
ABSTRACT
Inflammatory chemokines are critical contributors in attracting relevant immune cells to the tumor microenvironment and driving cellular interactions and molecular signaling cascades that dictate the ultimate outcome of host anti-tumor immune response. Therefore, rational application of chemokines in a spatial-temporal dependent manner may constitute an attractive adjuvant in immunotherapeutic approaches against cancer. Existing data suggest that the macrophage inflammatory protein (MIP)-1 family and related proteins, consisting of CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES), can be major determinant of immune cellular infiltration in certain tumors through their direct recruitment of antigen presenting cells, including dendritic cells (DCs) to the tumor site. In this study, we examined how CCL3 in a murine colon tumor microenvironment, CT26, enhances antitumor immunity. We identified natural killer (NK) cells as a major lymphocyte subtype that is preferentially recruited to the CCL3-rich tumor site. NK cells contribute to the overall IFNγ content, CD103+ DC accumulation, and augment the production of chemokines CXCL9 and CXCL10 for enhanced T cell recruitment. We further demonstrate that both soluble CCL3 and CCL3-secreting irradiated tumor vaccine can effectively halt the progression of established tumors in a spatial-dependent manner. Our finding implies an important contribution of NK in the CCL3 – CD103+ DC – CXCL9/10 signaling axis in determining tumor immune landscape within the tumor microenvironment.
Introduction
Tumor intrinsic and extrinsic factors within the tissue microenvironment dictate the ultimate function, timing and robustness of local and systemic host immune responses, thereby tipping the balance between effective tumor elimination and immune escape.Citation1 Chemokine content within the tumor microenvironment (TME) constitutes one of the important factors contributing to the orchestration and modulation of cellular trafficking, interaction and delivery of effector function among relevant immune cells and tumor cells.Citation2,3 The effect of such chemokine networks on the intensity of tumor-infiltrating lymphocytes (TIL) within TME is distinct from that arising from tumor-intrinsic mutational load, which is thought to correlate with the spectrum of antigenic peptides and responding effector cells. In support of this view, recent studies in melanoma implicate that tumors modulate intra-tumoral T cell density in part by regulating inflammatory chemokine productions in the TME via a WNT/β-catenin-dependent mechanism.Citation4,5 In these studies, melanoma tumor cells that harbor genetic alterations in the WNT/β-catenin pathway could up-regulate inflammatory chemokine, CCL4, which attract dermal-resident CD103+ dendritic cells (DCs). The CD103+ DCs elevated the production of CXCL9 and CXCL10, resulting in attracting T cells to infiltrate the tumor.Citation4 Although these studies were primarily focused on CCL4, the production of CCL3 was also significantly increased in their tumor system.Citation4 We and other investigators have previously reported a critical role for both CCL3 and CCL4 in maximizing antigen scanning by naïve CCR5+ CD8+ T cells on activated DCs undergoing antigen-specific interactions with CD4+ or CD8+ T cells in the vaccine-draining lymph nodes (LN). These interactions are a necessary process for maximal helper CD4+ T cell-dependent memory CD8+ T cell generation.Citation6–8
For these and other reasons, CCL3 has been demonstrated to diminish tumor growth in mouse models.Citation9 In particular, reports have shown that irradiated wild type CT26 tumors (WTTU) and those engineered to secrete CCL3 (L3TU) can be used as vaccines to reduce growth under some circumstances.Citation2,3 In another study, CCL3 was shown to enhance the abscopal effect following local radiation.Citation10 These studies were followed by a clinical trial utilizing CCL3 as an immune adjuvant (clinicaltrials.gov: NCT01441115).
The afore-mentioned studies were focused primarily on evaluating the function of the adoptive immune responses that mediate the primary tumor rejection, but the molecular and cellular mechanisms by which CCL3 exerts its affect was not described in detail. In this current study, we examined the local and systemic effects of CCL3 on cellular and cytokine composition within TME. We employed CT26, a highly immunogenic murine colon tumor derived from BALB/c mice,Citation11,12 and compared the host immune responses between CCL3-negative wild-type (WTTU) CT26 and CT26 engineered to constitutively secrete CCL3 (L3TU). We hypothesize that tumor-derived CCL3 could maximize inflammatory responses in the TME by soliciting the recruitment of innate immune cells, particularly NK cells, that may contribute to the subsequent recruitment of CD103+ DC and T cells as described by others.Citation4,5 We demonstrate that CCL3 production by CT26 significantly slowed in vivo tumor growth, a process partially driven by CCL3-dependent accumulation of NK cells that supply the critical IFNγ in the TME. In turn, enhanced NK and IFNγ accumulation resulted in increased CXCL9 and CXLC10 production as well as CD103+ CD11c+ DCs (CD103+ DCs) to the primary tumor site (PTS). Finally, we demonstrate therapeutic efficacy of recombinant CCL3 (rCCL3) and irradiated whole-cell L3TU vaccine in blunting established CT26 tumor growth in a site-dependent manner.
Results
CCL3 facilitates tumor rejection via thymic-dependent and thymic-independent mechanisms
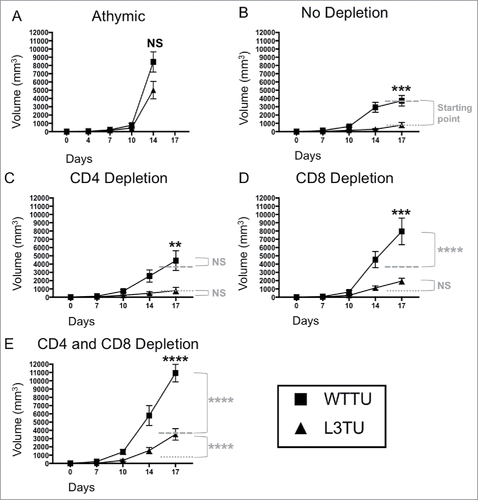
In order to examine how conventional T cell responses affect the growth of aggressive murine colon tumor CT26 (WTTU) and CT26 engineered to secrete CCL3 (L3TU), we measured tumor growth rates in both athymic nude and immunocompetent mice. At baseline, WTTU secretes CCL5 but not CCL3 and CCL4 (Supplemental Fig. S1A).Citation13,14 L3TU produces similar levels of CCL4 and CCL5 as WTTU, with CCL3 production at an average of 1000 pg (ranges ∼350–1300 pg / 1 × 106 cells / ml in 24 hours) as measured from 3 independent studies. Similar to tumor growth rates in vitro (Supplemental Fig. S1B), WTTU and L3TU grew at a similar rate in vivo in athymic nude mice (), suggesting that introducing CCL3 into CT26 did not cause an intrinsic growth defect. Interestingly, L3TU grew significantly slower than WTTU over the course of 3 weeks in BALB/c mice (). The depletion of CD4+ cells did not significantly affect the growth of either L3TU or WTTU (). However, the depletion of CD8+ T cells alone or in conjunction with CD4+ T cells depletion resulted in a rapid tumor progression in WTTU, with tumor sizes approaching that in athymic nude mice. Although L3TU tumor sizes were also significantly increased in BALB/c mice depleted of CD8+ T cells alone or both CD4+ T cells and CD8+ T cell (, , ), the resulting tumors were smaller than those in athymic nude mice, suggesting an additional, non-CD4+/CD8+ T cell-dependent mechanism that is partially responsible for suppressing L3TU growth in vivo.
Figure 1. Autologous CCL3 retards CT26 growth in vivo with partial dependence on both CD8+ T cells and non-T cell sources. Mice were injected with 1 × 106 WTTU (square) or L3TU (triangle) tumor cells in the left flank. Average tumor growths were shown in BALB/c athymic mice (A), mice without antibody depletion (B), CD4+ depletion antibody (C), CD8+ depletion antibody (D), or in CD4+ and CD8+ double depleted mice (E). N = 12 mice/cohort for WTTU group and n = 15 mice/cohort for L3TU group. Data shown are combined results of 3 independent experiments. Black stars (*) compare WTTU and L3TU groups within each graph. Gray dashed (—-) and dotted (….) lines compare the growth shift of each graph to no depletion WTTU and L3TU in . Gray stars (*) compare the significance between the growth shifts of the gray dashed and dotted lines compared to no depletion WTTU and L3TU in . NS: not significant; *, P = 0.01 to 0.05; **, P = 0.001 to 0.01; ***, P = 0.0001 to 0.001; ****, P < 0.0001. Error bars are shown as standard error of mean (SEM).
CCL3 enhances CD4+ and CD8+ T cell infiltration to the primary tumor site
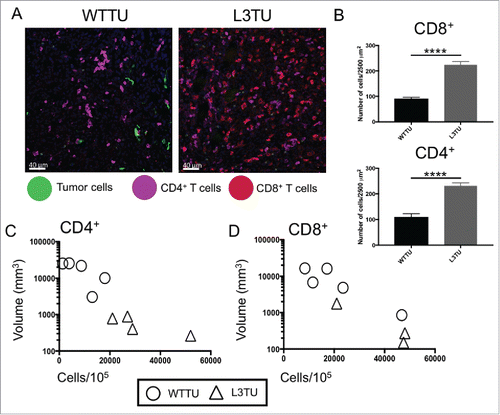
As the presence of CCL3 enhanced CD4+ and CD8+ T cell-dependent rejection of CT26, we sought to verify whether the presence of CCL3 promoted the infiltration of these T cell subsets into the PTS. Immunofluorescence (IF) analysis of tumor tissue sections at 21 days post inoculation revealed a significant increase in both CD4+ and CD8+ T cell infiltrations in the L3TU TME as compared to WTTU (). Interestingly, the degree of T cell infiltration was also inversely correlated with tumor size (), further strengthening the association between the intensity of T cell infiltration and tumor growth kinetics.
Figure 2. L3TU enhances CD4+ and CD8+ T cell infiltrations into the primary tumor site. Mice were injected with 1 × 106 WTTU or L3TU cells in the left flank. At day 21, mice were euthanized and only visible tumors were removed for FACS and IF analyses. A, Representative IF images of GFP-labeled tumors (green), CD4+ T cells (purple) and CD8+ T cells (red). B, Quantification of IF showing T cell number per 2500 um.Citation2 Shown are data obtained from a total of 4 tumors with 16 sections per tumor and 3 areas per section. C-D, Correlation of CD4+ or CD8+ T cell numbers per 1 × 105 total cells by FACS versus WTTU (circle) or L3TU (triangle) tumor volume; N = 5 experiments with 5 biological replicates for each group. ****, P < 0.0001. Error bars are shown as standard deviation (SD).
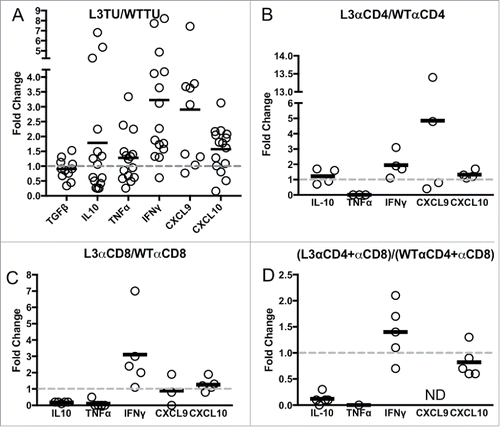
To further understand the molecular mechanisms affecting CCL3-promoted T cell accumulation in the TME, we analyzed cytokine and chemokine contents by qPCR.Citation15 Compared to WTTU, L3TU TME expressed similar levels of TGFβ and TNFα (0.9- and 1.2-fold, respectively; ). However, the amount of IL-10, IFNγ, CXCL9 and CXCL10 mRNAs were upregulated in the L3TU (1.8-, 3.2-, 2.9- and 1.6-fold, respectively; ). While the relative levels of CXCL9 mRNA were higher in L3TU, the overall mRNA abundance was low in both WTTU and L3TU TME relative to the other cytokines and chemokines. Next, we analyzed the contribution of CD4+ and CD8+ T cell subsets to the abundance of the cytokine mRNA contents. Depletion of CD4+ and CD8+ T cells in the L3TU resulted in an even more profound reduction in the overall TNFα content as compared to TNFα levels in WTTU after depletion of these T cell subsets (∼5-fold; , , and Supplemental Fig. S2), supporting the notion that the source of TNFα comes primarily from the T cell subsets with CCL3-enhanced CT26 tumor rejection. Indeed, CD4+ T cell depletion did not reduce TNFα abundance in the WTTU CD4+ depletion group, and CD8+ depletion or CD4+/CD8+ double depletion reduced TNFα only by ∼50% in the WTTU TME (Supplemental Fig. S2A, C, E). Next, we examined the effect of CD4+ and CD8+ T cell subsets on TME IFNγ levels. Both CD4+ and CD8+ T cells clearly contributed to the IFNγ content of the L3TU TME (Supplemental Fig. S2B, S2D, S2F). However, despite a reduction in TNFα, IL-10 and CXCL9 contents in L3TU TME relative to WTTU TME, depletion of these two T cell subsets failed to completely abrogate the total IFNγ content associated with CCL3 ().
Figure 3. IFNγ levels are sustained in L3TU tumors despite T cell depletions. Mice were injected with 1 × 106 WTTU or L3TU cells in the left flank. On day 21, mice were euthanized and tumors removed for qPCR analysis of TGFβ, IL-10, FOXp3, TNFα, IFNγ, CXCL9, and CXCL10. A, Expressions of various cytokine mRNAs in L3TU compared to WTTU. B-D, Relative cytokine mRNA levels of L3TU compared to WTTU in animals depleted of CD4+ cells (L3αCD4 or WTαCD4), CD8+ cells (L3αCD8 or WTαCD8), or both CD4+ and CD8+ cells (L3αCD4+αCD8 or WTαCD4+αCD8). Each symbol represents an individual animal. The expressions of TNFα and CXCL9 were below detection levels in some of the groups. Data represent a compilation of 3 independent experiments. ND: Not detected.
CCL3 recruits NK cells to promote CD103+ DCs infiltration and support T cell function within the primary tumor
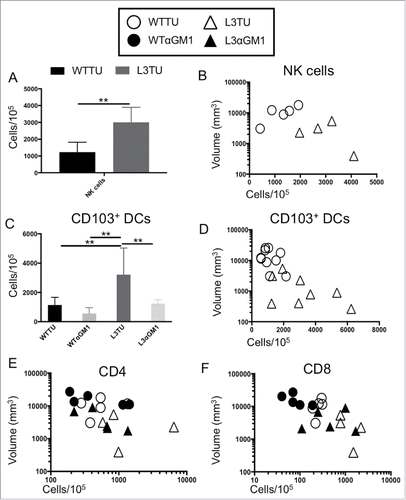
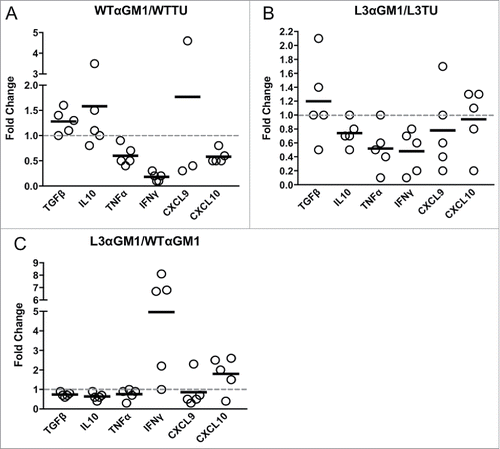
NK cells can serve as a predominant source of IFNγ16,17 and CD103+ DCs have been identified as a major source of CCL4-driven CXCL9 and CXCL10 production within TMEs.Citation4 Therefore, we examined the relative abundance of these important cell populations and their contributions to the differences observed between L3TU and WTTU that were not accounted for by the presence of CD4+ and CD8+ T cells (; Supplemental Fig. 2) A nearly 3-fold increase in the number of NK cells were observed within L3TU TME, and NK cell numbers correlated inversely with the tumor size (, ). Similarly, CD103+ DCs were found to be increased ∼3-fold in L3TU relative to WTTU, with a similar inverse correlation between the density of CD103+ DCs and tumor size (, ). In contrast, macrophage and neutrophil accumulation was not altered in the presence of CCL3 within TME (data not shown). Although the accumulation of CD103+ DC has been linked directly to tumor production of CCL4,Citation4 we wished to determine whether the presence of NK cells and their associated IFNγ production could affect local CD103+ DC accumulation, as IFNγ has been shown to induce other immune cells to upregulate CXCL9 and CXCL10, chemokines which are important for directing the homing of DCs and activated T cells to the TME.Citation5,18,19 Indeed, depletion of NK cells resulted in a dramatic reduction in the number of CD103+ DC within the L3TU TME to a level similar to WTTU (). Interestingly, depletion of NK cells also reduced the numbers of CD4+ and CD8+ T cells within L3TU and trended towards a larger overall tumor size (, ).
Figure 4. CCL3 recruits NK cells to drive infiltration of CD103+ DCs and support T cell function at tumor sites. Mice were injected with 1 × 106 WTTU (circles) or L3TU (triangles) cells in the left flank. At day 21, only visible tumor masses were removed for FACS analysis. A, Enumeration of NK cells (CD49+ CD3−). N = 5 biological replicates. B, Correlation of data in A with individual tumor volume. C, Enumeration of dermal-resident (CD103+ CD11c+) DC subset in tumor masses with or without NK cell depletion (WTαGM1 or L3αGM1). D, Correlation of data in C with individual tumor volume. E, Correlation of CD4+ T cell density in individual tumor mass with (black) or without (white) NK cell depletion. F, Correlation of CD8+ T cell density in individual tumor mass with (black) or without (white) NK cell depletion. N = 2 independent experiments with 5 biological replicates each. **, P = 0.001 to 0.01. Error bars denote standard deviation (SD).
Next, we assessed NK contribution to the global production of cytokines and chemokines within the TME. Depletion of NK cells lead to a reduction in IFNγ and TNFα mRNA in both the WTTU and L3TU TME (, ). NK depletion also resulted in an increase of TGFβ mRNA by 1.3- and 1.6-fold in WTTU and L3TU, respectively. Overall, removal of NK cells normalized the levels of TGFβ, IL-10, TNFα and CXCL9 in L3TU to that in WTTU (). IFNγ and CXCL10 remained elevated in L3TU relative to WTTU after NK depletion, suggesting additional sources, such as T cells, further contributed to the increased immune infiltration in L3TU (; ).
Figure 5. NK cells and CCL3 support inflammation and T cell homing. Mice were injected with 1 × 106 WTTU or L3TU cells in the left flank. At day 21, tumors were analyzed by qPCR for TGFβ, IL-10, TNF-α, IFNγ, CXCL9, and CXCL10. A, Relative change in cytokine expression in WTTU mass with or without NK cell depletion. B, Relative change in cytokine expression in L3TU tumor mass with or without NK cell depletion. C, Relative changes in cytokine production by L3TU and WTTU with NK cell depletion. Each symbol represents an individual animal.
Subcutaneously administered rCCL3 significantly slowed tumor growth in established tumors
To test the antitumor efficacy of CCL3 as a therapy, we performed two immunotherapeutic approaches to treat established CT26. First, we used irradiated L3TU whole cell tumor vaccines to treat established WTTU in vivo. Disappointingly, direct intra-tumoral injections of either irradiated WTTU (iWT) or L3TU (iL3) tumor cell starting on day 7 failed to significantly slow the aggressive WTTU growth in vivo (Supplemental Fig. S3). To avoid the tolerizing local LN microenvironment into which both established WTTU tumor and iL3 vaccines drain, we investigated whether administering irradiated tumor vaccine at a distal body site could potentially avoid the immune suppressing factors expressed by established tumor and to unmask the inflammatory effector functions of CCL3. Following injection of WTTU in the left flank, we waited 7 days and inoculated in the contralateral footpad twice weekly with 1 × 106 iWT (WT/iWT) or iL3 (WT/iL3). Interestingly, the WT/iL3 protocol significantly stunted tumor growth compared to WT/iWT or WTTU alone, even though the effect was not as robust as L3TU tumor alone (), suggesting measurable efficacy of irradiated L3TU as a vaccine in curbing WTTU growth when such vaccines were given at a site other than the primary WTTU site.
Figure 6. rCCL3 or irradiated L3TU significantly slows established tumor growth. Mice were injected with 1 × 106 WTTU cells in the left flank. 7 days later mice were injected in the footpad or intra-tumorally (i.t.) with whole tumor lysate from lethally irradiated WTTU (iWT) or L3TU (iL3), or recombinant CCL3 (rCCL3; 100 ng). A, WTTU tumor growth in mice that received whole cell lysates of iWT or iL3. B, Tumor growth in mice that received either i.t. or s.c. injections of rCCL3. Subcutaneous injections were administered either ipsilaterally (ipsi) or contralaterally (contra) relative to WTTU 7-days post tumor injection (R7). Data shown as compilation of 2 independent experiments with n = 5–10 mice per group. NS: not significant; *, P = 0.01 to 0.05; **, P = 0.001 to 0.01; ***, P = 0.0001 to 0.001; ****, P < 0.0001. Standard error is shown as SD.
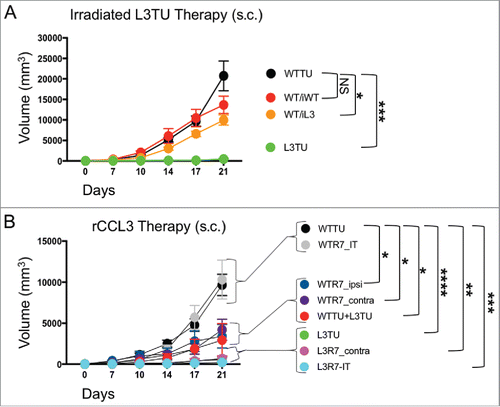
Next, we tested the efficacy of recombinant CCL3 (rCCL3) on established WTTU or L3TU tumor sites using high-dose (100 ng/dose) bolus s.c. administrations.Citation9,20 After injecting WTTU or L3TU in the left flank on day 0, mice received s.c. injections of rCCL3 starting on day 7 in either the ipsilateral footpad (WTR7_ipsi), contralateral footpad (WTR7_contra or L3R7_contra), or intra-tumorally (WTR7_IT or L3R7_IT). For comparison, additional cohorts of mice bearing WTTU or L3TU were injected with PBS starting on day 7, or inoculated with 1:1 mixture of live WTTU+L3TU on day 0. Interestingly, intra-tumoral injections of rCCL3 starting on day 7 failed to slow WTTU growth and in some of the mice even facilitated tumor growth when compared to WTTU group alone (; data not shown). However, rCCL3 administered distally either in the ipsilateral or contralateral footpad starting on day 7 resulted in a significant decrease in the overall WTTU tumor growth similar to 1:1 live WTTU+L3TU tumor mixed (). Although no significant differences were observed between L3R7_contra and L3R7_IT, these tumors were smaller when compared to L3TU alone (, ; Supplemental Fig. S4), suggesting a differential effect of CCL3 in the anti-tumor immune priming when administered on day 0 versus day 7.
Discussion
Colon cancer and melanoma have been associated with suppressed expressions of MIP-1 family of proteins that are typically upregulated by their respective normal tissue counterparts under stressed conditions.Citation21 CCL3, CCL4 and CCL5 share a high degree of homology, and bind to CCR5 (all) and CCR1 (CCL3 and CCL5) on many cell types including immature DCs and T cells in both humans and mice.Citation8,9 CCL3-transfected melanoma tumors can recruit adoptively transferred, antigen-primed bone marrow derived DCs (BMDCs) to PTS and drive tumor rejection.Citation3 CCL3 and CCL4 have also been implicated in directing CD8+ T cell infiltration into primary tumor sites in melanoma and colon cancers.Citation4,5 In particular, Spranger and colleagues demonstrated how melanoma tumors could prevent CD103+ DC infiltration though active suppression of CCL4 in a WNT/β-catenin-dependent pathway.Citation4 Alterations in the WNT/β-catenin pathway up-regulate CCL4 in melanoma, resulting in the recruitment of dermal-resident CD103+ DC to the PTS with subsequent migration to the tumor DLN (TDLN) for anti-tumor T cell priming.Citation4 In this paradigm, CCL4/CCL3-enhanced recruitment of CD103+ DCs are responsible for the production and release of CXCL9 and CXCL10, which facilitate subsequent accumulation of effector T cells to the tumor site. As the WNT/β-catenin pathway is also active in CT26,Citation22 similar molecular and cellular interplay may be at work in our current study. While multiple reports have suggested that an active WNT/β-catenin pathway suppresses the activation of CCL3 and CCL4 in some immune and tumor cell types,Citation4,23,24 it can also drive the production of CCL5 in other tumor models.Citation25 Indeed, despite evidence of baseline CCL5 production (Supplemental Fig. S1), engineered CCL3 over-expression by CT26 recapitulated similar findings as described by prior studies, resulting in both enhanced homing of CD103+ DCs and T cell infiltration into the primary tumors.Citation3,4 However, our current study further revealed an additional critical component in the CCL3-dependent cellular recruitment paradigm by identifying NK cells as a major contributor in blunting CT26 growth via production of IFNγ within the TME. Our NK cell findings add another player in the CCL3/4-directed, CD103+ DC-driven, CXCL9/CXCL10-induced mechanism of immune infiltration into tumors,Citation4 and recapitulates previous report showing IFNγ as being involved in the recruitment of CD103+ DCs.Citation19
We compared the global mRNA expressions of TNFα, IFNγ, TGFβ, and IL-10, as well as CXCL9 and CXCL10, which can be induced by IFNγ and are associated with improved prognosis in patients.Citation26–28 We showed that NK cells are critical for enhancing CD103+ DCs recruitment and augmenting proper effector function of T cells. We also identified IFNγ as contributing to CD103+ DC recruitment in L3TU, which correlated with the CCL3-directed homing of NK cells into the tumor sites (, ). Depletion of NK cells in L3TU not only significantly reduced CD103+ DC accumulation, but also decreased the abundance of IFNγ at the PTS, supporting the view that NK cells are important sources of IFNγ. Although T cells constitute another major source of IFNγ in the tumor rejection process, our data is further supported by previous studies using IFNγ-deficient T cells to demonstrate that NK cells could supply much of the IFNγ needed in effective tumor rejection.Citation16 However, NK cells also are dependent on T cells for full activation, thus underscoring the existing crosstalk between innate and adaptive anti-tumor responses.Citation16 The presence of IFNγ further prompts the production of CXCL9 and CXCL10 from the surrounding epithelial and immune cells, thereby augmenting T cell infiltration and enhancing DC accumulation though CXCR3.Citation5,29 Our study further demonstrated the functional importance of this molecular crosstalk by observing a drastic decrease in IFNγ and associated CXCL9 and CXCL10 levels after NK depletion, resulting in diminished T cell infiltration and accelerated tumor growth. Furthermore, our observation agrees with previous findings that the presence of IFNγ-producing cells in the primary tumor mass is inversely correlated with tumor growth,Citation30 and that IFNγ early in the TME activates DCs and T cells towards inflammatory phenotypes. Both CCL3 and IFNγ were implicated in endowing DCs the ability to polarize T cell inflammatory responses, proliferation, and immune memory generation.Citation6,31–35
Previous studies have attributed CCL3-promoted CT26 tumor rejection to increased macrophage and neutrophil infiltration at the PTS within the first 5 days following tumor inoculation as evidenced by histological examination.Citation2 However, our day 21 analysis of L3TU tumors by flow cytometry did not find such observation. The reason for this discrepancy is not entirely clear. One possibility is that the numbers of macrophages and neutrophils may have equilibrated between WTTU and L3TU by day 21. Another possibility could be the differences in the function, not the relative abundance, of these cells in the two different PTS. Ultimately, CT26 rejection is most effectively controlled by CD8+ T cells, which recognize the immuno-dominant antigen, AH1, on the H2-Ld haplotype.Citation12 Therefore, it was not a surprise that depletion of CD8+ T cells resulted in significant accelerated tumor growth in both WTTU and L3TU. CD4+ T cells have also been shown to modulate CT26 rejection.Citation36 Indeed, depletion of CD4+ T cells results in a significantly accelerated L3TU tumor growth when performed concurrently with CD8+ T cell depletion (). These effects are likely linked to increased cytokine production associated with these T cell populations in the L3TU TME (Supplemental Fig. S2F).
Chemokine-based immunotherapy has been investigated as a means to modulate anti-tumor immunity.Citation2,3 We provided exciting data demonstrating benefits in translational application of using iL3 vaccine or bolus rCCL3 therapy to significantly blunt the growth of established CT26 (, Supplemental Fig. S4). Despite reports supporting the benefits of intra-tumoral administration of cytokines as therapies,Citation37 we failed to observe notable effects of intra-tumoral iL3 or rCCL3 administration on slowing the growth of established WTTU. The exact mechanism for the failure of this approach is unclear. One possibility could be that day 7 WTTU TME and draining LN may be too immune tolerant to benefit from the therapeutic effects of iL3 or rCCL3. Most interestingly, however, we observed that contralateral or ipsilateral s.c. injections of iL3 and rCCL3 distal to the tumor site could efficiently blunt established CT26 growth. We hypothesize that that CCL3 administered at a distal site can either robustly stimulate immune cells within DLNs that are not exposed to the immediate suppressive factors derived from the tumor, or efficiently cause the mobilization of cells from the bone marrow to migrate into PTS or the LN. Supporting this hypothesis, CCL3 has been shown to enhance the release of innate responders such as NK cells and DCs from the marrow to the blood where they can home to tissues.Citation38,39 We are currently examining this fascinating aspect of CCL3-induced immune activation in the draining LN early in the therapeutic response.
In summary, we examined CCL3 contribution in facilitating effective cellular crosstalk between the innate and adaptive anti-tumor immune responses. We provided new insights into how this crosstalk supports the accumulation and effector function of T cells, NK cells and CD103+ DCs at the PTS. We also demonstrated the efficacy of CCL3 as a modulating immunotherapeutic for the treatment of established tumors. NK cells supporting T cell infiltration and function is a phenomenon that goes beyond mouse tumor models. NK cells involvement in the stepwise infiltration of lymphocytes has also been reported in human colon cancers.Citation40 In addition, CCL3 upregulation by IFNγ-producing NK cells has been implicated as an important component in the clinical efficacy of the FDA-approved monoclonal antibody, trastuzumab, for the rejection of HER2+ breast cancers.Citation41
Although our study focuses exclusively on CCL3 which showed a pronounced effect on CT26 rejection in vivo, similar contribution to tumor rejection has been ascribed to CCL4 in melanoma.Citation4 Indeed, CCL4-secreting CT26 (L4TU) also resulted in a significant production of IFNγ at the tumor site compared to WTTU (Supplemental Fig. S5). However, compared to L4TU, L3TU resulted in a more rapid rejection (data not shown). Future studies will exam the mechanisms of s.c. administered CCL3 in mobilizing marrow-derived immune cells to facilitate tumor rejection. Taken together, our current data further support the exploration of CCL3 as an adjuvant for enhancing antitumor immune response.
Material and methods
Mice
BALB/c mice were purchased from the Jackson Laboratory (Bar Harbor, ME) or bred in house. Both male and female 8- to 12-week-old BALB/c mice were used for all experiments. Mice were housed and handled according to National Institutes of Health institutional guidelines under approved protocols by Case Western Reserve University Institutional Animal Care and Use Committee (No. 2012-0126 and 2015-0118).
CT26 transfection, maintenance, and use
In order to create CCL3-secreting CT26 (L3TU), CT26 (WTTU) tumor cells (purchased from ATCC®) were stably transfected with a PCDNA3.1 plasmid vector that encodes mouse CCL3 under the CMV promoter and maintained under Hygromycin (150 µg / ml) selection. Before storage, cells are tested for mycoplasma contamination and stored as passage zero for experimental use later. Cells are stored in 10% dimethyl sulfoxide (DMSO) and 90% complete media (RPMI 1640 with 10% FBS, 1% HEPES, 1% non-essential amino acids, and 1% penicillin and streptomycin) in liquid nitrogen tanks when not in use. Cells are thawed and used for experiments [3-12 passages (∼4–6 weeks)] after in vitro growth is stabilized (∼3 passages). During use, cells are incubated at 37°C with 95%O2 and 5% CO2 infusion. Cell viability is measured using Trypan blue dye. Tumor cells are used for experiments only if cell viability is above 90% after counting. Prior to in vivo use, cells are washed with 1x PBS three times.
Tumor measurements
For tumor measurements, mice were injected s.c. with either WTTU or L3TU in the left flank. Tumors were inspected, palpated, and measured using electronic calipers twice weekly. Tumor volumes were calculated according to the formula, V = π x D x d2, where “D” is the larger diameter and “d” is the smaller diameter.Citation42
Tumor Isolation and tissue preparation for flow cytometry and qPCR analyses
Tumor masses were excised, weighed for comparison against volume measurements, and finely chopped with a razor blade. The tumor was then stirred thoroughly to mix cells into a uniform heterogeneous cell mixture and a small portion is then removed for qPCR analysis. The remaining sample was placed into a conical tube containing FACS buffer (0.5% fetal bovine serum (FBS) and 0.5% of pH 8 EDTA in 1x sterile PBS), collagenase D and DNase-1, then incubated at 37° C in a mixer. The mixture was then passed through a 40 µm strainer twice and prepared for antibody staining and analysis by fluorescence-activated cell sorting (FACS).
Flow cytometry, recombinant CCL3, and immunofluorescence materials and supplies
Antibodies for flow cytometry and immunofluorescence staining (IF) were purchased from eBioscience, BD Pharmingen, or BioLegend and included the following: rat anti-mouse CD4 FITC and PE (GK1.5), APC (RM4-5); CD8a FITC, PE, and APC (53-6.7); CD11c FITC, PE, and APC (N418); CD49b PE and APC (DX5); CD3 PE and APC (145-2C11); CD103 FITC (2E7). Analysis was performed using an Accuri C6 and FlowJo software against isotype controls and fluorescence minus one techniques. Recombinant murine CCL3 was purchased from PeproTech.
Tumor growth and antibody depletion experiments
For immune cell depletion studies, mice were injected with 100 µl of HBSS (control), 50 µg of αAsialo-GM1 (Poly21460), 75 µg anti-CD4 (GK1.5), 50 µg anti-CD8 (2.43), or both GK1.5 and 2.43 neutralizing antibodies given intraperitoneally to BALB/c mice on days −3, −1, 1, and then twice weekly thereafter. 1 × 106 WTTU, L3TU, or WTTU + L3TU cells were injected subcutaneously into the left flank on day 0. Tumor volumes were measured twice weekly as described above.
ELISA
1 × 106 WTTU or L3TU were incubated for 24 hours at 37oC in 5% CO2 in 1 ml of complete media (RPMI 1640 with 10% FBS, 1% HEPES, 1% non-essential amino acids, and 1% penicillin and streptomycin). The spent culture media or serum samples obtained from tumor-bearing mice were quantified for CCL3, CCL4 or CCL5 protein contents by ELISA in accordance with the manufacture's protocol (R&D systems, MMA00).
IFNγ ELISPOT
Single cell suspensions were made from harvested tumors at indicated times following tumor inoculation. Tumor infiltrating immune cells were isolated from tumor cells using Ficoll separation protocol. Immune cells were plated on IFNγ-coated plates in biological duplicates and IFNγ ELISPOT assays were performed as follows: Day 1, 96 well plates were coated with 1μg/ml of primary IFNγ overnight at 4oC. Day 2, 1 × 106 immune cell were added to the well along with 0.5 × 106 irradiate (2000 Rads) AH1-peptide-pulsed splenocytes (1ug/ml peptide for 1 hour at 37oC and 5% CO2) and incubated at 37oC and 5% CO2 for 24 hours. On day 3, biotinylated IFNγ secondary antibody (4ug/ml) was added to each welI and incubated overnight at 4oC, and 100ul/well of alkaline phosphatase strepavertin was added to each well in a humidity bag and incubate in the dark for 20 minutes or less (depending on the intensity of the forming spots). After drying, the plates were analyzed using an automatic C.T.L. ELISPOT plate reader.
Quantitative RT-PCR analysis
Total LN mRNA was isolated using TRIzol reagent in accordance with the manufacturer's protocol (Gibco BRL, Carlsbad, CA) and purified using an Illustra™ RNAspin Mini Kit (GE Healthcare Life Sciences). RNA quality was assessed by spectrophotometer absorption at 260/280 nm using the NanoDrop2000 spectrophotometer. RNA was converted to cDNA using the EasyScript™ Reverse Transcriptase protocol consisting of 200 U/µl Moloney murine leukemia virus reverse transcriptase incubated for 60 minutes at 42°C in the presence of 50mM Tris-HCl (pH 8.3), 100 mM NaCl, 0.1 mM EDTA, 5 mM DTT, 0.1% Triton X-100, 50% (v/v) glycerol, 10 µM of oligo (dT), 10 mM 29-deoxynucleoside 59-triphosphate, and 40 U/µl recombinant RNase inhibitor (Lamda BIOTECH, St. Louis, MO). cDNA was amplified in the presence of FAM-labeled gene-specific primers and Bullseye EvaGreen qPCR Mastermix (MIDSCI™; Saint Louis, MO) in a 96 well microtiter plate using the ABI Prism 7300 sequence detection system (Applied Biosystems). Each PCR reaction was performed in triplicate and compared to WTTU. Relative levels of mRNA were determined using the cycle threshold (Ct). The gene expression was standardized to cytochrome-c (CyC) expression within the tumor draining lymph node (TDLN). In order to compare the Ct values between target genes we normalized each Ct to the average of the WTTU Ct using the following equation: 2^−(Target gene – CyC – target normalizer).
Statistical analysis
Statistical analyses were performed using the standard one tailed unpaired t-test. All data presented as either +/- standard error of the mean ( only) or standard deviation.
Conflict of interest statement
The authors do not have any conflict of interest to declare.
Financial support
This work is supported by funding from the National Institutes of Health R01CA154656, R21CA181875, R21CA218790, R01HL111682, P30CA043703 (to AYH), NCI R01CA154656-S1 and F31CA192874 (to FA), R01AI091627 (to AYH and JM), the Rainbow Foundation at UH Rainbow Babies & Children's Hospital (to IDB), the St. Baldrick's Foundation (to NT and AYH), the Alex's Lemonade Stand Foundation, Hyundai Hope-on-Wheels Program, Pediatric Cancer Research Foundation, the Steven G. AYA Cancer Research Fund, the Samuel Szabo Foundation, the Keira Kilbane Cancer Discovery Fund, Errol's Cancer Discovery Fund, the Risman Family Philanthropic Funds, the Cleveland Foundation, the Harrington Discovery Institute, and the Theresia G. & Stuart F. Kline Family Foundation (to AYH).
Abbreviations
| BMDCs | = | bone marrow derived DCs |
| contra | = | contralateral |
| BMDCs | = | bone marrow derived DCs |
| DCs | = | dendritic cells |
| I.T. | = | intra-tumoral |
| IF | = | immunofluorescence |
| ipsi | = | ipsilateral |
| iWT | = | irradiated WTTU |
| iL3 | = | irradiated L3TU |
| L3R7 | = | L3TU with rCCL3 give at day-7 |
| L3/iL3 | = | WTTU with iL3 treatment |
| LNs | = | lymph nodes |
| L3TU | = | CCL3-secreting CT26 tumors |
| NK | = | natural killer |
| PTS | = | primary tumor site |
| rCCL3 | = | recombinant CCL3 |
| TME | = | tumor microenvironment |
| TILs | = | tumor-infiltrating lymphocytes |
| TDLN | = | tumor draining lymph node |
| WTTU | = | wild type CT26 tumors |
| WT/iWT | = | WTTU with iWT treatment |
| WTR7 | = | WTTU with rCCL3 give at day 7 |
supp_data.zip
Download Zip (554.5 KB)Acknowledgments
We are grateful for Drs. Mari Dallas, Suzanne Tomchuck, David Askew and R. Dixon Dorand for providing valuable reviews and critiques of this manuscript. We would like to thank Joseph Nthale for his contribution in the construction of both the GFP+ and GFP− CCL3-secreting CT26 tumor cells for this project; Saada Eid for help with IF sectioning and for generously sharing her expertise on various techniques used in this study; and Alexander Tong for his generous support in helping with the many labor-intensive components of this project.
References
- Dunn GP, Old LJ, Schreiber RD The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi:10.1146/annurev.immunol.22.012703.104803. PMID:15032581.
- Nakashima E et al. A candidate for cancer gene therapy: MIP-1 alpha gene transfer to an adenocarcinoma cell line reduced tumorigenicity and induced protective immunity in immunocompetent mice. Pharm Res. 1996;13:1896–1901. doi:10.1023/A:1016057830271. PMID:8987092.
- He S, Wang L, Wu Y, Li D, Zhang Y CCL3 and CCL20-recruited dendritic cells modified by melanoma antigen gene-1 induce anti-tumor immunity against gastric cancer ex vivo and in vivo. J Exp Clin Cancer Res. 2010;29:37. doi:10.1186/1756-9966-29-37. PMID:20420712.
- Spranger S, Bao R, Gajewski TF Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi:10.1038/nature14404. PMID:25970248.
- Harlin H et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi:10.1158/0008-5472.CAN-08-2281. PMID:19293190.
- Castellino F et al. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 2006;440:890–895. doi:10.1038/nature04651. PMID:16612374.
- Hugues S et al. Dynamic imaging of chemokine-dependent CD8+ T cell help for CD8+ T cell responses. Nat Immunol. 2007;8:921–930. doi:10.1038/ni1495. PMID:17660821.
- Askew D et al. Transient surface CCR5 expression by naive CD8(+) T cells within inflamed Lymph Nodes is dependent on high Endothelial Venule interaction and augments Th cell-dependent memory response. Journal of Immunology. 2016;196:3653–3664. doi:10.4049/jimmunol.1501176.
- Menten P, Wuyts A, Van Damme J Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 2002;13:455–481. doi:10.1016/S1359-6101(02)00045-X. PMID:12401480.
- Shiraishi K et al. Enhancement of antitumor radiation efficacy and consistent induction of the abscopal effect in mice by ECI301, an active variant of macrophage inflammatory protein-1alpha. Clin Cancer Res. 2008;14:1159–1166. doi:10.1158/1078-0432.CCR-07-4485. PMID:18281550.
- Corbett TH, Griswold DP, Jr., Roberts BJ, Peckham JC, Schabel FM, Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975;35:2434–2439. PMID:1149045.
- Huang AY et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci U S A. 1996;93:9730–9735. doi:10.1073/pnas.93.18.9730. PMID:8790399.
- Cambien B et al. CCL5 neutralization restricts cancer growth and potentiates the targeting of PDGFRbeta in colorectal carcinoma. PLoS One. 2011;6:e28842. doi:10.1371/journal.pone.0028842. PMID:22205974.
- Chang LY et al. Tumor-derived chemokine CCL5 enhances TGF-beta-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res. 2012;72:1092–1102. doi:10.1158/0008-5472.CAN-11-2493. PMID:22282655.
- Mocellin S et al. Use of quantitative real-time PCR to determine immune cell density and cytokine gene profile in the tumor microenvironment. J Immunol Methods. 2003;280:1–11. doi:10.1016/S0022-1759(03)00274-6. PMID:12972183.
- Li Z et al. Cross-talk between T cells and innate immune cells is crucial for IFN-gamma-dependent tumor rejection. J Immunol. 2007;179:1568–1576. doi:10.4049/jimmunol.179.3.1568. PMID:17641023.
- Cacalano NA Regulation of natural killer cell function by STAT3. Front Immunol. 2016;7:128. doi:10.3389/fimmu.2016.00128. PMID:27148255.
- Carter SL, Muller M, Manders PM, Campbell IL Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFN-gamma but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia. 2007;55:1728–1739. doi:10.1002/glia.20587. PMID:17902170.
- Lantier L et al. Intestinal CD103+ dendritic cells are key players in the innate immune control of Cryptosporidium parvum infection in neonatal mice. PLoS Pathog. 2013;9:e1003801. doi:10.1371/journal.ppat.1003801. PMID:24367259.
- Fahey TJ, 3rd et al. Macrophage inflammatory protein 1 modulates macrophage function. J Immunol. 1992;148:2764–2769. PMID:1573267.
- Mikucki ME et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6:7458. doi:10.1038/ncomms8458. PMID:26109379.
- Castle JC et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15:190. doi:10.1186/1471-2164-15-190. PMID:24621249.
- Oderup C, LaJevic M, Butcher EC Canonical and noncanonical Wnt proteins program dendritic cell responses for tolerance. J Immunol. 2013;190:6126–6134. doi:10.4049/jimmunol.1203002. PMID:23677472.
- Ma B, Hottiger MO Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during inflammation. Front Immunol. 2016;7:378. doi:10.3389/fimmu.2016.00378. PMID:27713747.
- Yasuhara R et al. The beta-catenin signaling pathway induces aggressive potential in breast cancer by up-regulating the chemokine CCL5. Exp Cell Res. 2015;338:22–31. doi:10.1016/j.yexcr.2015.09.003. PMID:26363360.
- Budi EH, Duan D, Derynck R Transforming growth Factor-beta Receptors and Smads: Regulatory complexity and functional versatility. Trends Cell Biol. 2017;27:658–672. doi:10.1016/j.tcb.2017.04.005. PMID:28552280.
- Sabat R et al. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010;21:331–344. doi:10.1016/j.cytogfr.2010.09.002. PMID:21115385.
- Monteagudo C, Martin JM, Jorda E, Llombart-Bosch A CXCR3 chemokine receptor immunoreactivity in primary cutaneous malignant melanoma: correlation with clinicopathological prognostic factors. J Clin Pathol. 2007;60:596–599. doi:10.1136/jcp.2005.032144. PMID:16522748.
- Groom JR, Luster AD CXCR3 in T cell function. Exp Cell Res. 2011;317:620–631. doi:10.1016/j.yexcr.2010.12.017. PMID:21376175.
- Nakajima C et al. A role of interferon-gamma (IFN-gamma) in tumor immunity: T cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sites in IFN-gamma-deficient mice. Cancer Res. 2001;61:3399–3405. PMID:11309299.
- Dorner BG et al. MIP-1alpha, MIP-1beta, RANTES, and ATAC/lymphotactin function together with IFN-gamma as type 1 cytokines. Proc Natl Acad Sci U S A. 2002;99:6181–6186. doi:10.1073/pnas.092141999. PMID:11972057.
- Zhang N et al. Type 1 T-cell responses in chlamydial lung infections are associated with local MIP-1alpha response. Cell Mol Immunol. 2010;7:355–360. doi:10.1038/cmi.2010.32. PMID:20622889.
- Martin-Fontecha A et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–1265. doi:10.1038/ni1138. PMID:15531883.
- Sercan O, Stoycheva D, Hammerling GJ, Arnold B, Schuler T IFN-gamma receptor signaling regulates memory CD8+ T cell differentiation. J Immunol. 2010;184:2855–2862. doi:10.4049/jimmunol.0902708. PMID:20164422.
- Whitmire JK, Tan JT, Whitton JL Interferon-gamma acts directly on CD8(+) T cells to increase their abundance during virus infection. Journal of Experimental Medicine. 2005;201:1053–1059. doi:10.1084/jem.20041463. PMID:15809350.
- Golgher D, Jones E, Powrie F, Elliott T, Gallimore A Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol. 2002;32:3267–3275. doi:10.1002/1521-4141(200211)32:11%3c3267::AID-IMMU3267%3e3.0.CO;2-1. PMID:12555672.
- Van der Jeught K. et al. Targeting the tumor microenvironment to enhance antitumor immune responses. Oncotarget. 2015;6:1359–1381. doi:10.18632/oncotarget.3204. PMID:25682197.
- Bernardini G et al. CCL3 and CXCL12 regulate trafficking of mouse bone marrow NK cell subsets. Blood. 2008;111:3626–3634. doi:10.1182/blood-2007-08-106203. PMID:18227348.
- Charmoy M et al. Neutrophil-derived CCL3 is essential for the rapid recruitment of dendritic cells to the site of Leishmania major inoculation in resistant mice. PLoS Pathog. 2010;6:e1000755. doi:10.1371/journal.ppat.1000755. PMID:20140197.
- Coca S et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79:2320–2328. doi:10.1002/(SICI)1097-0142(19970615)79:12%3c2320::AID-CNCR5%3e3.0.CO;2-P. PMID:9191519.
- Roda JM et al. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006;66:517–526. doi:10.1158/0008-5472.CAN-05-2429. PMID:16397268.
- Tomayko MM, Reynolds CP Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–154. doi:10.1007/BF00300234. PMID:2544306.