
ABSTRACT
GD2-redirected chimeric antigen receptor (CAR) T lymphocytes represent a promising therapeutic option for immunotherapy of neuroblastoma (NB). However, despite the encouraging therapeutic effects observed in some hematological malignancies, clinical results of CAR T cell immunotherapy in solid tumors are still modest.
Tumor driven neo-angiogenesis supports an immunosuppressive microenvironment that influences treatment responses and is amenable to targeting with antiangiogenic drugs. The latter agents promote lymphocyte tumor infiltration by transiently reprogramming tumor vasculature, and may represent a valid combinatorial approach with CAR T cell immunotherapy.
In light of these considerations, we investigated the anti-NB activity of GD2-CAR T cells combined with bevacizumab (BEV) in an orthotopic xenograft model of human NB. Two weeks after tumor implantation, mice received BEV or GD2-CAR T cells or both by single intravenous administration. GD2-CAR T cells exerted a significant anti-NB activity only in combination with BEV, even at the lowest concentration tested, which per se did not inhibit tumor growth. When combined with BEV, GD2-CAR T cells massively infiltrated tumor mass where they produced interferon-γ (IFN-γ), which, in turn, induced expression of CXCL10 by NB cells. IFN-γ, and possibly other cytokines, upregulated NB cell expression of PD-L1, while tumor infiltrating GD2-CAR T cells expressed PD-1. Thus, the PD-1/PD-L1 axis can limit the anti-tumor efficacy of the GD2-CAR T cell/BEV association.
This study provides a strong rationale for testing the combination of GD2-CAR T cells with BEV in a clinical trial enrolling NB patients. PD-L1 silencing or blocking strategies may further enhance the efficacy of such combination.
Introduction
Neuroblastoma (NB) is the most frequent extracranial solid tumor in children, accounting for about 15% of pediatric tumor deaths.Citation1 Despite advances in multimodal therapeutic approaches, the outcome of patients with metastatic disease at diagnosis still remains poor.Citation2 Immunotherapy represents a valid adjuvant to standard frontline therapies, and different antibody and cell-based immunotherapeutic protocols for NB have been developed and tested in preclinical and clinical studies.Citation3
Genetic engineering of peripheral blood T lymphocytes with chimeric antigen receptors (CAR) has allowed the development of immunotherapeutic approaches based on CAR-redirected T lymphocytes that recognize tumor associated antigens in MHC-independent manner, overcoming defects in antigen processing and presentation of tumor cells.Citation4 GD2 is a disialoganglioside highly expressed on human NB cells that represents a good target for NB immunotherapy.Citation5 GD2-redirected CAR T cells (GD2-CAR T) represent a new therapeutic option for GD2+ tumors, and different clinical trials with GD2-specific CAR T cells in NB patients have been published and are ongoing.Citation6-9
Clinical results of CAR T cell-based immunotherapy have been very good in selected hematological malignancies such as acute lymphoblastic leukemia and multiple myeloma, but modest in solid tumors.Citation10-12 GD2- CAR T cell infusion in NB patients with bulky disease resulted in marginal clinical responses, and similar results were reported in most CAR T cell treated patients with other solid malignancies.Citation7-10 Different barriers contribute to limit the potency of CAR T cells in solid tumors, including poor in vivo persistence and expansion, limited trafficking and penetration into tumor mass and CAR T cell dysfunctions induced by the immunosuppressive tumor milieu.Citation10
Neoangiogenesis, driven by proangiogenic factors secreted by tumor cells plays a fundamental role in tumor growth and metastasis.Citation13 Tumor vascularity and expression of different angiogenic factors by human NB cells correlates with more aggressive advanced stages and with MYCN amplification of tumor cells.Citation14-16 Structural and functional abnormalities of tumor vasculature support a microenvironment that favors tumor progression and metastasis, and sustains immunosuppression and chemo-resistance.Citation17 The tortuous course, the irregular diameter and the erratic pericyte coverage of tumor microvessels creates a shambolic vascular network where blood flow is compromised. This, in turn, creates a hypoxic and acidic milieu in which tumor responsiveness to irradiation as well as tissue penetration of chemotherapeutic drugs and antitumor lymphocytes are impaired.Citation17,18
Targeting tumor vasculature by antiangiogenic agents is a promising approach for cancer therapy originally developed with the aim of starving tumor cells by inhibition of new vessel growth and destruction of existing abnormal tumor vasculature.Citation19,20 Vascular endothelial growth factor (VEGF) is a central mediator of tumor angiogenesis, and drugs are being tested for its direct or indirect targeting in preclinical studies and clinical trials.Citation21-23 Bevacizumab (BEV) is a humanized monoclonal antibody to human VEGF-A, which has been clinically approved for cancer therapy, either as monotherapy or in combination with standard cytotoxic therapy.Citation24 Different anti-angiogenic strategies including VEGF-A targeting were shown to control tumor growth in xenograft models of human NB and are currently being tested in clinical trials.Citation25-27
Beside their vessel blocking activity, antiangiogenic agents have been shown to transiently repair and remodel tumor vasculature, reprogramming tumor microenvironment and creating a “vessel normalization window” that improves anti-tumor immunity and enhances tumor sensitivity to chemo- and radio-therapy.Citation28-31 Antiangiogenic agents promote leukocyte/endothelium interaction and sustain leukocyte extravasation and tumor infiltration, thus lending support to the use of such agents in combination with cellular immunotherapy of cancer.Citation32-34
We have here investigated the antitumor activity of GD2-CAR T lymphocytes in combination with BEV in an orthotopic xenograft model of cell-based immunotherapy for human NB. We used clinical grade, third generation GD2-CAR T cells containing CD28 and OX40 as costimulatory endodomains, that do not produce the same high levels of tonic CAR signaling and functional exhaustion reported for second generation GD2-CAR T cells.Citation35,36 Our results demonstrate that GD2-CAR T cells exerted a significant anti-tumor activity only when combined with BEV due to increased tumor infiltration.
Results
In vivo anti-NB activity of GD2-CAR T cells in combination with BEV
GD2-CAR T lymphocytes were expanded in vitro from the peripheral blood of normal individuals in the presence of IL-7 and IL-15 for 14 days, then checked for immunophenotype and cytotoxic activity before being used for in vivo experiments. GD2-CAR T cell fractions consisted of CD45RO+, CD62L+ central memory and CD45RO+, CD62L− effector memory CD4+ and CD8+ T cells, which specifically lysed the GD2+ HTLA-230 and IMR-32 human NB cell lines (Fig S1). In contrast, control non-transduced (NT) T cell blasts did not kill GD2+ target cell lines (Fig S1).
In vivo anti-NB activity of GD2-CAR T cells in combination with BEV was investigated using an orthotopic xenograft model by implanting Scid/Beige mice in the adrenal gland with GD2+ HTLA-230 or IMR-32 cell lines. Two weeks after HTLA-230 cell engraftment, mice received iv NT T cells (10 × 106/mouse), or BEV, or GD2-CAR T cells (10 × 106/mouse) either alone or 48 h after BEV infusion. As shown in , mice receiving GD2-CAR T cells after 5 mg/kg BEV infusion survived significantly longer than mice treated with NT cells or GD2-CAR T cells alone (P = 0.0002). Survival of mice treated with GD2-CAR T cells as single agent did not significantly differ from that of mice receiving control NT cells. BEV alone at 5 mg/Kg significantly improved survival of NB-bearing mice as compared with both NT- and GD2-CAR T cell treatment (P = 0.0044). In the attempt to reduce the antitumor effects of BEV while preserving BEV-mediated tumor microvessel remodelling, a second group of mice received a lower dose of BEV (2 mg/Kg). As shown in , only HTLA-230 tumor-bearing mice that received the combined treatment with GD2-CAR T cells + 2 mg/Kg BEV showed significantly increased survival compared to mice receiving NT cells or GD2-CAR T cells alone (P = 0.0006). BEV infusion at 2 mg/Kg did not exert any antitumor effect. Consistently with these results, significantly increased survival was observed in Scid/Beige mice orthotopically engrafted with a second NB cell line, i.e. IMR-32, that received GD2-CAR T cells and 2 mg/Kg BEV in combination ().
Figure 1. Survival of mice engrafted with HTLA-230 or IMR-32 NB cells and treated with GD2-CAR T cells and BEV as single agents or in combination. A-B. Six-week old female Scid/Beige mice were xenografted in the left adrenal gland with HTLA-230 NB cells (1x106 cells/mouse). Two weeks later, mice received human untransfected T cells (NT, 10x106/mouse), BEV(Avastin®, A: 5 mg/Kg; B: 2 mg/Kg) or GD2-CAR T cells (10x106 cells/mouse) given either alone or 48 hours after BEV. A single iv treatment was performed. Kaplan-Meier analyses from two independent experiments with T cells from two donors are shown. C. Six-week old female Scid/Beige mice were xenografted in the left adrenal gland with IMR-32 NB cells (1x106 cells/mouse). Two weeks later, mice received human untransfected T cells (NT, 10x106/mouse), BEV (Avastin®, 2 mg/Kg) or GD2-CAR T cells (10x106 cells/mouse) given either alone or 48 hours after BEV. A single iv treatment was performed. Kaplan-Meier analysis is shown.
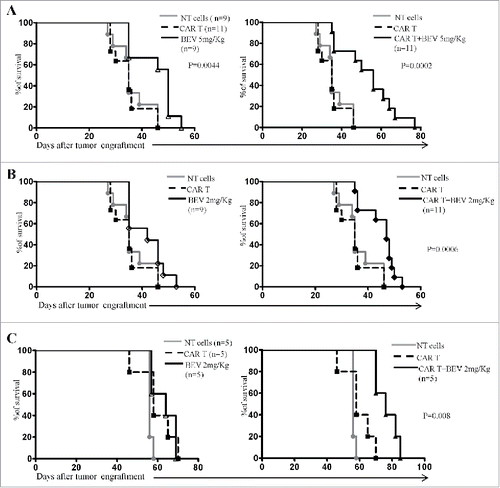
These results indicated that in Scid/Beige mice orthotopically engrafted with human NB cells, GD2-CAR T cells exerted their anti-NB activity exclusively when combined with the anti-angiogenic agent, pointing to synergistic anti-NB effects of GD2-CART cells and BEV.
BEV-mediated tumor microenvironment remodelling maximizes anti-NB functions of GD2-CAR T cells by supporting their tumor infiltration capacity
Orthotopic xenografts with human NB HTLA-230 cells in Scid/Beige mice gave rise to rapidly growing and well-vascularized tumors characterized by numerous mitotic and some apoptotic figures (). The vascular network was significantly reduced in tumors from 5 mg BEV-treated mice () in association with a marked reduction in VEGF-A expression by NB cells (Figure S2, a and c) and frequent apoptotic figures () along the vascular walls (), which resulted in small ischemic-hemorrhagic foci (). Morphological changes were observed in the surviving vessels, which frequently appeared elongated and less tortuous () than those of control tumors ().
Figure 2. Histologic and immunohistochemical features of tumors developed after orthotopic engraftment of HTLA-230 NB in Scid/Beige mice treated with BEV, GD2-CAR T cells, or GD2-CAR cells+BEV. H&E staining of tumors developed in untreated (CTR) mice (a) and in mice receiving 5 mg/Kg BEV (e), GD2-CAR T cells (i) or GD2-CAR T cells+BEV (m). Arrowheads in a: mitotic figures; arrows in i and m: lymphocyte infiltration. CD31 and CD3 immunostaining of tumors developed in untreated mice (b,c), BEV- (f,g), GD2-CAR T cell- (j,k) or GD2-CAR T cells+BEV- (n,o) treated mice. Tunel assay in tumors developed in untreated mice (d) and in mice treated with BEV (h), GD2-CAR T cells (l) or GD2-CAR T cells+BEV(p). Arrowheads in h: apoptotic figures along the vascular endothelial walls. NE: necrosis. (Magnification a-l: X400; m: X630; n-p: X400)
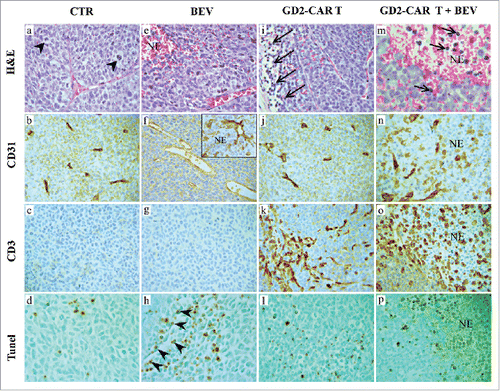
Table 1. Microvessel count and apoptotic index in tumors from Scid/Beige mice either untreated or treated with BEV, GD2-CAR T cells or both.
Tumors from GD2-CAR T-treated mice were vascularized such as were control tumors (, j and ). The former tumors were characterized by a distinct CD3+T cell infiltrate (), which was found mostly at the edge of the tumor mass, where some apoptotic events were observed ( and ). The combined treatment with GD2-CAR T and 5 mg BEV induced wide ischemic-hemorrhagic foci () in association with a rarefied ( and ) and severely damaged vascularization, and a massive infiltration of CD3+T cells into the tumor mass (), as confirmed by double CD31/CD3 immunostaining (). The apoptotic events ( and ), together with the ischemic-necrotic phenomena, resulted in multiple wide areas of tumor destruction. Later on, the tumors became small shrunken masses formed by sheets of ghost tumor cells mixed with rare lymphocytes within a rarefied and vessel-free stroma (Fig. S3, a, b).
Figure 3. Analysis of vascular network and T lymphocyte distribution in tumors developed in Scid/Beige mice after orthotopic engrafment of HTLA-230 NB cells and treated with GD2-CAR T cells alone or in combination with BEV. CD31 (red) and CD3 (blue) double immunostaining of tumors from mice treated with GD2-CAR T cells (a) as single agent or in combination with BEV at 2 mg/Kg (b,c) and 5 mg/Kg (d). (a, b, c: X400; d: X200).
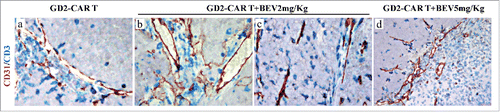
BEV alone at lower dose (2 mg/Kg) led to limited reduction in VEGF-A expression (Fig S2, b), without a significant decrease in microvessel number (7.2 ± 3.3 of 2 mg BEV-treated vs 10.6 ± 2.7 of CTR, ), which, as observed after 5 mg/Kg BEV treatment, appeared remarkably less winding than vessels of control tumors. The combined treatment with GD2-CAR T cells plus 2 mg/Kg BEV resulted in substantial extravasation of CD3+T lymphocytes () homogeneously penetrating the tumor (), which showed frequent (p < 0.05) apoptotic events (apoptotic index: 15.8 ± 3.8% of GD2-CAR T + 2 mg BEV-treated, vs 6.0 ± 2.2% of CTR, ), a compromised vascular network (blood vessels: 4.0 ± 2.5 of GD2-CAR T+2 mg BEV-treated, vs 10.6 ± 2.7 of CTR, ) and ischemic-hemorrhagic foci.
In summary, these experiments indicate that GD2-CAR T cells could efficiently infiltrate the inner tumor mass and cause significant tumor damage only when combined with BEV, even at a dose per se devoid of anti-tumor activity.
Interplay between NB tumor cells and GD2-CAR T cells sustains and amplifies GD2-CAR T cell recruitment into NB tumor
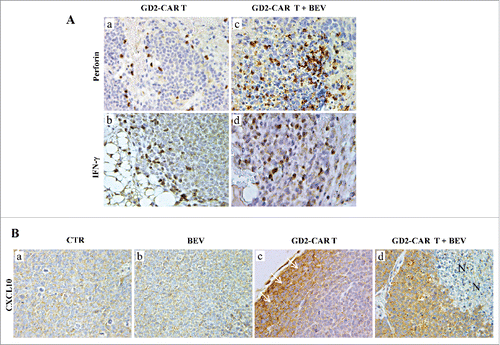
Next we investigated whether GD2-CAR T/NB cell interactions could reprogram tumor microenvironment and amplify GD2-CAR T cells recruitment inside the tumor mass. In tumors from GD2-CAR T-treated mice, perforin and IFN-γ were expressed by lymphoid cells confined at the edge of the tumor (, and ), while the IFNγ -inducible chemokine CXCL10 was detected only in the adjacent tumor cells (). In contrast, tumors from mice receiving GD2-CAR T cells in combination with BEV were heavily infiltrated with lymphoid cells mostly expressing perforin and IFN-γ (, and ), and widespread cancer cell expression of CXCL10 was observed within the tumor core (). These results support the hypothesis that GD2-CAR T cells producing IFN-γ and perforin induce tumor cell expression of CXCL10, which recruits additional GD2-CAR T cells. This model predicts that GD2-CAR T cells express functional CXCR3.
Figure 4. Perforin, IFN-γ and CXCL10 expression in tumors developed in Scid/Beige mice orthotopically engrafted with HTLA-230 NB cells and treated with GD2-CAR T cells alone or combined with BEV. A.Perforin and IFN-γ immunostaining of tumor masses from mice receiving GD2-CAR T cells either alone (a,b) or in combination with BEV (c,d). (a, b:X400; c, d: X630). B.CXCL10 immunostaining of tumors from untreated mice (a, CTR) and from mice treated with BEV (b), GD2-CAR T cells alone (c) or in combination with BEV (d). (c, white arrows indicate CXCL10+ cancer cells at the edge; d, N: necrosis) (a-d:X400).
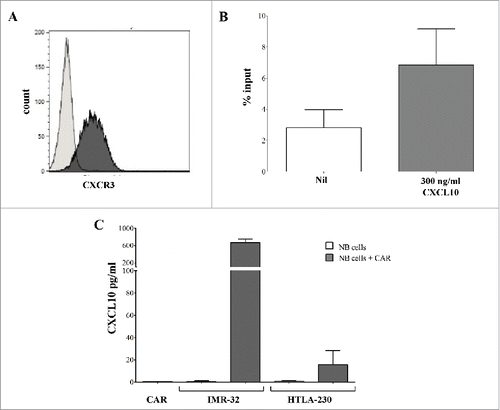
To test this hypothesis, we next performed in vitro studies addressing GD2-CAR T cell expression of CXCR3 and migration induced by CXCL10. As shown in and , GD2-CAR T cells expressed CXCR3 and migrated in response to its ligand CXCL10, while co-culture of GD2-CAR T cells and NB cells induced CXCL10 chemokine release by tumor cells ().
Figure 5. Chemotactic properties of GD2-CAR T cells and their interplay with NB tumor cells. A. Flow cytometric analysis of CXCR3 expression on GD2-CAR T cells after 12-14 days of culture. A representative staining is shown. Black profile indicates staining with specific mAb, gray profile indicates staining with irrelevant isotype-matched mAb. B. Chemotaxis of GD2-CAR T cells to CXCL10 was tested in vitro by transwell. Results are expressed as % of input (see Methods). Means of 3 different experiments +SD are shown. C. CXCL10 production by IMR-32 and HTLA-230 NB cell lines cultured alone or in the presence of GD2-CAR T cells for 24 h. Supernatants of GD2-CAR T cells (CAR) cultured alone for 24h were used as controls. Means of 3 different experiments +SD are shown.
We finally investigated whether IFN-γ-producing GD2-CAR T cells could induce PD-L1 upregulation in NB cells. Preliminary in vitro experiments showed that PD-1 expression level on GD2-CAR T cells before infusion was low (n = 3, median CD4+: 29%; CD8+: 3,5%, data not shown), but increased with CAR T cell activation after co-culture with tumor cells (n = 3, median CD4+: 46%; CD8+: 29%, data not shown), likely mimicking the sequence of events which occur in vivo following infusion of GD2-CAR T cells. Similar results have been reported previously by other investigators.Citation7
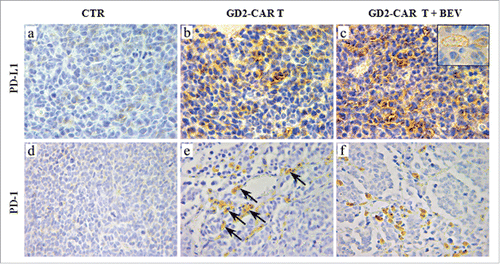
As shown in scattered and slight expression of PD-L1, which appeared mainly on tumor cells (and sometimes on endothelial cells) was observed at the edge of tumors in GD2-CAR T cell-treated mice (), whereas it was widely distributed in tumors from GD2-CAR T plus BEV-treated mice (). Expression of PD-1 by tumor infiltrating cells with lymphocyte morphology was detected in both experimental conditions, but it was evidenced at the tumor edge in the former, while it was evenly distributed in the latter (). Thus, IFN-γ and possibly other cytokines secreted by tumor infiltrating GD2-CAR T cells, could sustain the recruitment of GD2-CAR T cells inside the tumor, but also contribute to the generation of immunosuppressive tumor microenvironment.
Figure 6. PD-L1 and PD-1 expression in tumors developed in Scid/Beige mice orthotopically engrafted with HTLA-230 NB cells and treated GD2-CAR T cells alone or combined with BEV. PD-L1 and PD-1 immunostaining of tumors from mice either untreated (a, d, CTR) or treated with GD2-CAR T cells alone (b, e) or in combination with BEV (c, f). PD-L1 expression involved tumor cells and, sometimes, vascular endothelial cells (c, inset panel), whereas PD-1 was expressed by tumor infiltrating lymphocyte-like cells. PD-1+ cells were detected mainly at the tumor edge after administration of GD2-CAR T cells alone (arrows in e), while they were observed penetrating throughout the tumor after the combined treatment with GD2-CAR T cells and BEV. (a, b, c and inset: X630; d, e, f: X400).
Discussion
Major determinants that impact on the efficacy of CAR T cells are in vivo expansion, persistence and trafficking/penetration into the tumor, as well as physical and immunosuppressive barriers opposed by the tumor microenvironment.Citation10 In spite of the attempts to overcome these hurdles, treatment of solid tumor-bearing patients remains problematic, and the results of the clinical trials so far conducted have been generally poor.Citation10 Some encouraging results have been reported in a small study with nineteen NB patients who were treated with GD2-CAR T cells, 2 of whom achieved complete remission, and in another trial with 17 sarcoma patients receiving HER2-CAR T cells, 4 of whom showed stable disease.Citation8,37
A strategy to improve CAR T cell trafficking to the tumor rests upon the enhancement of extracellular matrix (ECM) degradation. In this respect, recently it was reported that in vitro cultured T cells lose the expression of the enzyme heparanase, which degrades heparan sulfate proteoglycans of the extracellular matrix and subendothelial basement membrane. In a xenograft model of human NB, engineering of GD2-CAR T cells with heparanase gene allowed higher infiltration of the genetically modified cells into the tumor mass and superior anti-tumor activity.Citation38 CAR T cells trafficking to the tumor may be also enhanced by engineering of these cells with chemokine or chemokine receptor encoding genes.Citation39-41 Alternatively, the migratory capacity of CAR T cells to the tumor mass can be improved by targeting and reprogramming the tumor microenvironment with anti-angiogenic agents. These molecules, which include the anti VEGF-A antibody BEV and multi-target tyrosine kinase inhibitors, reduce the number of tumor microvessels and modify transiently the tumor vasculature, improving perfusion and, consequently, reducing hypoxia and metabolic acidosis. These changes allow a better infiltration of cells and drugs into the tumor.Citation30
Here we have investigated whether the combined administration of BEV and human GD2-CAR T cells to immunodeficient mice xenografted with human NB cells improved T cell migration and invasion to the tumor site.
BEV is highly specific for human VEGF-A.Citation19,22,24 BEV showed an extremely weak interaction with murine VEGF-A and failed to neutralize it in different bio-assays, including proliferation of mouse endothelial cells stimulated by murine VEGF.Citation42 These findings support our working hypothesis that VEGF-A blockade occurred almost exclusively at the tumor site where malignant cells were the only source of human VEGF-A.
We used clinical grade, third generation GD2-CAR T cells containing CD28 and OX40 as costimulatory endodomains, that are less prone to the functional exhaustion reported for second generation GD2-CAR T cells.Citation35,36 A costimulatory domain alternative to OX40 used for CAR T cell production is 4-1BB. In T cells engineered with anti-CEA CAR, the CD28 endodomain was superior to initiate the T cell response, while OX40 or 4-1BB sustained the long term response, with OX40 being most effective.Citation43 So far, no data were reported on the best combination between CD28-OX40 and CD28-4.1BB, respectively, in third generation GD2-CAR T cell setting.
In this study, treatments with BEV and/or human GD2-CAR T cells were administered to mice two weeks after orthotopic NB implantation. GD2-CAR T cells alone did not exert any anti-tumor activity, whereas BEV administered alone at 5 mg/Kg inhibited tumor growth, and the combination of BEV with GD2-CAR T cells was one log more effective than treatment with BEV alone. We eliminated the intrinsic anti-tumor activity of BEV as single agent by decreasing the dose to 2 mg/Kg. In this experimental condition BEV retained the ability to synergize with GD2-CAR T cells in the inhibition of NB growth. Histologic and immunohistochemical analyses of NB tumors showed that BEV at 5 mg/Kg, but not at 2 mg/Kg, reduced the number of tumor microvessels and induced tumor cell apoptosis, whereas both BEV concentrations reduced VEGF-A expression in the tumor, although at different extent, and normalized the structure of the vasculature. Similar results have been reported previously in a pre-clinical model of breast cancer in which an anti-VEGFR2 antibody was administered at doses lower than those endowed with anti-angiogenic activity. Such low doses induced vascular normalization, reprogrammed macrophages from a M2-like to a M1 phenotype and facilitated T cell tumor infiltration.Citation34
Failure of GD2-CAR T cells administered alone to mediate anti-NB activity was found to depend on defective migration inside the tumor mass. In contrast, GD2-CAR T cells administered after BEV infusion infiltrated massively the core of the tumor, where extensive areas of necrosis were detected, conceivably as a consequence of high expression of perforin and IFN-γ by T cells. Perforin is a cytotoxic molecules closely involved in tumor cell killing, while IFN-γ induces apoptosis of NB cells through multiple signaling pathways, including the CD95/CD95 L autocrine circuit and the Stat1/IRF1 pathway.Citation44,45 In addition, IFN-γ induces the expression of the anti-angiogenic chemokines CXCL9 and CXCL10.Citation46 The latter was abundantly expressed in NB tumors infiltrated with IFN-γ producing GD2-CAR T cells, pointing to a contribution of CXCL10 to inhibition of tumor angiogenesis and growth. Finally, in our experiments, CXCL10 attracted CXCR3+ GD2-CAR T cells in vitro, thus supporting an amplification model of recruitment of the latter cells to the tumor mass. Although these results point to a key role of IFN-γ in our model, other cytokines released by GD2-CAR T cells may have contributed to the therapeutic activity of the latter cells administered together with BEV.
Involvement of CXCL10 in immunotherapy NB models was previously demonstrated in another study from our group.Citation47 Human Vγ9Vδ2 T cells infused in combination with zoledronic acid in immunodeficient mice orthotopically xenografted with human NB cells infiltrated the tumor mass, where they expressed IFN-γ and the cytotoxic molecule TIA-1, and induced tumor cell expression of CXCL10. The latter chemokine played a dual role in this setting by inhibiting tumor angiogenesis and recruiting new waves of cytotoxic CXCR3+ Vγ9Vδ2 T cells to the tumor site.Citation47
The xenograft NB model used herein did not allow to evaluate the role of host immune system in the response to treatment. Nonetheless, it must be noted that pro-angiogenic factors, and in particular VEGF-A, have been shown to dampen the anti-tumor immune response through different mechanisms, including inhibition of dendritic cell maturation, increased accumulation of myeloid derived suppressor cells, generation of T regulatory cells, and induction of tumor associated-macrophages.Citation48 It is therefore conceivable that, in immunocompetent hosts, BEV-mediated inhibition of VEGF-A can further potentiate the therapeutic activity of GD2-CAR T cells by dampening or abrogating different immunosuppressive barriers in the tumor microenvironment.
One of the major immune checkpoints that dampen the anti-tumor immune response is the PD-1/PD-L1 axis.Citation49 PD-1 is expressed by infiltrating immune cells, while PD-L1 is expressed, either constitutively or inducibly, by tumor cells. IFN-γ is the best-characterized, although not the only, inducer of PD-L1 expression.Citation50 Our immunohistochemical analyses revealed that GD2-CAR T cells upregulated the expression of PD-L1 on NB cells, likely as a consequence of their ability to produce IFN-γ, which has been previously shown to upregulate PD-L1 in primary NB cells and NB cell lines.Citation51 On the other hand, GD2-CAR T cells expressed PD-1. Both PD-L1 and PD-1 expression were detected mainly at the tumor periphery following infusion of GD2-CAR T cells alone, and within the tumor mass upon administration of GD2-CAR T cells and BEV. Thus, the PD-1/PD-L1 axis may have limited the therapeutic efficacy of the latter combination that improved significantly the survival of NB-bearing mice but did not eradicate the tumor. Different strategies can be envisaged to silence or dampen PD-L1 at the tumor site or PD-1 expression in CAR T cells in order to overcome the immunosuppressive interactions between PD-1 and its ligand PD-L1 in the tumor microenvironment.Citation35,52,53 Previously it was shown that second-generation GD2-CAR T cells underwent potent activation and PD-1-mediated deletion following antigen encounter.Citation52 Furthermore, a small phase I study showed that lymphodepletion with cyclophosphamide and fludarabine (Cy/Flu) increased CAR T cell expansion in patients with neuroblastoma by up to 3 logs.Citation7 PD-1 inhibition with pembrolizumab did not further enhance CAR T cell expansion or persistence, and a striking expansion of CD45/CD33/CD11b/CD163+ myeloid cells was detected, that may have contributed to the modest clinical responses.Citation7
In conclusion, the results of this study are translationally relevant, since GD2-CAR T cells have been administered as single agent to pediatric NB patients and BEV safety has been established in a Children Oncology Group phase I trial enrolling pediatric patients with resistant solid tumors.Citation54 Toxicity of the combination of GD2-CAR T cells with BEV must be evaluated in an ad hoc designed clinical trial. PD-L1/PD-1 silencing or blocking strategies may further help enhance the efficacy of such a combination.
Materials and methods
Cell lines
The human NB cell lines HTLA-230 (kindly provided by Professor Emil Bogenmann, Children's Hospital Los Angeles, Los Angeles, California, USA) and IMR-32 (kindly provided by Professor Carol J. Thiele, National Cancer Institute, Bethesda, Maryland, USA) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), HEPES buffer, L-glutamine, penicillin-streptomycin and nonessential aminoacids (EuroClone, Milano, Italy). All cell lines were used between passages 25 and 50. Frozen aliquots were thawed and cultured for no more than 4 weeks, tested for mycoplasma and authenticated by STR (BMR Genomics S.r.l., Padova, Italy).
Plasmid construction and retrovirus production
The cassette encoding the single-chain antibody targeting GD2, CD28, OX40 endodomains and the ζ chain of the T-cell receptor complex was cloned into the SFG retroviral backbone to generate the third-generation CAR.GD2 retroviral vector.Citation55
Generation of CAR-modified T cells
Peripheral blood mononuclear cells (PBMC) were obtained from five healthy donors following protocol approval by Institutional Review Board (IRB) of Ospedale Pediatrico Bambino Gesù, Roma, Italy. PBMC were activated with OKT3 (Ortho Biotech, Bridgewater, NJ) and CD28 (Becton Dickinson, Mountain View, CA) antibodies and recombinant human interleukin-7 (IL7) (10 ng/mL) and IL15 (5 ng/ml) (Proleukin; Chiron, Emeryville, CA) in complete media [RPMI 1640 (Gibco-BRL) 45%, Click medium (Irvine Scientific, Santa Ana, CA) 45%, supplemented with 10% FBS (HyClone, GE Healthcare Life Sciences, Marlborough, MA) and 2 mM L-glutamine (GIBCO-BRL)]5656. Activated T cells were transduced with retroviral supernatants on day 3 in plates coated with recombinant fibronectin fragment (FN CH-296; Retronectin; Takara Shuzo, Otsu, Japan).Citation56 After transduction, T cells were expanded using IL7-IL15 and then used for the experiments described below.
Flow cytometry
The following monoclonal antibodies were used: CD3, CD4, CD8, CD45RO, CD62L, CD45RA (BD Biosciences, San Jose, CA), CXCR3 (R&D Systems, Minneapolis, MN) conjugated with FITC, PE, PE-Cy7 or APC. Isotype matched fluorochrome-conjugated murine Ig were used as negative controls. Expression of GD2- specific CAR was analyzed with the specific anti-idiotype antibody 1A7. Samples were analyzed with FACSCalibur (BD Biosciences) or Gallios (Beckman-Coulter, Marseille, France) cytometers and CellQuest (BD Biosciences) or Kaluza (Beckman-Coulter) softwares.
Cytotoxicity assays
Cytotoxicity assays were performed using a standard 4 h 51Cr-release test. GD2-CAR T cells or NT activated T lymphocytes were used as effectors. Human GD2+ NB cell lines or Raji Burkitt's lymphoma cell line were used as targets. Different effector to target ratio were tested. Results were expressed as percent specific lysis, according to the formula: %lysis = 100 × (experimental release − spontaneous release)/(maximum release − spontaneous release).
Chemotactic assays
Chemotaxis was investigated using 5 µm pore-size transwell plates (Costar, Cambridge, MA). Five × 105 cells were dispensed in the upper chamber, chemokines or medium alone were added to the lower chamber. Recombinant CXCL10 (Immunotools, Friesoythe, Germany) were tested at 300 ng/ml following preliminary titration experiments. Plates were incubated 2 h at 37°C. Migrated cells were collected and counted, and migration index was calculated as follows: (n° of migrated cells/n° of dispensed cells) x100. Migration index obtained with medium alone was subtracted from each value.
ELISA
Chemokine release by NB cells was tested after 24 h culture either in the presence or in the absence of GD2-CAR T cells. NB cells were plated in 6 well plate (5 × 105 cells/well) either in the presence or in the absence of GD2-CAR T cells at a NB/CAR T cell ratio of 10:1. GD2-CAR T cells cultured alone (5 × 104/well in 24 well plate) were used as controls. After 24 h culture, supernatants were collected and stored at -80° until tested using a standard ELISA (R&D Systems).
In vivo studies
Six-week-old female Scid/Beige mice (Envigo S.r.l., S.Pietro al Natisone, Italy) were anesthetized with ketamine (Imalgene 1000, Merial Italia S.p.A, Milano, Italy), subjected to laparotomy and orthotopically xenografted in the left adrenal gland with human NB cells (1 × 106 cells/mouse). Two weeks later, mice were allocated into different groups: (i) control group, receiving either human NT T cells (10 × 106cells/mouse) or normal saline; (ii) mice treated with BEV (Avastin®, Roche S.p.A, Italia) at 5 mg/Kg; (iii) mice treated with BEV at 2 mg/Kg; (iv) mice treated with GD2-CAR T cells (10 × 106 cells/mouse); (v) mice treated with GD2-CAR T cells (10 × 106 cells/mouse) 48 hours after BEV 5 mg/Kg injection and (vi) mice treated with GD2-CAR T cells (10 × 106 cells/mouse) 48 hours after BEV 2 mg/Kg injection. All treatments were administered once intravenously. Mice were inspected three times a week and were euthanized when signs of discomfort were detected. The protocol was reviewed and approved by Ethical Committee of IRCCS Azienda Ospedaliera Universitaria San Martino-IST (Genova-Italy) and by the Italian Ministry of Health.
Histology, immunohistochemistry and morphometric analyses
These studies were performed on tumor masses explanted from mice 48 hours after BEV or normal saline infusion or 72 hours after GD2-CAR T cell infusion. For histology, paraffin-embedded samples were sectioned at 3 µm and stained with hematoxylin and eosin (H&E). Single and double immunohistochemistry was done on paraffin-embedded sections. Single immunohistochemistry was performed using anti-human CD3 (A0452, Dako, Glostrup, DK), anti-human PD-L1 (Clone 22C3, Dako, Carpinteria, CA), anti-mouse CD31 (clone SZ31, Dianova, Hamburg, Germany), anti-human perforin (Clone 5B10, Novocastra, Newcastle Upon Tyne, UK), anti-human PD-1 (Clone NAT105), anti-human interferon-gamma (rabbit polyclonal, AB9657) and anti-human CXCL10 (rabbit polyclonal, AB9807) all from Abcam (Cambridge, UK), anti-human VEGF-A (rabbit polyclonal, RB-9031-P1, Thermo Scientific, Waltham, MA) Abs in a fully automated Leica Bond-Max instrument (Leica Biosystems, Nussloch GmbH, Germany). The immune complexes were detected using the Bond Polymer Refine Detection Kit (Leica Biosystems) according to the manufacturer's protocol. Negative controls were performed by replacing the primary Ab with 10% non-immune serum. Further controls were performed by omitting the secondary Ab. Controls were always negative.
For CD31/CD3 double immunostaining of formalin-fixed paraffin-embedded samples, sections were deparaffinized, subjected to heat-induced epitope retrieval with tris-EDTA pH 9.0 for 20 minutes, treated with H2O2/3% for 5 min. to inhibit endogenous peroxidase, and then washed in tris buffer. The slices were then incubated for 30 min with the first primary antibody (rat anti-mouse CD31, Dianova, Hamburg, Germany) followed by detection with biotinylated goat anti-rat Ab (Vector Laboratories, Burlingame, CA) and stained with Streptavidin/HRP (Lab Vision, Thermo Fisher Scientific, Fremont, CA)) followed by VECTOR NovaRED Peroxidase (HRP) Substrate (Vector Laboratories), according to the manufacturer's protocol. Then, sections were incubated for 30 min with the second primary Ab (rabbit anti-CD3, Dako) followed by detection with alkaline phosphatase (AP)-linked polymer (Leica Biosystems) and Ferangi Blue Chromogen (Biocare Medical, Concord, CA), according to the manufacturer's protocol.
Hematic microvessels were identified as small tubes or circles marked by anti-CD31 mAb. Counts of microvessels and apoptotic cells were performed at X400 in a 0.180 mm2 field. At least 3 samples (1 sample/tumor growth area) and 6–8 (depending on tumor width) randomly chosen fields/sample were evaluated. Results are expressed as mean ± SD of CD31 positive microvessels per field, or as mean ± SD percentage of TUNEL positive cells/number of total cells evaluated on formalin-fixed sections by immunohistochemistry or TUNEL assay, respectively.
Terminal deoxynucleotidyl transferase–mediated dutp nick end labeling (TUNEL) assay
DNA fragmentation associated with apoptosis was detected in 4 μm sections by TUNEL staining with the ApopTag plus Peroxidase in Situ Apoptosis kit (Millipore, Billerica, MA) according to the manufacturer's protocol.
Statistical analyses
In vitro and in vivo studies. Survival curves were constructed using the Kaplan-Meier method and compared by the log-rank test. In vitro data were from three independent experiments, results were expressed as mean ±SD. Graphs and statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Immunohistochemical studies. Differences among tumors from BEV, GD2-CAR T, GD2-CAR T + BEV and PBS-treated mice were assessed by one-way analysis of variance (ANOVA) test. The difference between each pair of means was evaluated using the Tukey pairwise multiple comparisons test. All statistical tests were evaluated using an α level of 0.05. Statistical analysis was carried out with SPSS software, version 11.0 (SPSS Inc, Chicago, IL).
Financial support
This work was supported by grants from Fondazione Italiana per la Lotta al Neuroblastoma, Associazione Italiana per la Ricerca sul Cancro (A.I.R.C.) (IG 17273 to V. Pistoia) and Italian Ministry of Health (RF-2013-02357552 to E. Di Carlo). Dr. Fabio Morandi was a recipient of a fellowship from Fondazione Italiana per la Lotta al Neuroblastoma and of Excellence in Research fellowship from Istituto G. Gaslini, Genova, Italy. Dr. Fabio Morandi is now a recipient of a fellowship from Fondazione Umberto Veronesi.
supp_data.zip
Download Zip (2.8 MB)Acknowledgments
We thank Fondazione Italiana per la Lotta al Neuroblastoma for having supported this study. We are grateful to Dr. Fabio Pastorino (Laboratory of Oncology, Istituto G.Gaslini) for his helpful suggestions about in vivo studies and to Mrs.Camilla Valentino (Laboratory of Oncology, Istituto G.Gaslini) for secretarial assistance.
Disclosure statement
The authors do not have any conflict of interest to disclose.
References
- Cheung NKV, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi:10.1038/nrc3526. PMID:23702928
- Maris JM. Medical Progress: Recent Advances in Neuroblastoma. New Engl J Med. 2010;362:2202–11. doi:10.1056/NEJMra0804577. PMID:20558371
- Seeger RC. Immunology and immunotherapy of neuroblastoma. Semin Cancer Biol. 2011;21:229–37. doi:10.1016/j.semcancer.2011.09.012. PMID:21971567
- Turtle CJ, Hudecek M, Jensen MC, Riddell SR. Engineered T cells for anti-cancer therapy. Curr Opin Immunol. 2012;24:633–9. doi:10.1016/j.coi.2012.06.004. PMID:22818942
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. New Engl J Med. 2010;363:1324–34. doi:10.1056/NEJMoa0911123. PMID:20879881
- De Angelis B, Caruana I, Orlando D, Boffa I, Guercio M, Sinibaldi M, Polito V, Pagliara D, Moseley A, Brenner MK, et al. Generation of T lymphocytes genetically modified to express third generation GD2-specific chimeric antigen receptor (CAR) with CD28/4.1BB costimulation to improve anti-tumor efficacy of adoptive T cell therapy for patients with neuroblastoma (NB). Bone Marrow Transpl. 2016;51:S58–S9.
- Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, et al. CAR T cells administered in combination with Lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017;25(9):2214–2224. doi:10.1016/j.ymthe.2017.05.012. PMID:28602436
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu EL, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6. doi:10.1182/blood-2011-05-354449. PMID:21984804
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu EL, Gee AP, Mei Z, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. doi:10.1038/nm.1882. PMID:18978797
- Beatty GL, O'Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: defining the challenges and next steps. Pharmacol Therapeut. 2016;166:30–9. doi:10.1016/j.pharmthera.2016.06.010.
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang XY, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-Targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra3. doi:10.1126/scitranslmed.3005930.
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. New Engl J Med. 2013;368:1509–18. doi:10.1056/NEJMoa1215134. PMID:23527958
- Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi:10.1146/annurev.med.57.121304.131306. PMID:16409133
- Meister B, Grunebach F, Bautz F, Brugger W, Fink FM, Kanz L, Mohle R. Expression of vascular endothelial growth factor (VEGF) and its receptors in human neuroblastoma. Eur J Cancer. 1999;35:445–9. doi:10.1016/S0959-8049(98)00387-6. PMID:10448297
- Meitar D, Crawford SE, Rademaker AW, Cohn SL. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J Clin Oncol. 1996;14:405–14. doi:10.1200/JCO.1996.14.2.405. PMID:8636750
- Ribatti D, Raffaghello L, Pastorino F, Nico B, Brignole C, Vacca A, Ponzoni M. In vivo angiogenic activity of neuroblastoma correlates with MYCN oncogene overexpression. Int J Cancer. 2002;102:351–4. doi:10.1002/ijc.10742. PMID:12402304
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi:10.1038/nature10144. PMID:21593862
- Potente M, Gerhardt H, Carmeliet P. Basic and Therapeutic Aspects of Angiogenesis. Cell. 2011;146:873–87. doi:10.1016/j.cell.2011.08.039. PMID:21925313
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–74. doi:10.1038/nature04483. PMID:16355214
- Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumor-growth Invivo. Nature. 1993;362:841–4. doi:10.1038/362841a0. PMID:7683111
- Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–91. doi:10.1038/nrc2403. PMID:18596824
- Ferrara N. VEGF as a therapeutic target in cancer. Oncology-Basel. 2005;69:11–6. doi:10.1159/000088479.
- Jayson GC, Hicklin DJ, Ellis LM. Antiangiogenic therapy-evolving view based on clinical trial results. Nat Rev Clin Oncol. 2012;9:297–303. doi:10.1038/nrclinonc.2012.8. PMID:22330688
- Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov. 2016;15:385–403. doi:10.1038/nrd.2015.17. PMID:26775688
- Roy Choudhury S, Karmakar S, Banik NL, Ray SK. Targeting angiogenesis for controlling neuroblastoma. J Oncol. 2012;2012:782020. doi:10.1155/2012/782020. PMID:21876694
- Segerstrom L, Fuchs D, Backman U, Holmquist K, Christofferson R, Azarbayjani F. The anti-VEGF antibody bevacizumab potently reduces the growth rate of high-risk neuroblastoma xenografts. Pediatr Res. 2006;60:576–81. doi:10.1203/01.pdr.0000242494.94000.52. PMID:16988184
- Shusterman S, Maris JM. Prospects for therapeutic inhibition of neuroblastoma angiogenesis. Cancer Lett. 2005;228:171–9. doi:10.1016/j.canlet.2005.01.049. PMID:15927358
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–27. doi:10.1038/nrd3455. PMID:21629292
- Dickson PV, Hamner JB, Sims TL, Fraga CH, Ng CY, Rajasekeran S, Hagedorn NL, McCarville MB, Stewart CF, Davidoff AM. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13:3942–50. doi:10.1158/1078-0432.CCR-07-0278. PMID:17606728
- Jain RK. Antiangiogenesis Strategies Revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–22. doi:10.1016/j.ccell.2014.10.006. PMID:25517747
- Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, Hernandez G, Mier J, He X, Hodi FS, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624. doi:10.1038/ncomms12624. PMID:27571927
- Shrimali RK, Yu ZY, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–80. doi:10.1158/0008-5472.CAN-10-0153. PMID:20631075
- Huang YH, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73:2943–8. doi:10.1158/0008-5472.CAN-12-4354. PMID:23440426
- Huang YH, Yuan JP, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. P Natl Acad Sci USA. 2012;109:17561–6. doi:10.1073/pnas.1215397109.
- Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, Lewis ID, Brenner MK, Brown MP. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 Blockade. Mol Ther. 2016;24:1135–49. doi:10.1038/mt.2016.63. PMID:27019998
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–90. doi:10.1038/nm.3838. PMID:25939063
- Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015;33:1688–96. doi:10.1200/JCO.2014.58.0225. PMID:25800760
- Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–U158. doi:10.1038/nm.3833. PMID:25849134
- Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother. 2010;33:780–8. doi:10.1097/CJI.0b013e3181ee6675. PMID:20842059
- Moon EK, Carpenito C, Sun J, Wang LCS, Kapoor V, Predina J, Powell DJ, Riley JL, June CH, Albelda SM. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T cells Expressing a Mesothelin-Specific Chimeric Antibody Receptor. Clin Cancer Res. 2011;17:4719–30. doi:10.1158/1078-0432.CCR-11-0351. PMID:21610146
- Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015;4:e988098. doi:10.4161/21505594.2014.988098. PMID:25949885
- Yu LL, Wu XM, Cheng ZY, Lee CV, LeCouter J, Campa C, Fuh G, Lowman H, Ferrara N. Interaction between bevacizumab and murine VEGF-A: A reassessment. Invest Ophth Vis Sci. 2008;49:522–7. doi:10.1167/iovs.07-1175.
- Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited: the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. 2011;129:2935–44. doi:10.1002/ijc.25960. PMID:22030616
- Bernassola F, Scheuerpflug C, Herr I, Krammer PH, Debatin KM, Melino G. Induction of apoptosis by IFN gamma in human neuroblastoma cell lines through the CD95/CD95 L autocrine circuit. Cell Death Differ. 1999;6:652–60. doi:10.1038/sj.cdd.4400537. PMID:10453076
- Fulda S, Debatin KM. IFN gamma sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene. 2002;21:2295–308. doi:10.1038/sj.onc.1205255. PMID:11948413
- Pistoia V, Bianchi G, Borgonovo G, Raffaghello L. Cytokines in neuroblastoma: from pathogenesis to treatment. Immunotherapy-Uk. 2011;3:895–907. doi:10.2217/imt.11.80.
- Di Carlo E, Bocca P, Emionite L, Cilli M, Cipollone G, Morandi F, Raffaghello L, Pistoia V, Prigione I. Mechanisms of the Antitumor Activity of Human V gamma 9 V delta 2 T cells in combination with zoledronic acid in a preclinical model of neuroblastoma. Mol Ther. 2013;21:1034–43. doi:10.1038/mt.2013.38. PMID:23481325
- Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, Terme M. Control of the immune response by pro-angiogenic factors. Front Oncol. 2014;4:70. doi:10.3389/fonc.2014.00070. PMID:24765614
- Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology. 2012;1:1223–5. doi:10.4161/onci.21335. PMID:23243584
- Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7 H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8(+) T cells. Cancer Res. 2004;64:1140–5. doi:10.1158/0008-5472.CAN-03-3259. PMID:14871849
- Dondero A, Pastorino F, Della Chiesa M, Corrias MV, Morandi F, Pistoia V, Olive D, Bellora F, Locatelli F, Castellano A, et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology. 2016;5:e1064578. doi:10.1080/2162402X.2015.1064578. PMID:26942080
- Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–44. doi:10.1172/JCI83092. PMID:27454297
- Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, Brenner M, Suzuki M. Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77:2040–51. doi:10.1158/0008-5472.CAN-16-1577. PMID:28235763
- Modak S, Kushner BH, Basu E, Roberts SS, Cheung NV. Combination of bevacizumab, irinotecan, and temozolomide for refractory or relapsed neuroblastoma: Results of a phase II study. Pediatr Blood Cancer. 2017;64. doi:10.1002/pbc.26448.
- Yvon E, Del Vecchio M, Savoldo B, Hoyos V, Dutour A, Anichini A, Dotti G, Brenner MK. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin Cancer Res. 2009;15:5852–60. doi:10.1158/1078-0432.CCR-08-3163. PMID:19737958
- Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–70. doi:10.1038/leu.2010.75. PMID:20428207