically significant.
Results
Long-term protection following treatment with TA99 relies on a specific host immune memory response that requires the innate immunity
The TA99 mAb targets TRP-1 (known also as gp75/TYRP-1), a glycoprotein synthesized by pigmented melanocytes, strongly expressed in B16F10 melanoma cells in vivo (Fig. S1A) and recognized as a dominant melanocytic antigen expressed by both primary and metastatic human melanoma tumors (Fig. S2A and B). First, we analyzed TA99 biodistribution in mice bearing B16F10 melanoma tumors by SPECT-CT imaging. I125-labeled TA99 specifically targeted tumor cells with a maximal signal intensity 3 d post-injection and was still detectable in tumors one week after injection (Fig. S1B). Conversely, the IgG2a isotype control antibody (IC) was never detected in tumors. Then, to evaluate TA99 immune cell-mediated effects we grafted immunocompetent C57BL/6 mice with B16F10 melanoma cells and treated them with TA99, IC antibody or PBS (6 i.p. injections starting at day 2 after graft). IC treatment did not have any effect, and IC-treated mice as well as PBS-treated mice died within 3 weeks (mean of 21 ± 3 days) after tumor cell graft ( and ). On the other hand, the TA99 mAb significantly delayed tumor growth and prolonged mice survival compared with IC or PBS ( and ). Moreover, about 30% of TA99-treated mice were still tumor-free after 60 d (). At that time, we could not detect TA99 any longer in the blood (data not shown), suggesting that tumor eradication or control can be obtained with TA99 monotherapy and maintained beyond persistence of the therapeutic mAb. To determine the immune mechanisms underlying TA99-mediated tumor control, we used the same treatment protocol in B16F10-engrafted athymic nude mice that cannot produce T cells and in NSG mice that lack functional macrophages, T, B, natural killer (NK) and DCs. Results in athymic nude mice were similar to those obtained in immunocompetent mice, with a significant delay of tumor progression only after TA99 treatment and the presence of a group of mice that were tumor-free 50 d after tumor engraftment ( and ). In NSG mice, TA99 treatment delayed tumor growth and increased overall survival by 5 d compared with IC-treated mice; however, all mice developed tumors ( and ). Altogether, these data clearly demonstrate that T cells are not required for TA99-mediated early control of primary B16F10 tumor growth and emphasize the role of NK and myeloid cells, present in athymic nude but not in NSG mice, in TA99 anti-tumor effect. To determine whether the innate immune system is involved in the early tumor control by TA99, we evaluated the effect of TA99 treatment on innate immune cell activation by analyzing the cytokine profile of myeloid and NK cells after TA99 treatment of C57BL/6 mice with established tumors (50 mm3). Two injections of TA99 (n = 8), but not of IC (n = 8), were sufficient to significantly increase the fraction of IL12-producing macrophages and DCs as well as IFNγ secretion by activated NK cells in the tumor (Fig. S3). These results illustrate the specific effect of TA99 on tumor infiltrating innate immune cells, and strengthen previous findings on the crucial role of FcγR-expressing immune cells for TA99 efficacy.Citation20-22
Figure 1. Long-term anti-tumor immunity in TA99-treated mice relies on a specific T-cell memory response. C57BL/6N (A, B), athymic nude (C, D) and NSG (E, F) mice engrafted with 5.104 B16F10 melanoma cells received 6 injections (i.p.) of TA99 mAb (TA99), isotype control (IC) or PBS (PBS). The mean tumor sizes ± SEM (A, C, E) and Kaplan-Meier survival curves (B, D, F) are shown. Kaplan-Meier survival curves for tumor-free TA99-treated C57BL/6N (G) and athymic nude (H) mice after a second injection (s.c.) of 5.104 B16F10 cells on the opposite flank (challenge). (I) Tumor-free TA99-treated C57BL/6N mice were challenged (s.c.) with 5.105 MC38 colon adenocarcinoma cells (MC38). For G, H and I, a group of newly grafted C57BL/6N or athymic nude mice was used as control. *p < 0.05; **p < 0.01; ****p < 0.0001 (log-rank test for Kaplan Meier survival curves and non-parametric Mann-Whitney test for tumor growth).
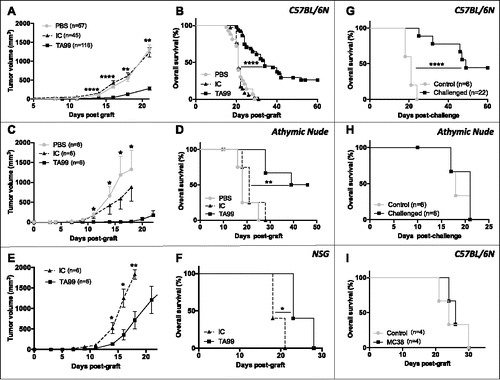
To determine whether TA99 treatment resulted in durable tumor immunity, we challenged C57BL/6 (n = 22) and athymic nude (n = 6) mice that remained tumor-free after TA99 treatment (tumor-free TA99-treated mice, hereafter) with a second s.c. injection of B16F10 cells at day 50 to 60 after the primograft. Half of the C57BL/6 mice developed tumors with a significant delay compared with control (untreated, newly grafted mice) and half of them remained tumor-free (). Conversely, all athymic nude mice developed tumors without any delay and died within 21 days, like control mice (). This indicates that T cells are required for the long-term anti-tumor immunity. Moreover, all tumor-free TA99-treated C57BL/6 mice grafted with the colon adenocarcinoma cell line MC38 (n = 4) developed tumors and died with a similar kinetics as control mice (). This demonstrates that the immune response established after TA99 treatment is B16F10-specific.
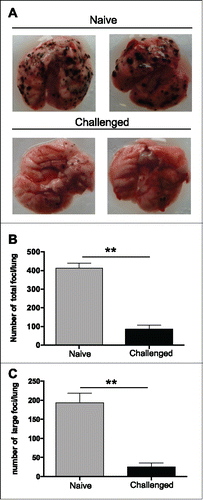
Finally, we analyzed whether tumor-free TA99-treated C57BL/6 mice could control tumor cell dissemination in the lung after i.v. challenge with B16F10 cells at day 60 post-primograft. Twelve days post-injection, we counted the tumor foci in the lungs and classified them according to their size (small foci: <3 mm, and large foci: >3 mm). The total number of foci and the number of large foci were significantly lower in TA99-treated mice (n = 10) than in controls (untreated and never grafted s.c.) (n = 8) ().
Figure 2. Long-term anti-tumor immunity in TA99-treated mice reduces B16F10 lung metastasis formation. 105 B16F10 cells were injected (i.v.) in age-matched naive or tumor-free TA99-treated C57BL/6N mice that were killed 12 d later to quantify tumor foci in the lungs. Representative images of lungs (A) and quantification of total foci (B) and foci larger than 3 mm (C) in the lungs of naive (gray bars, n = 8) and tumor-free TA99-treated mice (black bars, n = 10). In B and C, data are the mean ± SEM. **p < 0.01 (non-parametric Mann-Whitney test).
Altogether these data demonstrate that a short-term monotherapy with the TA99 TA-targeting mAb stimulates innate immune cells and induces, in a fraction of mice, long-term protective immunity through the development of a specific T-cell-dependent memory immune response.
TA99 mAb treatment induces specific cellular and humoral memory anti-tumor immune responses
We then analyzed the specific cellular and humoral anti-tumor immune responses in tumor-free TA99-treated mice. To assess the presence of a functional memory CD8 T cell response, we challenged tumor-free TA99-treated C57BL/6 mice by s.c. injection of B16F10 cells between day 50 and 60 after the primograft. Twelve days later, we injected i.v. splenocytes loaded with a mix of gp 100 and TRP2 peptides or the irrelevant peptide OVA and assessed the specific cytotoxic T lymphocyte (CTL) activity in the spleen and draining and non-draining lymph nodes 5 d later (i.e., 17 d post challenge). CTL activity was significantly increased (from 5 to 25%) in the spleen ( and ), but not in draining and non-draining lymph nodes (Fig. S4), of TA99-treated animals (n = 9) compared with naive mice (n = 9).
Figure 3. TA99 treatment induces cellular and humoral anti-tumor immune responses. Tumor-free TA99-treated C57BL/6N mice received a second s.c. injection of 5.104 B16F10 cells on the opposite flank (challenged) and the CTL activity of CD8 splenocytes was evaluated 17 d later and compared with that of age-matched untreated and never grafted controls (naive). Representative flow cytometry histograms (A) and percentages of CTL activity (B) in naive (n = 9) and challenged (n = 9) mice. (C) Serum samples were collected from the same naive (n = 9) and TA99-treated mice before (pre-chall) and after challenge (post-chall) and anti-B16F10 IgGs quantified by ELISA. (D, E) Serum samples from naive mice (non-immune sera) or from tumor-free TA99-treated mice (immune sera) were used to treat C57BL/6N mice grafted with 5.104 B16F10 cells for the first time (n = 6/group). The mean tumor size ± SEM (D) and Kaplan-Meier survival curves (E) are indicated. *p < 0.05; **p < 0.01 (log-rank test for Kaplan Meier survival curves, nonparametric Mann-Whitney test for tumor growth and non-parametric Kruskal-Wallis test for ELISA).
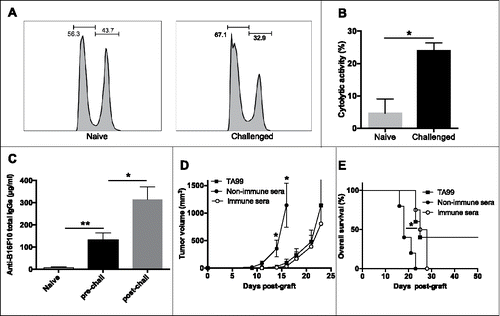
We also evaluated the endogenous antibody response in tumor-free TA99-treated mice just before and 12 d after the challenge. Before the new injection of melanoma cells, we detected an average of 130 μg/ml of anti-B16F10 IgGs in serum samples of TA99-treated mice, but anti-B16F10 IgGs were not detected in serum samples from naive or B16F10 grafted but non-treated mice ( and data not shown). The concentration of anti-B16F10 IgGs was significantly increased (mean value: 315 μg/ml; p < 0.05) after the challenge with melanoma cells, suggesting a memory B cell response in tumor-free TA99-treated mice (). To test whether this specific antibody response could contribute to TA99 protective effect, we collected serum from tumor-free TA99-treated mice (immune serum) and from naive control mice (non-immune serum). Administration of immune serum to mice grafted with B16F10 cells (same therapeutic protocol as for TA99) delayed tumor growth, as observed with the TA99 mAb, whereas non-immune serum had no effect (n = 6/group) (). Similarly, overall survival was significantly increased in mice treated with immune serum compared with animals that received non-immune serum (+ 5 days; p < 0.05). Nevertheless, all mice treated with immune serum developed tumors ().
This demonstrates that tumor-free TA99-treated mice have developed cellular and humoral anti-tumor responses to control tumor progression. These responses could explain the long-lasting tumor control and protection against tumor challenge.
TA99 treatment increases the fraction of effector T cells at tumor sites but does not counteract their exhausted phenotype
Although TA99 treatment led to tumor eradication in about 30% of C57BL/6 mice, in all other mice, it could not efficiently control tumor progression on the long-term (). To understand the immunomodulatory effects associated with TA99 immunotherapy and the reasons for tumor escape, we analyzed the effector T lymphocyte phenotype in mice treated with PBS, IC or TA99 (n = 9/group) when tumors reached a volume of 300–400 m3. In each group, we isolated lymphocytes from spleens, lymph nodes and tumors and evaluated the ratio between effector (CD44highCD62Llow) and naive (CD44lowCD62Lhigh) CD8 T cells and the percentages of effector (CD25+FoxP3−) and regulatory (CD25+FoxP3+) CD4 T cells (). The ratio between effector and naive CD8 T cells was very low in spleen (, left) and lymph nodes (data not shown) in all treatment groups. Similarly, the percentage of effector CD4 T cells in spleen and lymph nodes was not modified by TA99 immunotherapy (, left, and data not shown). Conversely, TA99 treatment strongly increased the percentage of CD8 and CD4 effector T cells in tumors compared with IC and PBS ( and , right), highlighting the specific immune modulation of the tumor microenvironment by TA99 toward a strong intra-tumor effector T cell recruitment. Immunophenotyping of the infiltrating T cell populations revealed a high frequency of regulatory T cells whatever the treatment (). The presence of these immunosuppressive cells suggests that effector T cells recruited by TA99 might be functionally impaired, thus explaining the absence of tumor control.
Figure 4. TA99 treatment increases the percentage of tumor infiltrating effector lymphocytes, but does not prevent their exhaustion. (A-C) Immune cells were isolated from spleens (white symbols) and tumors (black symbols) of mice treated with PBS (PBS, n = 9), IgG2a isotype control (IC, n = 9) or TA99 (TA99, n = 9). The ratio between effector memory (CD44+CD62L−) and naive (CD44−CD62L+) CD8+ T cell (A), and percentage of CD25+FOXP3− effector CD4+ T cells (B) and CD25+FOXP3+ CD4+ Tregs (C) are indicated. (D, E) Expression of PD-1 and TIM3 in CD8 (D) and CD4 (E) T cells isolated as indicated in A and B. Top, representative dot-plots; bottom, individual data from 9 mice/group (same mice as in A-C). (F) Expression of PD-1 and TIM3 in PBMCs and TILs from 15 untreated patients with metastatic melanoma. The percentages of PD1+ and PD1+TIM3+ in the CD4 and CD8 subpopulations are shown. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant; determined with the non-parametric Kruskal-Wallis test (A, B, C, D, E) or the non-parametric Wilcoxon matched-pairs rank test (F).
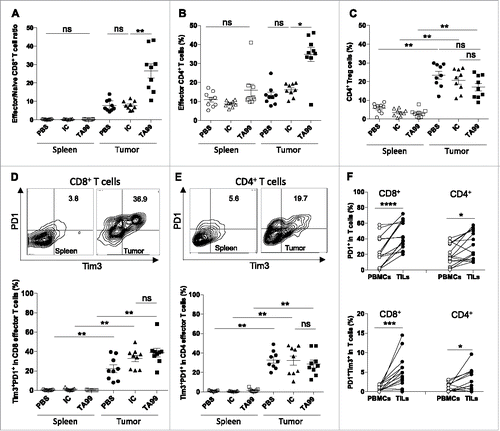
Effector T cell dysfunction has been associated with the upregulation of inhibitory receptors, such as CTLA-4, PD-1 and TIM3.Citation23 In patients with melanoma, several studies have associated PD-1 and TIM3 upregulation in circulating TA-specific CD8 T cells with their exhaustion/dysfunction.Citation24-27 We thus assessed PD-1 and TIM3 expression in effector T lymphocytes from B16F10 tumor-bearing mice when tumors reached a volume of 300–400 mm3. The percentage of CD4 and CD8 effector cells that expressed the exhaustion markers PD-1 and TIM3 was significantly higher within the tumor (right) than in the spleen (left), whatever the treatment ( and ). In agreement, the percentage of tumor-infiltrating CD4 and CD8 cells that produced IFNγ alone or IFNγ and IL-2 was not affected by TA99 treatment (Fig. S5). We obtained similar results when we monitored the expression of PD-1 and TIM3 in biopsies and blood samples from untreated patients with stage IV melanoma (n = 15). Specifically, the percentage of cells that expressed PD-1 alone or with TIM3 was significantly higher in tumor-infiltrating CD4 and CD8 T lymphocytes (TILs) than in paired PBMCs (), a phenotype associated with a decreased production of IFNγ or/and IL-2 following PMA-ionomycin stimulation in vitro in TILs than in PBMCs (Fig. S6). Of note that although TRP-1 is a dominant melanocytic antigen, we observed no correlation between TRP-1 expression and the percentage of TILs expressing TIM3 and PD-1 (data not shown).
Collectively these data show that TA99 treatment is associated with a specific increase of effector T lymphocytes in the tumor. However, in 70% of mice, TA99 immunotherapy was not sufficient to prevent the anti-tumor immunity alteration associated with the recruitment of regulatory CD4 T cells and the exhaustion of effector lymphocytes.
The anti-PD-1 antibody synergizes with TA99 immunotherapy to potentiate the cellular anti-tumor immunity
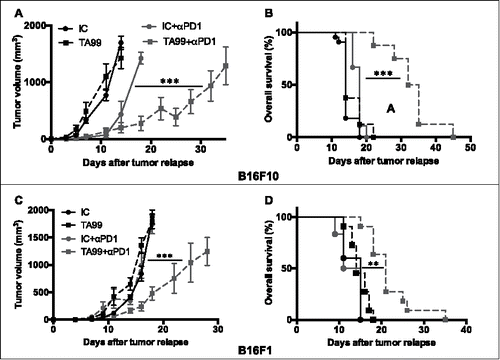
As PD-1 expression in tumor-infiltrating effector T cells was high at the time of tumor emergence, we then asked whether PD-1 blockade could prolong the beneficial effect of TA99 treatment. To this aim, we grafted immunocompetent C57BL/6 mice with B16F10 melanoma cells and treated them with TA99 or the IC antibody, as before. At the time of tumor emergence (i.e., from day 14 to day 31 post-graft; 50 mm3), mice were left un-treated or treated with anti-PD-1 antibodies twice a week for 3 weeks, and we followed tumor progression. In the absence of anti-PD-1 treatment, tumor progression after emergence was similar in both isotype control (IC) and TA99-treated (TA99) groups (n = 6/group). In the isotype control group (IC+αPD1; n = 8), PD-1 therapy slightly delayed tumor growth, but had no effect on survival ( and ). Conversely, in TA99-treated mice (TA99+αPD1; n = 8), we observed a significant delay of tumor progression throughout the anti-PD-1 treatment () associated with a significant increase in overall survival (). Similar results were observed in mice grafted with the B16 variant B16F1 ( and ), despite a weaker effect of TA99 treatment alone on disease-free survival (Fig. S7A) compared with mice grafted with B16F10 melanoma cells ().
Figure 5. Anti-PD-1 treatment slows down tumor progression and increases survival of TA99-treated mice. C57BL/6N mice were grafted (s.c.) with 5.104 B16F10 (A and B) or B16F1 (C and D) cells and treated with the TA99 mAb or the isotype control (IC). Upon tumor appearance (50 mm3), mice received anti-PD-1 antibodies (i.p.) twice a week (IC+αPD-1, n = 8 or TA99+αPD-1, n = 8) or PBS (IC, n = 6 or TA99, n = 6) for 3 weeks. The mean tumor sizes ± SEM (A and C) and Kaplan-Meier survival curves (B and D) are indicated. Day 0 stands for the day of tumor appearance. ***p < 0.001; **p < 0.01 determined with mixed-effects ML regression test for tumor growth (A and C) and the log-rank test for Kaplan Meier survival curves (B and D).
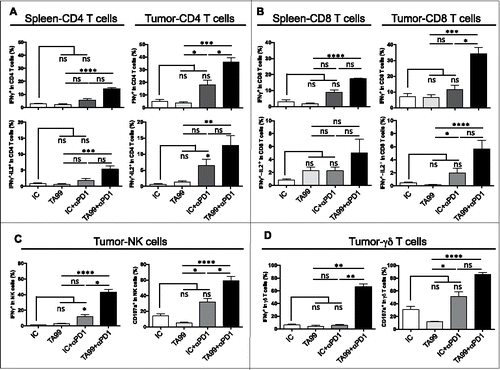
Although it was not concomitant to the TA99 treatment, we hypothesized that the anti-PD-1 therapy might strengthen the beneficial effect of TA99 (increased effector fraction in the tumor) by restoring the function of effector TILs. We thus studied the effect of PD-1 blockade on the functionality of peripheral and tumor-infiltrating CD4 and CD8 effector T cells. As in preliminary experiments we obtained similar results in spleen and lymph nodes (data not shown), we then used spleen as representative of the periphery for all the experiments. After, graft of B16F10 melanoma cells and treatment with TA99 or IC followed by anti-PD-1 antibodies (TA99 + αPD1, n = 9; IC+αPD1, n = 9) or not (TA99, n = 7; IC, n = 7), we analyzed cytokine production when tumors reached 200–300 mm3. The percentage of CD4 and CD8 T cells that produced IFNγ or/and IL-2 was very low in spleens and tumors from mice treated with TA99 or IC only ( and and Fig. S8A). Conversely, in TA99+αPD1 mice, IFNγ and IFNγ plus IL-2-producing CD4 and CD8 T cells were significantly increased in the tumor (up to 36.15 ± 2.17% and 34.24 ± 3.91% of IFNγ producing tumor-infiltrating CD4 and CD8 T cells respectively, compared with 3.83 ± 0.46% and 6.6 ± 1.52% in mice that received only TA99) ( and and Fig. S8A). Anti-PD1 treatment alone (IC+αPD1) significantly increased only the percentage of IFNγ plus IL-2-producing CD4 cells in the tumor ( and Fig. S8A). These data clearly demonstrate the synergistic effect between TA99 therapy and PD-1 blockade on the activation of effector T lymphocytes ( and and Fig. S8A). We then found that this synergistic effect was not restricted to CD4 and CD8 effector T cells because IFNγ-producing NK cells in the tumor were also increased in TA99+αPD1 mice compared with animals that received TA99 or anti-PD-1 antibody alone (42.95 ± 3.12% vs 2.73 ± 0.25% and 11.83 ± 2.97%, respectively) ( and Fig. S8B). Similarly, the percentage of IFNγ-producing γδ T cells was higher in TA99+αPD1 mice (66.35 ± 3.79% vs 4.18 ± 0.46% in animals that received one single antibody) ( and Fig. S8B). Moreover, in TA99+αPD1 mice, increased IFNγ production by NK and γδ T cells was associated with a significant increase of CD107a expression, reflecting a higher degranulation activity ( and and Fig. S8B). We also observed similar synergistic effects on cytokine production in B16F1 grafted mice (Fig. S7B).
Figure 6. PD-1 blockade synergizes with TA99 therapy to enhance effector T cell functions. Upon tumor appearance, TA99-treated and isotype control (IC)-treated mice received anti-PD-1 antibodies (i.p.) twice per week (TA99+αPD1, n = 9; IC+αPD1, n = 9) or PBS (TA99, n = 7; IC, n = 7). When tumors reached 200–300 mm3,cytokine production was analyzed in spleen and tumor by flow cytometry. (A and B) Percentage of IFNγ (top) and IFNγ plus IL-2 (bottom)-producing CD4 (A) and CD8 (B) T cells isolated from spleens and tumors. (C and D) Percentage of NK (C) and γδ T cells (D) that produce IFNγ (left) and expressing CD107a (right) in tumors; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant (non-parametric Kruskal-Wallis test).
Altogether, these data clearly show that the synergistic effect between TA99 and anti-PD-1 treatments leads to a significant increase of Th1 cytokine production (IFNγ and IL2) in the tumor through the restoration of both innate and adaptive immune cell functions. Despite increased immune response rates, we never detected any systemic toxicity upon TA99 plus anti-PD-1 treatment.
PD-1 blockade potentiates the endogenous memory responses
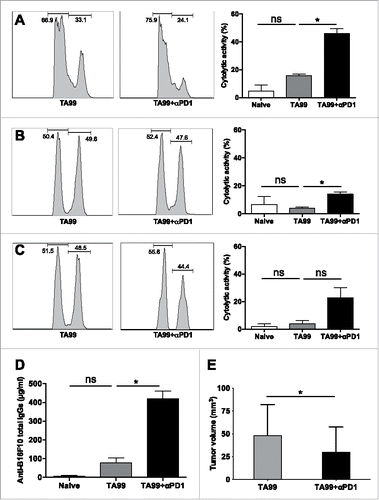
As 50% of tumor-free TA99-treated mice developed a tumor after the second melanoma cell graft (), we investigated whether treatment with the anti-PD-1 antibody after the challenge could also have an impact on the cellular and humoral memory responses. Thus, we treated or not challenged tumor-free TA99-treated mice with the anti-PD-1 antibody at the time of tumor escape, and assessed CTL activity (same method as before). In spleen, CTL activity increased by 2-folds after anti-PD-1 treatment (TA99+αPD1) compared with mice that did not received anti-PD1 (TA99) (). Similarly, CTL activity could be measured in draining and non-draining lymph nodes after anti-PD-1 treatment (TA99+αPD1) but not in mice treated with TA99 alone (TA99) ( and ). In parallel, we quantified endogenous anti-B16F10 IgGs by ELISA. We observed a 4-fold increase of anti-B16F10 IgGs in the serum of mice that received anti-PD-1 compared with mice that did not received anti-PD1 (416.11 ± 42.06 µg/ml vs 72.39 ± 25.4 µg/ml) (). Altogether, these data suggest that PD-1 blockade potentiates the TA99-induced endogenous memory responses. This translates into a significant delayed tumor progression in TA99+αPD1-treated mice (n = 5) compared with TA99-treated mice (n = 7) ().
Figure 7. PD-1 blockade synergizes with TA99 treatment to enhance the cellular and humoral adaptive memory responses. Tumor-free TA99-treated mice were challenged (s.c.) with 5.104 B16F10 cells. Upon tumor appearance, mice received 2 i.p. injections, anti-PD-1 antibodies (TA99+αPD1, n = 5) or PBS (TA99, n = 7). The CTL activity of CD8 splenocytes was evaluated 17 d after the challenge and compared with that of splenocytes from age-matched non-grafted and untreated mice (naive, n = 8). Representative flow cytometry histograms (left) and percentages of CTL activity (right) in spleen (A), draining (B) and non-draining lymph nodes (C) from naive and TA99-treated mice treated or not with anti-PD-1 antibodies. (D) Concentration of anti-B16F10 IgGs determined by ELISA in serum samples from naive, TA99-treated and TA99+αPD1-treated mice on the day of sacrifice for the CTL assay. (E) Tumor growth in TA99-treated and TA99+αPD1-treated animals. *p < 0.05; ns, not significant; determined with the non-parametric Kruskal-Wallis (A, B, C, D) and the non-parametric Mann-Whitney test (E).
Discussion
We previously demonstrated in a mouse model of virus-induced leukemia that immune complex formation between infected cells and an anti-viral mAb improves DC maturation and antigen presentation, thus resulting in a sustained memory immune response and long-lasting protective effects.Citation10 This was followed by several preclinical studies showing that the host adaptive immunity is essential for tumor control after treatment with TA-targeting mAbs.Citation9,12,14,28 Innate immune cells, especially NK cells and DCs, also are needed for the efficacy of TA-targeting-based immunotherapies, notably anti-ERBB-2 and anti-CD20 therapies, in preclinical models.Citation13,15 In the case of TA99-based immunotherapy in the B16F10 melanoma preclinical model, activating FcγRs, especially FcγRI together or not with FcγRIII or FcγRIV,Citation20,22 are crucial for TA99 protective effect. It was proposed that among FcγR-expressing cells, neutrophils are required and sufficient to mediate the anti-tumor effect of the TA99 mAb.Citation29 Here, we observed a similar TA99-mediated tumor growth control in athymic nude mice, which lack T lymphocytes, and in immunocompetent C57BL/6 mice, resulting in 30–50% of tumor-free mice after the treatment. Conversely, we never observed tumor eradication in the severely immunocompromised NSG mice after TA99 treatment, although neutrophils are present and functional in this strain.Citation30 These results suggest that in the absence of other functional immune cells, neutrophils are not sufficient to control B16F10 tumor progression.
Thus, our results strengthen the hypothesis that the control and eradication of the primary tumor by TA99 can be achieved in the absence of T cells and confirm the key implication of innate immune cells in this model.Citation22,31 This conclusion is further supported by the early and significant increase of IL12-producing macrophages and DCs as well as of IFNγ-producing NK cells observed in the tumor of TA99-treated mice. This TA99 short-term effect was also associated with a decrease in the tumors of CD11b+Gr1int monocytic-myeloid-derived suppressor cells (M-MDSC) (data not shown), which have been described as the most suppressive MDSC subset in the tumor microenvironment.Citation32 However, despite the clear contribution of the innate immune cells to TA99 anti-tumor effect, our data also show that the establishment of a long-term protective anti-tumor immunity definitely requires TA99-dependent development of T cell cytotoxic activity to efficiently control both subcutaneous and intravenous tumor challenges.
We also showed that TA99 treatment induces a humoral immune response characterized by the presence of endogenous anti-B16F10 IgGs in the serum that displayed a significant anti-tumor activity when transferred to mice engrafted with B16F10 tumor cells. However in contrast to TA99 immunotherapy, serum transfer was not sufficient to induce long-term tumor-free survival in our experimental settings. It was previously reported that the B cell population is essential for T cell priming during the early steps of the anti-tumor immune response and for optimal cellular immunity in the B16F10 melanoma model.Citation33 Here, we showed that TA99 stimulates the innate immunity at the tumor site. We can thus speculate that endogenous anti-B16F10 antibodies may keep stimulating DCs, macrophages and NK cells through FcR-dependent mechanisms, and therefore maintain the immunomodulatory effects beyond the persistence of the therapeutic mAb. We previously showed such a mechanism in a mouse model of virus-induced leukemia.Citation10 Although our results demonstrate that tumor-specific antibodies are generated following TA99 monotherapy, B cell contribution to treatment efficacy remains to be fully determined. A similar frequency of tumor inhibition was observed in B16F10 tumor-bearing µMT mice that lack mature B cells after combined treatment with TA99, extended half-life IL-2, anti-PD-1 antibody and TRP2 vaccine, suggesting that the host antibody response is not essential for the therapeutic effect of this combination.Citation34 However, one can speculate that with this complex therapeutic combination, the contribution of B cells could be compensated by a very robust T cell response induced by the T cell vaccine together with the anti-PD-1 antibody.
A crucial clinical issue is tumor escape in patients treated with TA-targeting mAbs. Our model, in which 30% of mice remained tumor-free and 70% developed tumors after a significant delay following TA-targeting mAb-based therapy, represents a suitable preclinical model to study tumor escape following anti-tumor antibody treatment. Analysis of the tumor immune microenvironment in TA99-treated mice at the time of tumor detection clearly shows that TA99 monotherapy specifically increases CD4 and CD8 effector cells in the tumor, but not in the spleen. The B16F10 melanoma is known to exploit escape mechanisms, particularly low expression of MHC class I antigens and promotion of a strong immunosuppressive microenvironment.Citation35 In line with these observations, immune infiltrate analysis in progressing B16F10 tumors revealed an increase of the Treg fraction and upregulation of TIM3 and PD-1 in both CD4 and CD8 effector T cells at the tumor site, a phenotype associated with defective secretion of IFNγ and IL-2 by these effector cells after in vitro stimulation.
Cancer immunotherapies that target immune checkpoints, including anti-PD-1/anti-PD-L1 antibodies have shown clinical activity in melanoma and also in other cancer types.Citation18 Several clinical trials are currently assessing the potential of combining immune checkpoint inhibitors, such as anti-PD-1 or anti-CTLA-4 molecules, with various anti-cancer treatments (radiotherapy,Citation36,37 chemotherapy,Citation38-41 targeted therapies including mAbsCitation42,43). Recent preclinical studies clearly demonstrated the benefit of adding immune checkpoint inhibitors to increase the anti-tumor effect of TA-targeting mAbs.Citation15,34,44-46 In all these studies, the TA-targeting mAb-immune checkpoint inhibitor combination was designed to prevent tumor growth and/or improve regression of established tumors. Specifically, in the B16F10 model, PD-1/PD-L1 blockade with an anti-PD-L1 mAb increases TA99 anti-tumor effect; however, durable anti-tumor responses require the concomitant neutralization of CD47, an anti-phagocytic ligand exploited by tumor cells to evade antibody-dependent clearance by phagocytes.Citation45
To our knowledge, the effect of immune checkpoint blockade has never been studied in conditions of tumor escape after TA-targeting monotherapy. Here, we clearly show that anti-PD-1 treatment twice a week soon after primary tumor escape induces a strong Th1 cytokine production by effector T CD4 and CD8 lymphocytes and also γδ T cells and NK cells. This reduces tumor progression and increases mice survival in the absence of any immune-related adverse events. Even if B16F10 melanoma tumors respond better to TA99 mAb monotherapy, we observed similar synergistic effects in mice grafted with the B16F1 variant, described as less metastatic than the B16F10 cells, but without any quantitative or qualitative differences in the patterns of exposed surface proteins including TRP-1. Combined therapies to sensitize non-responder or resistant patients are currently tested in clinical trials and one of the challenges is to identify the best timing for therapeutics association. Our findings show that by blocking PD-1 it is possible to boost the host anti-tumor immune response initiated several weeks before by treatment with a TA-targeting mAb. However, in our model the synergy observed between TA99 and anti-PD-1 mAbs was not sufficient to induce complete tumor regression, suggesting the implication of other immune suppressive pathways. Our study also demonstrates, for the first time, that PD-1 inhibitors can potentiate the cellular and humoral memory anti-tumor responses and delay tumor progression in challenged mice. Altogether, these promising results could pave the way for the development of novel therapeutic strategies, particularly for patients with acquired resistance and/or tumor relapse because of the development of an exhausted phenotype after the first therapeutic approach. These results are also of particular interest following the report of a phase 1 clinical trial using the anti-human TRP-1 mAb IMC-20D7S in 27 advanced melanoma patients with evidence of antitumor activity, since one complete response was observed and 10 patients achieved stable disease (Khalil DN et al. Clin Cancer Research 2016).Citation47
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Authors contribution
N. Bonnefoy and L. Gros conceived, supervised the study and wrote the manuscript. L. Gros, L. They and H-A. Michaud designed and performed experiments and analyzed the data. B. Guillot and O. Becquart coordinated the melanoma patients' surgery and collected tumor and blood samples. C. Mollevi and M. Jarlier gave expertise for statistical analysis. L. They, H-A. Michaud, V. Lafont and J.F Eliaou discussed results and reviewed the manuscript.
KONI_A_1353857_Supplementary_materials.zip
Download Zip (7.1 MB)Acknowledgments
The authors thank the staff of the IRCM animal facility for their technical assistance and Joëlle Simony-Lafontaine for her helpful expertise in the analysis and interpretation of IHC data.
Funding
This work was supported by institutional grants from INSERM, Université de Montpellier and additional support from the Labex MabImprove, (LG), the Canceropole Grand Sud Ouest (projets emergence NB and LG), the Canceropole Grand Sud Ouest and the GIRCI SOOM (API-K, OB) and the Société Française de Dermatologie (OB). L. They is supported by a Ph. D. studentship and H-A. Michaud by a post-doctoral fellowship from the Labex MabImprove. O. Becquart is supported by a fellowship from the Fondation pour la recherche Nuovo Soldati.
References
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015; 348:56-61; PMID:25838373; https://doi.org/10.1126/science.aaa8172
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13:1050-9; PMID:17704786; https://doi.org/10.1038/nm1622
- Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 2008; 26:1789-96; PMID:18347005; https://doi.org/10.1200/JCO.2007.14.8957
- Musolino A, Naldi N, Dieci MV, Zanoni D, Rimanti A, Boggiani D, Sgargi P, Generali DG, Piacentini F, Ambroggi M, et al. Immunoglobulin G fragment C receptor polymorphisms and efficacy of preoperative chemotherapy plus trastuzumab and lapatinib in HER2-positive breast cancer. Pharmacogenomics J 2016; 16:472-7; PMID:27378608; https://doi.org/10.1038/tpj.2016.51
- Burkhardt B, Yavuz D, Zimmermann M, Schieferstein J, Kabickova E, Attarbaschi A, Lisfeld J, Reiter A, Makarova O, Worch J, et al. Impact of Fc gamma-receptor polymorphisms on the response to rituximab treatment in children and adolescents with mature B cell lymphoma/leukemia. Ann Hematol 2016; 95:1503-12; PMID:27376362; https://doi.org/10.1007/s00277-016-2731-x
- Hilchey SP, Hyrien O, Mosmann TR, Livingstone AM, Friedberg JW, Young F, Fisher RI, Kelleher RJ, Jr, Bankert RB, Bernstein SH. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood 2009; 113:3809-12; PMID:19196657; https://doi.org/10.1182/blood-2008-10-185280
- Gros L, Dreja H, Fiser AL, Plays M, Pelegrin M, Piechaczyk M. Induction of long-term protective antiviral endogenous immune response by short neutralizing monoclonal antibody treatment. J Virol 2005; 79:6272-80; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15858011; PMID:15858011; https://doi.org/10.1128/JVI.79.10.6272-6280.2005
- Gros L, Pelegrin M, Michaud HA, Bianco S, Hernandez J, Jacquet C, Piechaczyk M. Endogenous cytotoxic T-cell response contributes to the long-term antiretroviral protection induced by a short period of antibody-based immunotherapy of neonatally infected mice. J Virol 2008; 82:1339-49; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18032505; PMID:18032505; https://doi.org/10.1128/JVI.01970-07
- Nasser R, Pelegrin M, Michaud HA, Plays M, Piechaczyk M, Gros L. Long-lasting protective antiviral immunity induced by passive immunotherapies requires both neutralizing and effector functions of the administered monoclonal antibody. J Virol 2010; 84:10169-81; PMID:20610721; https://doi.org/10.1128/JVI.00568-10
- Michaud HA, Gomard T, Gros L, Thiolon K, Nasser R, Jacquet C, Hernandez J, Piechaczyk M, Pelegrin M. A crucial role for infected-cell/antibody immune complexes in the enhancement of endogenous antiviral immunity by short passive immunotherapy. PLoS Pathog 2010; 6:e1000948; PMID:20548955; https://doi.org/10.1371/journal.ppat.1000948
- Stagg J, Sharkey J, Pommey S, Young R, Takeda K, Yagita H, Johnstone RW, Smyth MJ. Antibodies targeted to TRAIL receptor-2 and ErbB-2 synergize in vivo and induce an antitumor immune response. Proc Natl Acad Sci U S A 2008; 105:16254-9; PMID:18838682; https://doi.org/10.1073/pnas.0806849105
- Abes R, Gelize E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 2010; 116:926-34; PMID:20439625; https://doi.org/10.1182/blood-2009-10-248609
- Deligne C, Metidji A, Fridman WH, Teillaud JL. Anti-CD20 therapy induces a memory Th1 response through the IFN-gamma/IL-12 axis and prevents protumor regulatory T-cell expansion in mice. Leukemia 2015; 29:947-57; PMID:25231744; https://doi.org/10.1038/leu.2014.275
- Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK, Greene M, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010; 18:160-70; PMID:20708157; https://doi.org/10.1016/j.ccr.2010.06.014
- Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H, Teng MW, Smyth MJ. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A 2011; 108:7142-7; PMID:21482773; https://doi.org/10.1073/pnas.1016569108
- Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, Fumagalli D, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Desmedt C, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol 2014; 25:1544-50; PMID:24608200; https://doi.org/10.1093/annonc/mdu112
- Bianchini G, Pusztai L, Pienkowski T, Im YH, Bianchi GV, Tseng LM, Liu MC, Lluch A, Galeota E, Magazzu D, et al. Immune modulation of pathologic complete response after neoadjuvant HER2-directed therapies in the NeoSphere trial. Ann Oncol 2015; 26:2429-36; PMID:26387142; https://doi.org/10.1093/annonc/mdv395
- Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol 2016; 13:143-58; PMID:26598942; https://doi.org/10.1038/nrclinonc.2015.209
- Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest 2015; 125:3338-46; PMID:26241053; https://doi.org/10.1172/JCI80004
- Albanesi M, Mancardi DA, Macdonald LE, Iannascoli B, Zitvogel L, Murphy AJ, Daeron M, Leusen JH, Bruhns P. Cutting edge: FcgammaRIII (CD16) and FcgammaRI (CD64) are responsible for anti-glycoprotein 75 monoclonal antibody TA99 therapy for experimental metastatic B16 melanoma. J Immunol 2012; 189:5513-7; PMID:23150715; https://doi.org/10.4049/jimmunol.1201511
- Clynes R, Takechi Y, Moroi Y, Houghton A, Ravetch JV. Fc receptors are required in passive and active immunity to melanoma. Proc Natl Acad Sci U S A 1998; 95:652-6; PMID:9435247; https://doi.org/10.1073/pnas.95.2.652
- Otten MA, van der Bij GJ, Verbeek SJ, Nimmerjahn F, Ravetch JV, Beelen RH, van de Winkel JG, van Egmond M. Experimental antibody therapy of liver metastases reveals functional redundancy between Fc gammaRI and Fc gammaRIV. J Immunol 2008; 181:6829-36; PMID:18981101; https://doi.org/10.4049/jimmunol.181.10.6829
- Fuertes Marraco SA, Neubert NJ, Verdeil G, Speiser DE. Inhibitory receptors beyond T cell exhaustion. Front Immunol 2015; 6:310; PMID:26167163; https://doi.org/10.3389/fimmu.2015.00310
- Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 2010; 207:2175-86; PMID:20819923; https://doi.org/10.1084/jem.20100637
- Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Olive D, Kuchroo V, Zarour HM. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res 2012; 72:887-96; PMID:22205715; https://doi.org/10.1158/0008-5472.CAN-11-2637
- Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, Tarhini AA, Tawbi HA, Kirkwood JM, Moschos S, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8(+) T cells induced by melanoma vaccines. Cancer Res 2014; 74:1045-55; PMID:24343228; https://doi.org/10.1158/0008-5472.CAN-13-2908
- Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009; 114:1537-44; PMID:19423728; https://doi.org/10.1182/blood-2008-12-195792
- Nasser R, Pelegrin M, Plays M, Gros L, Piechaczyk M. Control of regulatory T cells is necessary for vaccine-like effects of antiviral immunotherapy by monoclonal antibodies. Blood 2013; 121:1102-11; PMID:23264590; https://doi.org/10.1182/blood-2012-06-432153
- Albanesi M, Mancardi DA, Jonsson F, Iannascoli B, Fiette L, Di Santo JP, Lowell CA, Bruhns P. Neutrophils mediate antibody-induced antitumor effects in mice. Blood 2013; 122:3160-4; PMID:23980063; https://doi.org/10.1182/blood-2013-04-497446
- Racki WJ, Covassin L, Brehm M, Pino S, Ignotz R, Dunn R, Laning J, Graves SK, Rossini AA, Shultz LD, et al. NOD-scid IL2rgamma(null) mouse model of human skin transplantation and allograft rejection. Transplantation 2010; 89:527-36; PMID:20134397; https://doi.org/10.1097/TP.0b013e3181c90242
- Bevaart L, Jansen MJ, van Vugt MJ, Verbeek JS, van de Winkel JG, Leusen JH. The high-affinity IgG receptor, FcgammaRI, plays a central role in antibody therapy of experimental melanoma. Cancer Res 2006; 66:1261-4; PMID:16452176; https://doi.org/10.1158/0008-5472.CAN-05-2856
- Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol 2010; 40:22-35; PMID:19941314; https://doi.org/10.1002/eji.200939903
- DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol 2010; 184:4006-16; PMID:20194720; https://doi.org/10.4049/jimmunol.0903009
- Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med 2016; 22:1402-10; PMID:27775706; https://doi.org/10.1038/nm.4200
- Ly LV, Sluijter M, van der Burg SH, Jager MJ, van Hall T. Effective cooperation of monoclonal antibody and peptide vaccine for the treatment of mouse melanoma. J Immunol 2013; 190:489-96; PMID:23203930; https://doi.org/10.4049/jimmunol.1200135
- Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res 2013; 1:365-72; PMID:24563870; https://doi.org/10.1158/2326-6066.CIR-13-0115
- Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, Scher HI, Chin K, Gagnier P, McHenry MB, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol 2013; 24:1813-21; PMID:23535954; https://doi.org/10.1093/annonc/mdt107
- Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517-26; PMID:21639810; https://doi.org/10.1056/NEJMoa1104621
- Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Neal J, Lu H, Cuillerot JM, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol 2012; 30:2046-54; PMID:22547592; https://doi.org/10.1200/JCO.2011.38.4032
- Kanda S, Goto K, Shiraishi H, Kubo E, Tanaka A, Utsumi H, Sunami K, Kitazono S, Mizugaki H, Horinouchi H, et al. Safety and efficacy of nivolumab and standard chemotherapy drug combination in patients with advanced non-small-cell lung cancer: a four arms phase Ib study. Ann Oncol 2016; 27:2242-50; PMID:27765756; https://doi.org/10.1093/annonc/mdw416
- Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Jr, Lao CD, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015; 16:375-84; PMID:25795410; https://doi.org/10.1016/S1470-2045(15)70076-8
- Amin A, Lawson DH, Salama AK, Koon HB, Guthrie T, Jr, Thomas SS, O'Day SJ, Shaheen MF, Zhang B, Francis S, et al. Phase II study of vemurafenib followed by ipilimumab in patients with previously untreated BRAF-mutated metastatic melanoma. J Immunother Cancer 2016; 4:44; PMID:27532019; https://doi.org/10.1186/s40425-016-0148-7
- Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, Zeng W, Giobbie-Hurder A, Atkins MB, Ibrahim N, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res 2014; 2:632-42; PMID:24838938; https://doi.org/10.1158/2326-6066.CIR-14-0053
- Linch SN, Kasiewicz MJ, McNamara MJ, Hilgart-Martiszus IF, Farhad M, Redmond WL. Combination OX40 agonism/CTLA-4 blockade with HER2 vaccination reverses T-cell anergy and promotes survival in tumor-bearing mice. Proc Natl Acad Sci U S A 2016; 113:E319-27; PMID:26729864; https://doi.org/10.1073/pnas.1510518113
- Sockolosky JT, Dougan M, Ingram JR, Ho CC, Kauke MJ, Almo SC, Ploegh HL, Garcia KC. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci U S A 2016; 113:E2646-54; PMID:27091975; https://doi.org/10.1073/pnas.1604268113
- Charlebois R, Allard B, Allard D, Buisseret L, Turcotte M, Pommey S, Chrobak P, Stagg J. PolyI:C and CpG Synergize with Anti-ErbB2 mAb for Treatment of Breast Tumors Resistant to Immune Checkpoint Inhibitors. Cancer Res 2017; 77:312-9; PMID:27872096; https://doi.org/10.1158/0008-5472.CAN-16-1873
- Khalil DN, Postow MA, Ibrahim N, Ludwig DL, Cosaert J, Kambhampati SR, Tang S, Grebennik D, Kauh JS, Lenz HJ, et al. An Open-Label, Dose- Escalation Phase I Study of Anti-TYRP1 Monoclonal Antibody IMC-20D7S for Patients with Relapsed or Refractory Melanoma. Clin Cancer Res 2016; 22:5204-10; PMID:5117650; http://www.ncbi.nlm.nih.gov/pubmed/27797971