
ABSTRACT
Cancers frequently evade immune-recognition by lowering peptide:MHC-I complexes on their cell surface. Limited peptide supply due to TAP-deficiency results in such MHC-Ilow immune-escape variants. Previously, we reported on a category of TAP-independent self-peptides, called TEIPP, with selective presentation by these tumors. Here we demonstrate that in contrast to T cells specific for conventional tumor antigens, TEIPP-directed T cells remain naïve in mice bearing immune-escaped tumors. This unaffected state was caused by low levels of MHC-I on the tumors and the failure to cross-present low levels of antigenic protein by host APCs. Importantly, increased levels of MHC-I, antigen or co-stimulation resulted in potent activation of TEIPP-specific T cells via direct presentation. Genetic knockdown by CRISPR/Cas9 technology of the relevant MHC-I allele in tumor cells indeed abrogated T cell activation. Vaccine-mediated priming of TEIPP-specific T cells induced efficient homing to MHC-Ilow tumors and subsequently protected mice against outgrowth of their MHC-Ilow tumor. Thus, our data open up the search of TEIPP-specific T cells in cancer patients to explore their application against MHC-Ilow tumor cells.
Introduction
Cancer immunotherapy has reached major successes in recent years with the introduction of several new treatment options in the clinic. Especially immune checkpoint therapy with blocking antibodies to PD-1 and PD-L1, which prevent signalling of inhibiting co-receptors on T cells, are FDA- and EMA-approved in an increasing number of oncologic indications.Citation1,2 However, the majority of patients still succumb to their disease or relapse after initial successful treatment, highlighting the importance of further delineating critical factors of success. It has become clear that tumors can evade immune recognition by downregulation of MHC-I levels and thereby resist T cell immunity and checkpoint therapy. Two recent studies described that tumors from patients relapsing or not responding to checkpoint inhibitor therapy had mutations in genes encoding the IFNγ pathway, including JAK/STAT signalling.Citation3,4 This failure of tumor cells to respond to IFNγ affects levels of MHC-I as cells are impaired to induce expression of the peptide transporter associated with antigen presentation (TAP).Citation5 Also mutations or epigenetic silencing of components of the antigen-processing machinery, such as TAP lead to strong reduction of MHC-I on cancer cells.Citation6,7 All these alterations result in a general deficiency to present conventional tumor antigens to CD8+ T cells.
We study an alternative CD8+ T cell repertoire that specifically recognize peptides on cells deficient for the peptide transporter TAP. Due to this TAP deficiency, cells express strongly reduced levels on MHC-I, but in conjunction an alternative peptide repertoire is presented on residual MHC-I molecules, called TEIPP: T cell epitopes associated with impaired peptide processing.Citation8,9 TEIPP-specific T cells are therefore a potential candidate to treat immune-escaped, MHC-Ilow tumors. Of note, as TEIPP antigens are presented in MHC-I molecules, tumor cells with a complete loss of MHC-I molecules due to a mutation in β2 m or the heavy chain, will not present TEIPP antigens and are therefore not targetable by TEIPP-specific T cells.
The first identified mouse TEIPP was a C-terminal peptide of Trh4, a ceramide synthase spanning the ER membrane.Citation8,10 The protein is ubiquitously expressed in all somatic cells, but its peptide epitope is surprisingly only presented on TAP-deficient cells.Citation9 Antigen processing and presentation of the epitope is independent of the proteolytic enzyme complex proteasome and the TAP transporter. Instead, release of the epitope depended on intramembrane cleavage by signal peptide peptidase (SPP).Citation10 Using a T cell receptor-transgenic (TCR tg) mouse based on a Trh4-specific CD8+ T cell clone, we previously demonstrated that these TCR tg T cells (‘LnB5 tg’) undergo normal, efficient thymic selection and are not hampered by central or peripheral tolerance,Citation11 most likely since the Trh4 self-peptide is only MHC-I presented in TAP-deficient cells. Upon transfer of naïve LnB5T cells in wildtype, tumor-free B6 mice, cells remain naïve as expected. In contrast, transfer of LnB5 T cells to TAP-deficient mice resulted in vigorous proliferation and strong activation, especially under inflammatory conditions.Citation11
In the present study, we aimed to understand the behaviour of naïve TEIPP T cells in mice bearing a MHC-Ilow tumor and, secondly, the molecular requirements for their optimal priming. In contrast to T cells against conventional tumor antigens, we found that naïve TEIPP-specific T cells were not activated by resident tumors and hardly infiltrated MHC-Ilow tumors, despite the fact that the antigen was presented there. Sufficient TEIPP T cell activation was only reached by tumor cells with high levels of MHC-I as well as cognate antigenic protein. Strikingly, this manipulated activation was operated via direct priming and not via cross-priming of TEIPP antigens and resulted in strong influx in wild type MHC-Ilow tumors. Importantly, it prevented outgrowth of such immune escape tumors. These results imply that the TEIPP-specific CD8+ T cell subset remains naïve even in tumor-bearing mice harboring MHC-Ilow tumors and indicate that vaccination strategies may optimally exploit these immune cells for immunotherapy.
Results
Activation of TEIPP T cells requires high antigen and MHC-I levels on tumor cells
Previously, we have shown using a TCR transgenic mouse model that TEIPP T cells efficiently egress from the thymus and arrive in the periphery with an antigen-unexperienced phenotype.Citation11 To study the potential of MHC-Ilow tumor cells to activate naive TEIPP T cells in vivo, we applied a model in which congenic naïve TCR-transgenic TEIPP T cells (‘LnB5 tg’) were transferred to recipient mice that were subsequently injected with irradiated MHC-Ilow RMA-S cells, which display the cognate Trh4/Db complex. Previously, we and others showed that RMA-S tumor injection failed to induce TEIPP T cell immunity.Citation12,13 Therefore, we overexpressed the Trh4 antigen and/or the co-stimulatory molecule B7.1 (CD80) in RMA-S cells and analysed T cell activation (). Neither enhanced levels of Trh4, nor B7.1 or the combination of these two resulted in strong expansion of LnB5tg T cells, albeit that CD62 L downregulation and IFNγ production was detected in some animals as signs of activation.
Figure 1. High antigen and MHC-I levels are required for TEIPP T cell priming. CFSE labelled TCR-transgenic TEIPP T cells were transferred to recipient mice and T cell activation and proliferation were measured after challenge with indicated irradiated tumor cells. (A) Blood samples were analysed for the presence and activation status of T cells by flow cytometry five days after the second injection of MHC-Ilow RMA-S cell lines. ‘Trh4’ indicates cells transfected with full length cDNA of the cognate antigen and ‘B7’ indicated RMA-S cells transfected with the mouse CD80 gene. IFNγ production by TEIPP T cells was measured in blood after the second injection, upon overnight stimulation with short Trh4 peptide. (B) Blood samples from mice challenged with MHC-Ihigh RMA cell lines. Means and SEM are plotted from one (RMA-S.B7, RMA-S.Trh4.B7), two (RMA and RMA-S.Trh4) or five experiments (RMA.Trh4). Student T-test compared to T cells only: *P < 0.05, **P < 0.01, ***P < 0.001.
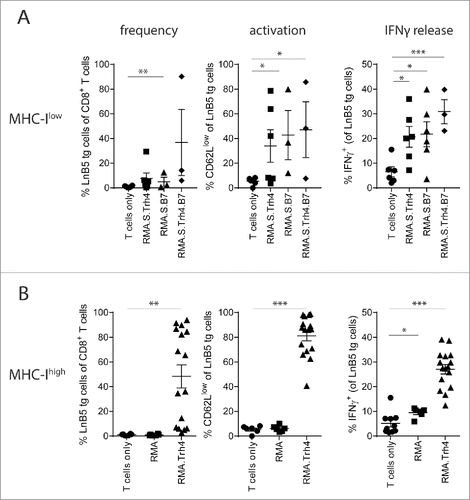
We recently showed that dendritic cells pulsed with long peptides comprising the Trh4 TEIPP epitope induced potent T cell activation, suggesting that high levels of antigen and MHC-I are necessary for efficient priming.Citation11 The fact that RMA-S cells were generally poor in activating TEIPP T cells in vivo () could be related to the low MHC-I levels, leading to poor TCR:MHC-I interactions crucial for proper T cell activation. We therefore made advantage of the TAP-proficient RMA.Trh4 cells, in which the Trh4 antigen was overexpressed to similar levels as in RMA-S.Trh4, but clearly expressed higher total levels of MHC-I (Supplementary Figure S1). Notably, wild type RMA cells fail to present Trh4 peptides due to competition with the TAP-mediated repertoire, but we have shown that overexpression of the Trh4 antigen overcomes this TAP barrier and leads to efficient presentation of the Trh4 epitope in MHC-I at the cell surface.Citation9 Indeed, parental RMA cells failed to prime TEIPP T cells (). Strikingly, RMA.Trh4 cells induced a strong expansion of TEIPP T cells, comprising in half of the mice more than 60% of the peripheral CD8+ T cell population (). On average, 80% of the LnB5 T cells displayed an activated CD62Llow phenotype. In addition, an increase in the percentage of IFNγ-producing cells was observed after a brief in vitro stimulation with Trh4 peptide (). The more homogeneous activation of TEIPP T cells by RMA.Trh4 was in sharp contrast to the very heterogeneous activation found with RMA-S.Trh4 and highlights the importance of high general level of MHC-I, since overexpression of Trh4 was comparable in both cell lines (Supplementary Figure S1). So, under normal conditions TEIPP antigens only emerge on the surface of TAP-deficient cells, but overexpression of the antigen can also lead to TEIPP presentation in TAP-proficient cells. Together, our data show that high MHC-I antigen presentation and strong expression of the TEIPP antigen are important for the in vivo activation of TEIPP T cells.
TEIPP T cell activation is mediated by direct priming on tumor cells
The fact that RMA.Trh4 cells induced a surprisingly strong TEIPP T cell activation in vivo prompted us to study how this priming of naïve TEIPP-specific T cells took place. Either via direct interaction with the RMA.Trh4 cells or indirectly via cross-priming a process by which professional antigen-presenting host cells ingest, process and present Trh4 antigen to T cells.Citation14,15 To test the capacity of cross-priming, we overexpressed Trh4 in allogeneic P815 cells (Supplementary Figure. S2A), a mastocytoma cell line from a DBA/2 mouse on H-2d background, lacking the Db-restricting element for direct presentation to TEIPP T cells. Injection of P815 or P815.Trh4 cells did not elicit accumulation of TEIPP T cells in the blood of mice (). Some T cell activation was measured in both groups compared to mice that only received T cells, however, these T cells failed to produce IFNγ after a brief in vitro stimulation with peptide (). In contrast, a strong response to MHC-I allo-antigens was detected in these same mice by the endogenous T cell repertoire (Supplementary Figure S2B). So in this setting, injection of allogeneic P815.Trh4 cells did not lead to cross-priming of TEIPP T cells whereas these cells were immunogenic enough to trigger alloreactivity.
Figure 2. TEIPP T cell activation is mediated by direct priming on tumor cells. Mice received naïve LnB5 tg T cells and were injected with irradiated tumor cells. (A) Analysis of phenotype of T cells in blood of mice injected with allogeneic P815 or P815.Trh4 cells, five days after the second injection. IFNγ production by TEIPP T cells was measured by overnight stimulation with short Trh4 peptide. Data pooled from two independent experiments, with 4 mice per group, shown as mean and SEM. (B) Expression of H2-Db and H2-Kb molecules on RMA.Trh4 cells generated by Crispr/CAS9 technology: wildtype (wt), Db-/− or Kb-/− cells. Plots representative for at least two experiments. (C) IFNγ release by the LnB5 T cell clone upon in vitro co-culture with the decreasing amounts of cells from the RMA.Trh4 cell panel. Data shown as mean and SD, from one of two experiments with comparable results. (D) Naïve LnB5 tg T cells were transferred to recipient mice that were then injected twice with irradiated RMA.Trh4, RMA.Trh4 Db-/− or RMA.Trh4 Kb-/− cells. LnB5 T cell activation was measured in blood after the second injection. IFNγ production by TEIPP T cells in blood, upon overnight stimulation with short Trh4 peptide. Data pooled from two independent experiments, with 4 mice per group, shown as mean and SEM. Student T-test: n.s. = not significant, **P < 0.01, ***P < 0.001.
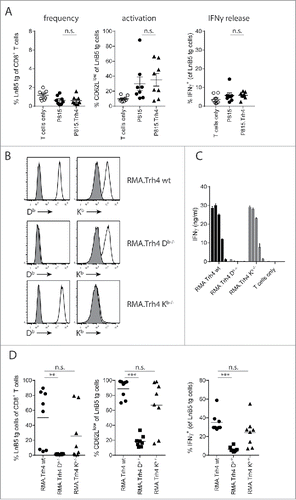
Next, we examined direct priming by tumor cells using the CRISPR/CAS9 technology to knock-out the H2-Db gene in RMA.Trh4 cells. As control, we knocked-out the irrelevant H2-Kb gene. Gene knock-out phenotypes were verified at the protein level by flow cytometry and cells were sorted twice to obtain pure populations (). Indeed, RMA.Trh4.Db-/− cells failed to present the Trh4 epitope to a Trh4-specific T cell clone in vitro, whereas strong T cell recognition was observed when wildtype RMA.Trh4 cells and RMA.Trh4 Kb-/− cells were tested (). Importantly, when irradiated RMA.Trh4 Db-/− or -Kb-/− cells were injected in mice to study the effect on priming of naïve TEIPP T cells in vivo, the lack of Db molecules caused a complete loss of activation capacity, while removal of the Kb molecule did not result in decreased priming efficiency (). Ex vivo analysis of IFNγ release by in vivo activated TEIPP T cells corroborated these results (). Of note, RMA.Trh4.Db-/− and RMA.Trh4.Kb-/− cells overexpressed the Trh4 transcript to comparable degree (Supplementary Figure S2C). Moreover, the applied in vivo model required two injections with tumor cells for clear results, but heterologous prime-boost schedules with the RMA.Trh4 panel demonstrated that the first injection was responsible for the priming event of TEIPP T cells (Supplementary Figure S2D). Together, these data show a critical role for direct priming by tumor cells of TEIPP T cells and elucidate why MHC-Ilow RMA-S tumor cells fail to activate this T cell specificity, leaving this subset ‘untouched’.
TEIPP-T cells are not activated in tumor-bearing mice
Since the thus far applied model with irradiated tumor cells does not precisely reflect the situation of tumor-bearing mice, we subcutaneously inoculated mice with progressively growing RMA-S tumors, with or without overexpression of the Trh4 antigen or the co-stimulatory molecule B7.1, and transferred naïve, congenic and CFSE-labelled LnB5 T cells at the time tumors were palpable (). Seven days after T cell transfer, tumor-draining lymph nodes (dLN), non-draining lymph nodes (ndLN) and tumors were removed, dispersed and analysed for the presence and activation status of the transferred TEIPP T cells. Although TEIPP T cells were still detectable in dLN and ndLN and comprised 2–4% of the CD8+ T cell population, hardly any cells infiltrated the RMA-S tumors (). Moreover, we did not observe any division of the T cells nor loss of the CD62 L marker, in these mice. Since RMA-S cells are optimal targets for in vitro pre-activated LnB5 tg T cells,Citation11 the lack of TEIPP T cell activation in RMA-S-tumor bearing mice underlined our earlier conclusion on the failure of T cell priming by this MHC-Ilow tumor, despite presentation of the cognate peptide-epitope.
Figure 3. Tumor infiltration by tumor-specific TCR-transgenic T cells. LnB5 transgenic T cells (CD90.1+) were labelled with CFSE and transferred to (CD90.1−) mice bearing palpable RMA-S (A), RMA-S.Trh4 (D) or RMA.S.B7 (E) tumors. (B) Melanoma-specific pmel transgenic T cells (CD90.1+) were transferred to B16F10-tumor bearing mice. (C) OVA-specific OT-I transgenic T cells (CD90.1+) were transferred to B16F10.OVA-tumor bearing mice. After seven days, frequency and phenotype of transferred T cells was analysed in tumor, tumor-draining lymphnode (dLN) and non-draining lymphnodes (ndLN). Representative dot plots are shown, gated on live cells (upper row), or gated on CD90.1+ T cells (lower row). Data is pooled from 2 or 3 independent experiments with 3 mice per group. Student T-test: *P<0.05, **P < 0.01, ***P < 0.001, n.s.: non-significant.
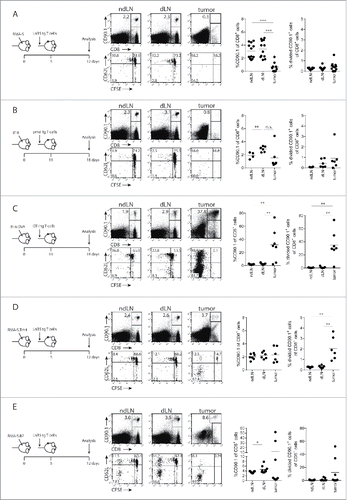
To compare these data on LnB5tg T cells to other TCR tg CD8+ T cells in tumor models, we examined priming efficiency of pmel-1 T cells (specific for the gp100 melanocyte differentiation self-antigen) and OT-I T cells (specific for the OVA foreign antigen).Citation16,17 Pmel-1 T cells were transferred in mice harbouring B16F10 melanomas and OT-I T cells were tested in B16F10 tumors with transgenic ovalbumin. In tumor-draining LN, a significantly increased frequency of transgenic pmel-1 T cells was observed compared with contralateral non-draining LN, coinciding with a modest increase of divided T cells, although tumor infiltration was modest (). This is in line with recent data demonstrating cross-presentation of tumor antigens in the draining LN by host dendritic cells.Citation18 In contrast, transfer of naïve OT-I cells in B16F10.OVA tumor-bearing mice resulted in strikingly high numbers of activated OT-I cells in the tumors, comprising up to 70% TCR transgenic OT-I cells (). Apparently, the pmel and OT-I transgenic T cells were primed in the presence of a tumor, whereas the RMA-S tumor did not induce priming of naïve TEIPP T cells.
We then tested mice with RMA-S tumors that overexpressed Trh4 or B7.1. Overexpression of Trh4 in RMA-S did not enhance the frequencies or the percentage of dividing lymph node resident TEIPP T cells when compared to RMA-S tumor-bearing mice (). However, the frequency of intratumoral TEIPP T cells was slightly increased when compared to RMA-S tumors. Additionally, the majority of tumor-infiltrating TEIPP T cells had proliferated and displayed an CD62Llow activated phenotype (). Thus, overexpression of the Trh4 antigen mildly improved the activation and number of TEIPP T cells in tumors. Of note, MHC-I surface levels of RMA-S and RMA-S.Trh4 tumor cells were comparably low, suggesting that higher Trh4 expression can result in modestly improved priming (Supplementary Figure S1A and 1B).
Transfer of naïve TEIPP T cells in RMA-S.B7 tumor-bearing mice resulted in a slightly increased frequency of TEIPP T cells in the dLN when compared to ndLN, albeit that the percentage of dividing cells was low in both cases (). Tumor-infiltration of RMA-S.B7 was heterogeneous with only half of the tumors displaying high numbers of dividing TEIPP T cells ().
We concluded that TEIPP-specific T cells are not primed by MHC-Ilow RMA-S tumors and therefore fail to infiltrate these lesions. Consequently, the TEIPP T cell repertoire remains ‘untouched’ in tumor-bearing mice and might be optimally primed by immunotherapeutic strategies.
Successful MHC-Ilow tumor-infiltration and prevention of outgrowth by RMA.Trh4-induced priming
To study if activated TEIPP T cells could migrate to MHC-Ilow tumors, naive mice received LnB5tg T cells and were injected twice with irradiated RMA.Trh4 to allow T cells to become activated. Mice were inoculated with RMA-S cells after the first injection of RMA.Trh4 cells (). Activated TEIPP T cells strongly infiltrated MHC-Ilow RMA-S tumors, in that more than 50% of the intratumoral CD8+ T cell population represented LnB5tg cells in the majority of mice (). This was in contrast to the very few tumor-infiltrating TEIPP T cells in mice receiving T cells only. Of the infiltrating TEIPP T cells in the RMA.Trh4-injected mice, all had an activated phenotype as measured by CD62 L downregulation (). These results show that TEIPP T cells are capable to infiltrate MHC-Ilow tumors once they are properly activated.
Figure 4. TEIPP T cell activation promotes tumor infiltration and protection against tumor outgrowth. (A and B) Naïve mice received LnB5 transgenic T cells and injection with irradiated RMA.Trh4 cells, and were inoculated with a subcutaneous RMA-S tumor. At day 20 after T cell injection, tumor, tumor-draining lymphnode (dLN) and non-draining lymphnodes (ndLN) were analysed. Data pooled from two independent experiments with four mice per group. Student t-test: **P < 0.01. (C) Mice received T cells and were immunized twice with irradiated RMA.Trh4 cells and one week after the second injection challenged with a RMA-S tumor. (D) Individual tumor outgrowth curves and (E) Kaplan-Meier survival plot. Pooled means and SEM from three independent experiments are shown. Log-rank test: *P < 0.05, ***P < 0.001.
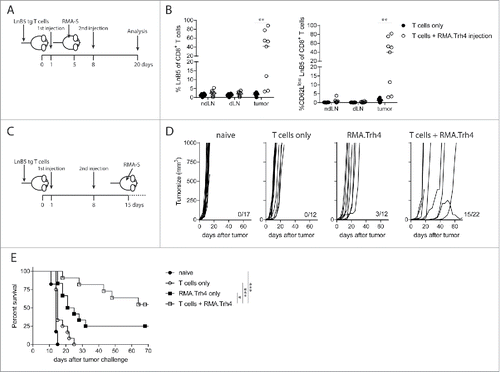
Finally, we examined the efficacy of these activated TEIPP T cells to control outgrowth of tumors. Therapeutic setup of this experiment was not successful, since RMA-S tumor growth was too fast to allow for full activation of TEIPP T cells using a prime-boost scheme (Supplementary Figure S3). Therefore, a prophylactic setting was chosen in which mice with activated TEIPP T cells were challenged with RMA-S tumors. The combination of T cell transfer and in vivo activation by RMA.Trh4 injections resulted in a strong prevention of tumor outgrowth ( and ). More than seventy percent of the challenged mice were still alive at day 65 after tumor challenge, whereas naïve mice or mice only receiving T cells or only RMA.Trh4 injections succumbed to tumor outgrowth ().
Overall, this study showed that TEIPP-specific T cells remain naïve in the presence of MHC-Ilow tumors, but can efficiently be activated by cells expressing high levels of the Db/Trh4 complex. Once activated, TEIPP T cells strongly infiltrate MHC-Ilow tumors and control further outgrowth of the malignant lesions. Our data highlight the potential of TEIPP antigens and TEIPP-specific T cells to target immune-escaped tumors.
Discussion
There is an urgent need to target tumors with low MHC-I expression which are not responsive to conventional T-cell based immunotherapies. Natural killer cells are well known to target MHC-Ilow cells,Citation19 and NK cell transfer in cancer patients has proven to be feasible and show promising results.Citation20,21 but have had little success in clinical trials yet. Here, we show that CD8+ TEIPP T cells, specific for TAP-independently processed, non-mutated self-antigens, can be effectively exploited for the treatment of these aggressive tumors. Naïve TEIPP T cells remain ‘untouched’ in tumor-bearing mice, and as a consequence do not infiltrate these tumors. Potent activation of TEIPP T cells resulted in a strong influx in these non-immunogenic tumors and, consequently, efficiently protected mice against a tumor outgrowth. Importantly, since TEIPP T cells only recognize TAP-deficient cells and remain naïve in wildtype mice, there is no risk for autoimmunity.
TEIPP antigens are unusual in their intracellular processing mechanism as they are MHC-I presented independent of the peptide transporter TAP and have to compete with TAP-mediated peptides for their loading on MHC-I in the endoplasmic reticulum.Citation9 We previously described that the here studied Trh4-derived peptide-epitope is intramembraneously cleaved by the enzyme Signal Peptide Peptidase (SPP) at the C-terminus and does not require the proteasome.Citation10 These unusual features of TEIPP antigens might impact the priming of the cognate CD8+ T cell repertoire in tumor-bearing mice.
Using an artificial model in which irradiated tumor cells were used to study the requirements for TEIPP T cell activation, we showed that high levels of both MHC-I and Trh4 by tumor cells were needed to induce potent T cell priming. Interestingly, this activation did not depend on the most common pathway of cross-priming via host dendritic cells but in fact required direct priming by tumor cells engineered to present high MHC-I and antigen levels ().
Cross-priming has been described by many studies to induce an anti-tumor T cell response, in which tumor-antigens are taken up by dendritic cells (DCs) and ‘crossed’ in the endogenous MHC-I pathway to be presented in the context of MHC-I to CD8+ T cells.Citation14,15,22 Indeed, the importance of cross-priming has been described in several tumor models, including a recent study showing that CD103+ DCs in lymphnodes of mice bearing a TAP-deficient melanoma, overexpressing OVA (B78.OVA), could induce proliferation of both pmel and OT-I transgenic T cells.Citation18,23,24 The lack of cross-priming as a pathway for CD8+ T cell induction in the TEIPP model could be related to the nature of the peptide. An elegant study demonstrated that signal peptides, which are small peptides liberated by the SPP enzyme, are less efficiently presented through cross-presentation by host APC, whereas efficient priming is induced through direct presentation.Citation25 Such small peptide intermediates might not be suitable to picked up by dendritic cells. Notably, the effective TEIPP T cell priming could also be mediated via a process called cross-dressing, in which peptide:MHC complexes are transferred from the surface of tumor cells to professional APCs in lymphnodes, thereby inducing T cell activation.Citation26,27 Interestingly, we showed before that TEIPP T cells can be efficiently activated in vivo upon vaccination with a long synthetic peptide containing the Trh4 epitope, most likely via cross-priming by host DCs, suggesting that large quantities of the peptide-epitope are able to reach host DC in the animals and be loaded in the MHC-I processing pathway.Citation11
One of the important implications of the poor priming capacity of MHC-Ilow tumors, due to their low general levels of MHC-I and lack of co-stimulatory ligands, is the naïve status of the TEIPP T cell repertoire even in tumor-bearing mice. Obviously, the T cells fail to home and infiltrate tumors and therefore not experience tumor-induced tolerance or exhaustion.Citation28,29 As infiltration of T cells in the tumor is clearly one of the requirements for a good protective anti-tumor response,Citation30 TEIPP T cells need to be primed and activated for optimal exploitation in immunotherapeutic strategies. The expectation is that simply blocking inhibitory receptors will not suffice for this T cell subset. Also the blocking TAP function in DCs or tumor cells by for example an oncolytic virus, might not induce a potent TEIPP T cell response due to lack of high antigen:MHC-I complexes. As mentioned, vaccination with long synthetic peptides is effective in inducing potent TEIPP T cell priming, and is therefore a suitable way to prime TEIPP T cells and recruit them for immunotherapy. Moreover engineered RMA.Trh4 cells are also potent inducers of TEIPP-specific T responses, resulting in a subsequent influx in MHC-Ilow tumors, which are hardly immunogenic. The surprising data in our study that MHC-Ihigh RMA.Trh4 tumor cells could efficiently prime TEIPP T cells, whereas RMA-S.Trh4 cells failed, even though Trh4 peptide:MHC-I surface levels were similar between these two cell lines,Citation9 might be explained by essential non-cognate peptide/MHC interactions with the TCR.Citation31 Once TEIPP T cells were efficiently activated by synthetic long peptide or engineered tumor cells, mice were capable to control MHC-Ilow tumors, the majority of which remaining tumor-free for more than two months (). However, peptide vaccination represents a much better controllable platform compared to engineered tumor cells that need to be fine-tuned and genetically expressed for each antigen.
The importance to target epitopes on MHC-Ilow tumors to counteract immune evasion was recently highlighted by a study in IFNγ-unresponsive tumors.Citation32 Here, they showed that T cells specific for an IFNγ-independently processed epitope were potent in eradicating MHC-Ilow, IFNγ-unresponsive tumors in mice, whereas T cells targeting a conventional epitope of the same antigen requiring IFNγ for its presentation failed to do so.Citation32 As TEIPP antigens are selectively presented on TAP-deficient cells and do not depend on IFNγ signalling for their presentation, TEIPPs and their cognate T cell receptors might effectively be exploited for immunotherapy of MHC-Ilow tumors which have escaped from conventional immunotherapies.
Materials and methods
Cell lines and mice
The tumor cell lines RMA, RMA-S (TAP2-deficient), RMA-S.B7.1 (RMA-S transfected with mouse CD80 gene), RMA-S.Trh4 and RMA.Trh4 cell lines have been described before.Citation8,9 B16 and B16.OVA cells were also described before.Citation33 RMA-S and RMA cells were originally derived from Klas Kärre (Karolinska Institutet, Sweden) and B16 cells were bought from ATCC (Manassas, Virginia, USA). All cells were cultured no longer than one month and regularly tested by flow cytometry for MHC class I expression. Mycoplasma testing for all cell lines was performed every 2 months by PCR. P815.Trh4 cells were generated by retroviral transduction of P815 cells with the long Trh4 transcript as previously performed.Citation9 The generation and culture of TEIPP T cell clone ‘LnB5’ specific for the Trh4 derived peptide MCLRMTAVM in the context of H2-Db (hereafter named Db) has been previously described.Citation8,10 All cells were cultured in complete IMDM medium (Invitrogen, Carlsbad, CA) containing 8% heat-inactivated FCS (Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (Invitrogen) at 37° C in humidified air with 5% CO2. C57 BL/6 mice were purchased from Charles River (L'Arbresle, France). OT-I TCR transgenic mice, transgenic for the OVA257–264/Kb-restricted T cell receptor were derived from Jackson's Laboratory (stock no. 003831). The pmel-1 TCR transgenic mice, containing gp10025–33/ Db- specific T cells, were a gift from Dr. N.P. Restifo (National Cancer Institute, Bethesda, Maryland). Generation and phenotype of the LnB5 TCR transgenic mouse model has been described before.Citation11 Mice were housed in individually ventilated cages and used at 6 to 12 weeks of age. All animal experiments were approved by the Central Committee Animal Experiments of the Netherlands (AVD116002015271).
Generation of RMA.Trh4 Db or Kb knock-out cells using CRISPR/Cas9 system
CRISPR/CAS9 sgRNA's targeting both Db and Kb were designed using online CRISPR Design software (http://crispr.mit.edu). The sgRNA sequence (5′- AGATGTACCGGGGCTCCTCG-3′) was cloned into a sgRNA expression vector (Addgene 41824) using a Gibson In-fusion kit. RMA-Trh4 cells were transfected with the vector containing the sgRNA and a plasmid containing Cas9 WT (Addgene 41815), using lipofectamine 2000. Flow cytometry analysis of cells transfected with the sgRNA/CAS9WT plasmids generated both Db and Kb deficient cell populations, in line with homology between the genes. From these transfected cells, Db or Kb –deficient cells were FACS-sorted and used for further experiments.
Tumorinoculation and adoptive T cell transfer
For tumor cell inoculation, 0.1 × 106 (B16 and B16.OVA), or 2 × 106 cells (RMA-S, RMA-S.B7 and RMA.S-Trh4) were injected in 200 μl 0.1% BSA/PBS subcutaneously. After 5 days (RMA-S, RMA-S.B7 or RMA-S.Trh4) or 11 days (B16 or B16.OVA), when a palpable tumor was present, CFSE labeled T cells were injected intravenously. For T cell transfers, lymph nodes and spleen were isolated from the TCR transgenic mice and mechanically disrupted. Cells were passed through nylon wool to enrich for T cells and 3 × 106 cells were injected in 200 µl PBS intravenously in recipient mice. For tumor homing experiments, cells were labeled with 5 μM CFSE (Invitrogen) prior to transfer. Injection of irradiated tumor cells was performed at day one and day eight after T cell transfer. These tumor cell were harvested, washed twice with PBS and irradiated at 60 Gy. Five million irradiated cells were injected i.p. per mouse. At day eight and nine after T cell transfer, mice received 600,000 IU recombinant human IL-2 (proleukin, Novartis) intraperitoneally in 100 μl PBS. To deplete NK cells, mice were given 100 μg anti-NK1.1 antibody (PK136), intraperitoneally in 200 μl PBS, every 3–4 days. Blood was taken from mice five days after the second injection and analysed for the frequency and phenotype of transgenic T cells.
Flow cytometry analyses
For flow cytometry analysis, tumor-draining lymphnode (dLN) and non-draining (mesenteric) lymphnode (ndLN) were isolated and mechanically disrupted. The tumor was cut in small pieces and treated with liberase (Roche) for 15 minutes at 37°C and then put over a cell strainer. Single cell suspensions were stained in 0.1% BSA/PBS with antibodies from Biolegend specific for CD4 (clone RM4–5), CD8 (53.6–7), CD3 (145–2C11), CD62 L (MEL–14), H2-Db (28–14–8), H2-Kb (AF6–88.5), eBioscience specific for NK1.1 (PK136) and CD90.1 (HIS51). Intracellular cytokine staining was performed using the ICS kit from BioLegend according to manufactures protocol. In short, cells were permeabilized for 20 min with the fixation buffer on ice, washed twice in 1× permeabilization/washing buffer and thereafter stained for IFNγ (XMG1.2, Biolegend). Cells were analyzed on a FACS Calibur or Fortessa (BD) and all analysis was performed using FlowJo (Treestar).
qPCR analysis
Cell pellets were washed twice with PBS and snapfrozen in liquid nitrogen. RNA was isolated using the RNAeasy kit (Qiagen), according to manufactures protocol. cDNA was synthesized using the High capacity RNA-to-cDNA kit (Applied Biosystems). qPCR analysis were performed using the SybrGreen supermix (Bio-Rad) and Ct values were normalized to the expression levels of housekeeping gene GAPDH (fw primer: 5′-GTGCTGAGTATGTCGTGGAGTCTAC-3′, rev: 5′GGCGGAGATGATGACCCTTTTGG −3′. For the Trh4 transcript, the common forward primer was used: 5′-GCAGACCCCTTACTGGAAGCTGCC-3′ and reverse: 5′- CGGTCATCCTTAGACACATGCAAAGG-3′. For the splice variant, lacking an exon and therefore not encoding for the C-terminal TEIPP epitope, the reverse primer used was 5′-CTGCGGTCATCCTTAGACACCTTTCC −3′. Data was analyzed using Bio-Rad CFX software.
In vitro stimulations
To verify the recognition of the RMA.Trh4 knock-out variants, 3000 cells of the LnB5 T cell clone were co-cultured overnight with the RMA.Trh4 knock-out variants, at different cell concentrations. The next day, supernatant was harvested and IFNγ was measured by ELISA as described before.Citation11
Statistics
Statistical analysis was done in GraphPad Prism (version 6). The specific test is indicated in the Figure legends. P values below 0.05 were considered statistically significant.
Author contributions
EMD, KAM, SHVDB, and TVH developed the concept and designed experiments. EMD, MS, KAM, BJQ conducted experiments and analyzed data. EMD, SHVDB, and TVH interpreted results. EMD and TVH wrote the manuscript.
Financial support
This work was supported by the Dutch Cancer Society (UL2010-4785 and UL 2013–6142) to T. van Hall and S.H. van der Burg.
The authors declare no potential conflicts of interest.
supp_data.zip
Download Zip (1.6 MB)References
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23-34. doi:10.1056/NEJMoa1504030
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1803-13. doi:10.1056/NEJMoa1510665
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375(9):819-29. doi:10.1056/NEJMoa1604958
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167(2):397-404 e399. doi:10.1016/j.cell.2016.08.069
- Ma W, Lehner PJ, Cresswell P, Pober JS, Johnson DR. Interferon-gamma rapidly increases peptide transporter (TAP) subunit expression and peptide transport capacity in endothelial cells. J Biol Chem. 1997;272(26):16585-90. doi:10.1074/jbc.272.26.16585
- Johnsen AK, Templeton DJ, Sy M, Harding CV. Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J Immunol. 1999;163(8):4224-31
- van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16(4):219-33. doi:10.1038/nrc.2016.16
- van Hall T, Wolpert EZ, van Veelen P, Laban S, van der Veer M, Roseboom M, Bres S, Grufman P, de Ru A, Meiring H, et al. Selective cytotoxic T-lymphocyte targeting of tumor immune escape variants. Nat Med. 2006;12(4):417-24. doi:10.1038/nm1381
- Oliveira CC, Querido B, Sluijter M, Derbinski J, van der Burg SH, van Hall T. Peptide transporter TAP mediates between competing antigen sources generating distinct surface MHC class I peptide repertoires. Eur J Immunol. 2011;41(11):3114-24. doi:10.1002/eji.201141836
- Oliveira CC, Querido B, Sluijter M, de Groot AF, van der Zee R, Rabelink MJ, Hoeben RC, Ossendorp F, van der Burg SH, van Hall T. New role of signal peptide peptidase to liberate C-terminal peptides for MHC class I presentation. J Immunol. 2013;191(8):4020-8. doi:10.4049/jimmunol.1301496
- Doorduijn EM, Sluijter M, Querido BJ, Oliveira CC, Achour A, Ossendorp F, van der Burg SH, van Hall T. TAP-independent self-peptides enhance T cell recognition of immune-escaped tumors. J Clin Invest. 2016;126(2):784-94. doi:10.1172/JCI83671
- Li XL, Sluijter M, Doorduijn EM, Kale SP, McFerrin H, Liu YY, Li Y, Mottamal M, Yao X, Du F, et al. Limited density of an antigen presented by RMA-S cells requires B7–1/CD28 signaling to enhance T-cell immunity at the effector phase. PloS one. 2014;9(11):e108192. doi:10.1371/journal.pone.0108192. PMID:25383875
- Wolpert EZ, Petersson M, Chambers BJ, Sandberg JK, Kiessling R, Ljunggren HG, Kärre K. Generation of CD8+ T cells specific for transporter associated with antigen processing deficient cells. Proc Natl Acad Sci U S A. 1997;94(21):11496-501. doi:10.1073/pnas.94.21.11496
- Bevan MJ. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J Exp Med. 1976;143(5):1283-8. doi:10.1084/jem.143.5.1283
- Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47-64. doi:10.1146/annurev.immunol.19.1.47
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569-80. doi:10.1084/jem.20030590
- Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78(2):110-17. doi:10.1046/j.1440–1711.2000.00889.x
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer cell. 2016;30(2):324-36. doi:10.1016/j.ccell.2016.06.003
- Malmberg KJ, Sohlberg E, Goodridge JP, Ljunggren HG. Immune selection during tumor checkpoint inhibition therapy paves way for NK-cell “missing self” recognition. Immunogenetics. 2017;69(8–9):547-56. doi:10.1007/s00251-017-1011-9
- Curti A, Ruggeri L, D'Addio A, Bontadini A, Dan E, Motta MR, Trabanelli S, Giudice V, Urbani E, Martinelli G, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. 2011;118(12):3273-79. doi:10.1182/blood-2011-01-329508
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Trans Med. 2016;8(357):357ra123. doi:10.1126/scitranslmed.aaf2341. PMID:27655849
- den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192(12):1685-96. doi:10.1084/jem.192.12.1685
- van Mierlo GJ, Boonman ZF, Dumortier HM, den Boer AT, Fransen MF, Nouta J, van der Voort E, Offringa R, Toes RE, Melief CJ. Activation of dendritic cells that cross-present tumor-derived antigen licenses CD8+ CTL to cause tumor eradication. J Immunol. 2004;173(11):6753-59. doi:10.4049/jimmunol.173.11.6753
- van Mierlo GJ, den Boer AT, Medema JP, van der Voort EI, Fransen MF, Offringa R, Melief CJ, Toes RE. CD40 stimulation leads to effective therapy of CD40(-) tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc Natl Acad Sci U S A. 2002;99(8):5561-66. doi:10.1073/pnas.082107699
- Wolkers MC, Brouwenstijn N, Bakker AH, Toebes M, Schumacher TN. Antigen bias in T cell cross-priming. Science. 2004;304(5675):1314-17. doi:10.1126/science.1096268
- Dolan BP, Gibbs KD, Jr., Ostrand-Rosenberg S. Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J Immunol. 2006;177(9):6018-24. doi:10.4049/jimmunol.177.9.6018
- Wakim LM, Bevan MJ. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature. 2011;471(7340):629-32. doi:10.1038/nature09863
- Nurieva R, Wang J, Sahoo A. T-cell tolerance in cancer. Immunotherapy. 2013;5(5):513-31. doi:10.2217/imt.13.33
- Horna P, Sotomayor EM. Cellular and molecular mechanisms of tumor-induced T-cell tolerance. Curr cancer Drug Targets. 2007;7(1):41-53. doi:10.2174/156800907780006940
- Galon J, Fox BA, Bifulco CB, Masucci G, Rau T, Botti G, Marincola FM, Ciliberto G, Pages F, Ascierto PA, et al. Immunoscore and Immunoprofiling in cancer: an update from the melanoma and immunotherapy bridge. 2015. J Transl Med. 2016;14:273. doi:10.1186/s12967-016-1029-z
- Krogsgaard M, Li QJ, Sumen C, Huppa JB, Huse M, Davis MM. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434(7030):238-43. doi:10.1038/nature03391
- Textor A, Schmidt K, Kloetzel PM, Weissbrich B, Perez C, Charo J, Anders K, Sidney J, Sette A, Schumacher TN, et al. Preventing tumor escape by targeting a post-proteasomal trimming independent epitope. J Exp Med. 2016;213(11):2333-48. doi:10.1084/jem.20160636
- Schuurhuis DH, van Montfoort N, Ioan-Facsinay A, Jiawan R, Camps M, Nouta J, Melief CJ, Verbeek JS, Ossendorp F. Immune complex-loaded dendritic cells are superior to soluble immune complexes as antitumor vaccine. J Immunol. 2006;176(8):4573-80. doi:10.4049/jimmunol.176.8.4573