
ABSTRACT
Systemic treatment of malignant pleural mesothelioma (MPM) is moderately active for the intrinsic pharmacological resistance of MPM cell and its ability to induce an immune suppressive environment. Here we showed that the expression of bromodomain (BRD) proteins BRD2, BRD4 and BRD9 was significantly higher in human primary MPM cells compared to normal mesothelial cells (HMC). Nanomolar concentrations of bromodomain inhibitors (BBIs) JQ1 or OTX015 impaired patient-derived MPM cell proliferation and induced cell-cycle arrest without affecting apoptosis. Importantly, BBIs primed MPM cells for immunogenic cell death, by increasing extracellular release of ATP and HMGB1, and by promoting membrane exposure of calreticulin and ERp57. Accordingly, BBIs activated dendritic cell (DC)-mediated phagocytosis and expansion of CD8+ T-lymphocyte clones endorsed with antitumor cytotoxic activity. BBIs reduced the expression of the immune checkpoint ligand PD-L1 in MPM cells; while both CD8+ and CD4+ T-lymphocytes co-cultured with JQ1-treated MPM cells decreased PD-1 expression, suggesting a disruption of the immune-suppressive PD-L1/PD-1 axis. Additionally, BBIs reduced the expansion of myeloid-derived suppressor cells (MDSC) induced by MPM cells. Finally, a preclinical model of MPM confirmed that the anti-tumor efficacy of JQ1 was largely due to its ability to restore an immune-active environment, by increasing intra-tumor DC and CD8+ T-lymphocytes, and decreasing MDSC. Thereby, we propose that, among novel drugs, BBIs should be investigated for MPM treatment for their combined activity on both tumor cells and surrounding immune-environment.
Introduction
Malignant pleural mesothelioma (MPM) is an asbestos-related cancer characterized by an extremely long latency. Current classification is based on three main histological subtypes, epithelioid, sarcomatoid and biphasic, having respectively better, worse and intermediate prognosis.Citation1 Since MPM is usually diagnosed in advanced stages, chemotherapy usually remains the only therapeutic option, but it is only modestly effective, with a median overall survival of approximately 12 months.Citation1 This limited efficacy is also ascribed to the immune-evasive attitude of MPM that is characterized by a low antigenicity and by an immune-suppressive environment.Citation2–4 Molecular classification of MPM has lagged behind compared to other cancer types. Two recent high-throughput genomic analysesCitation5,6 and provisional data from The Cancer Genome Atlas TCGA (https://tcga-data.nci.nih.gov) indicate that MPM has a generally low mutational burden.Citation7 On the other hand, sporadic observations indicate that many genes involved in epigenetic modifications, such as BAP1, NF2, SPOP, NUTM1, LATS1/2 SMARCA4/1, SETDB1, SETD2, can be deleted, mutated or amplified,Citation7,8 while druggable kinases are not generally altered, thus limiting the use of existing targeted therapies.
BET-Bromodomain Inhibitors (BBIs) represent a new class of drugs that modulate the epigenetic and the transcriptional program of cancer cells exerting a very potent therapeutic action in several hematological and solid tumors.Citation9–11 Interestingly, it has been recently demonstrated that the BBI OTX015 decreases MPM cell proliferation by reducing c-Myc expression and delays MPM tumor growth with an efficacy comparable to standard chemotherapy.Citation12 However, the functional interactions between BBI and the host immune system of mesothelioma (MPM) tumors as well as the ability of BBIs to alter the immunogenicity of MPM cells remain therapeutically unexplored. Here, we show that BBIs act as multitasking agents that are able to interfere with MPM cell growth and to convert an immune-suppressive to an immune-active environment.
Results
BRDs are amplified or overexpressed in primary MPM samples and BBI treatment reduces cell growth of patient-derived MPM cells
We first interrogated through the cBioPortalCitation13,14 publicly available TCGA data of 87 MPM samples (MESO). Interestingly, BRD2, BRD3, BRD4 and BRD9 were either amplified or up-regulated in 6, 2, 9 and 13 cases, respectively (n = 87; ). Collectively, BRDs were up-regulated in 28/87 (32%) MPM samples. Thereby we extended BRD expression analysis to our series of 15 primary MPM samples (Tables S1 and S2). BRD2, BRD4 and BRD9 were significantly upregulated in tumors compared to primary not-transformed human mesothelial cells (HMC; ). Consistently with the high expression of BRDs in MPM, both BBIs JQ1 and OTX015 impaired cell proliferation in a dose-dependent manner in all histological subtypes of patient-derived MPM cells ( and , Fig. S1 A and B). Importantly, a concentration of 250 nM of BBIs was sufficient to interfere with cell cycle progression (, Fig. S1C, Fig. S2A and B). However, the anti-proliferative activity of JQ1 was not associated to apoptosis (), and OTX015 treatment was accompanied by a modest increase in cell death (about 15%; Fig. S1D).
Figure 1. BRD expression in MPM. (A) Oncoprint map of BRD gene amplification, up- and down-regulation in MPM samples analyzed by the TCGA-MESO database (n = 87). Data were obtained through the cBioPortal (http://www.cbioportal.org). (B) mRNA expression of BRD2, BRD3, BRD4 and BRD9 was detected in triplicates by real-time PCR in HMC and MPM cells. *p < 0.05: mean±SEM expression for BRD2 in epithelioid (epi), biphasic (bip) and sarcomatoid (sar) MPM samples vs mean±SEM expression in HMC (3.19±0.84 vs 1.29±0.08); not significant for BRD3 (6.52±2.92 vs 1.93±0.65); **p < 0.01 for BRD4 (4.56±1.06 vs 1.26±0.38); ***p < 0.001 for BRD9 (10.19±1.87 vs 1.83±0.39).
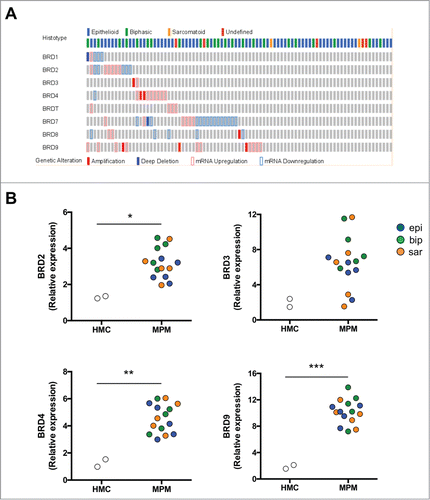
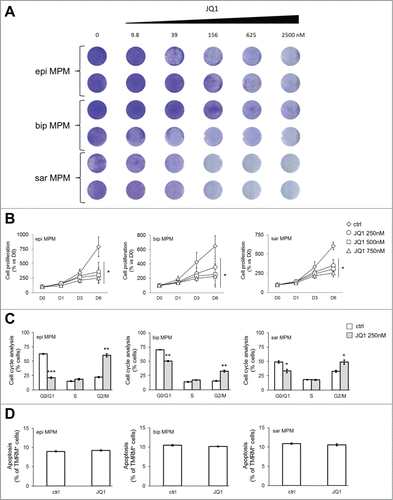
Figure 2. Antiproliferative effects of JQ1 on MPM patient derived cell lines. (A) MPM cells were incubated for 10 days at the indicated concentrations of JQ1, then stained with crystal violet solution (n = 3). Representative photographs of epithelioid (epi), biphasic (bip) and sarcomatoid (sar) MPM samples. (B) MPM cells were left untreated (ctrl) or incubated with JQ1 at the indicated concentrations. Proliferation rate was measured at day (D) 1, 3 and 6 in triplicates. Data of MPM samples (epi: epithelioid; bip: biphasic; sar: sarcomatoid) are means±SEM. *p < 0.05: JQ1-treated vs untreated MPM cells (D6). (C) Cells were incubated for 24 h (not shown) or 48 h in medium containing DMSO (ctrl) or 250 nM JQ1, then analyzed for cell cycle distribution in duplicates. Data of MPM samples are means±SEM. *p < 0.05; **p < 0.01; ***p < 0.001: JQ1-treated vs untreated MPM cells. The results after 24 h-treatment were superimposable (not shown). (D) MPM cells were incubated as reported in (C) for 72 h. The percentage of apoptotic cells was measured by TMRM assay in duplicates. Data of MPM samples (epi: epithelioid; bip: biphasic; sar: sarcomatoid) are means±SEM.
BBIs induce immunogenic cell death (ICD) along with adaptive immune response against MPM cells
Since inhibitors of chromatin-associated enzymes and BRDs can exert their therapeutic action also by modulating tumor cell immunogenicityCitation15,16 we investigated this aspect in our primary patient-derived MPM cells under BBI treatment. Intriguingly, JQ1 and OTX015 increased the release of ATP (, Fig. S3A) and High Mobility Group Protein 1 (HMGB1; , Fig. S3B) in the extracellular supernatant of MPM cells, as well as the exposure of the “eat-me signals” calreticulin (CRT; , Fig. S3C) and ERp57 (, Fig. S3D), without affecting these parameters in non-transformed HMC. All these findings are typical of immunogenic cell death (ICD), a process that promotes an anti-tumor adaptive response followed by expansion of T lymphocytesCitation17,18 with an increased percentage of cytotoxic CD8+CD107+ cellsCitation19 and secretion of IFN-γ.Citation17,18 Accordingly, BBIs significantly increased the DC-mediated phagocytosis of patient-derived MPM cells, which were more resistant to phagocytosis than HMC in untreated condition (, Fig. S3E). As we previously observed,Citation20 proliferation of co-cultured CD8+ T-lymphocytes was negatively affected by MPM cells respect to normal HMC (, Fig. S3F). This low expansion was associated with a lower IFN-γ secretion () and percentage of CD8+CD107+ cells (), after co-culture with DC that had phagocytized HMC or MPM cells. Conversely, BBIs increased all these parameters in patient-derived MPM cells, without significant differences across histotypes (; Fig. S3F-H).
Figure 3. JQ1 primes MPM cells for immunogenic cell death and raises an adaptive anti-tumor immune response. (A, B) ATP release was measured by a chemiluminescent-based assay, HMGB1 release was measured by ELISA in the supernatant of cells incubated for 6 days in medium containing DMSO (ctrl) or 250 nM JQ1. Pooled data of MPM samples (epi: epithelioid; bip: biphasic; sar: sarcomatoid) are means±SEM. For both panels: **p < 0.01: MPM cells vs HMC ctrl; °p < 0.05: JQ1-treated cells vs respective untreated cells. (C, D) Representative histograms of surface calreticulin (CRT) and ERp57, obtained by flow cytometry, in cells incubated as indicated in (A, B). The percentage of CRT/ERp57-positive cells treated with JQ1 is indicated. Mean fluorescence intensity±SEM of pooled data for CRT: HMC ctrl: 3.3±1.2; HMC JQ1: 5.1±2.1; epi MPM ctrl: 2.1±0.6; epi MPM JQ1: 18.9±1.9; bip MPM ctrl: 2.0±0.7; bip MPM JQ1: 25.7±2.5; sar MPM ctrl: 2.3±0.8; sar MPM JQ1: 21.3±2.1. Mean fluorescence intensity±SEM of pooled data for ERp57: HMC ctrl: 2.3±0.2; HMC JQ1: 3.3±1.0; epi MPM ctrl: 2.5±0.4; epi MPM JQ1: 15.9±2.9; bip MPM ctrl: 3.2±0.5; bip MPM JQ1: 21.4±2.8; sar MPM ctrl: 2.8±0.4; sar MPM JQ1: 18.9±3.4. Significance for both CRT and ERp57: p < 0.001: MPM cells vs HMC ctrl; p < 0.001: JQ1-treated cells vs respective untreated cells. Blank: non-immune isotypic antibody. (E) Cells were incubated as indicated in (A, B), then stained with PKH2-Green FITC, incubated at 1:1 ratio for 18 h at 37 °C with DC labelled with an anti-HLA-DR antibody (APC-conjugated). PKH2-Green FITC/HLA-DR APC-positive cells, i.e. DC that have phagocytized MPM cells, were counted by flow cytometry. Pooled data of MPM samples are means±SEM. *p < 0.05; **p < 0.01; ***p < 0.001: MPM cells vs HMC ctrl; °°°p < 0.001: JQ1-treated cells vs respective untreated cells. (F) The amount of proliferating T-cells co-cultured 6 days with HMC or MPM cells, treated as reported in (A, B), was measured by labelling them with [3H]-thymidine. As positive control of proliferation, PBMC were treated with the anti-CD3 and anti-CD28 antibodies; as negative control, the PBMC were grown in medium alone. Pooled data of MPM samples are means±SEM. *p < 0.05; **p < 0.01: MPM cells vs HMC ctrl; °°°p < 0.001: JQ1-treated cells vs respective untreated cells. (G) After 10 days of co-culture with DC that have phagocytized HMC or MPM cells, IFN-γ was measured by ELISA in the supernatant of CD3+ CD8+ T-cells. Pooled data of MPM samples are means±SEM. *p < 0.05; **p < 0.01; ***p < 0.001: MPM cells vs HMC ctrl; °°°p < 0.001: JQ1-treated cells vs respective untreated cells. (H) Representative dot plots of CD8+ CD107+ T-lymphocytes, isolated from T-cells co-cultured with DC as reported in (G), determined by flow cytometry. Mean fluorescence intensity±SEM of pooled data: HMC ctrl: 2.4±0.2; HMC JQ1: 3.0±0.3; epi MPM ctrl: 1.3±0.4; epi MPM JQ1: 6.3±0.5; bip MPM ctrl: 1.5±0.3; bip MPM JQ1: 6.2±0.8; sar MPM ctrl: 1.4±0.1; sar MPM JQ1: 8.2±1.2. Significance: p<0.001: MPM cells vs HMC ctrl; p < 0.001: JQ1-treated cells vs respective untreated cells.
![Figure 3. JQ1 primes MPM cells for immunogenic cell death and raises an adaptive anti-tumor immune response. (A, B) ATP release was measured by a chemiluminescent-based assay, HMGB1 release was measured by ELISA in the supernatant of cells incubated for 6 days in medium containing DMSO (ctrl) or 250 nM JQ1. Pooled data of MPM samples (epi: epithelioid; bip: biphasic; sar: sarcomatoid) are means±SEM. For both panels: **p < 0.01: MPM cells vs HMC ctrl; °p < 0.05: JQ1-treated cells vs respective untreated cells. (C, D) Representative histograms of surface calreticulin (CRT) and ERp57, obtained by flow cytometry, in cells incubated as indicated in (A, B). The percentage of CRT/ERp57-positive cells treated with JQ1 is indicated. Mean fluorescence intensity±SEM of pooled data for CRT: HMC ctrl: 3.3±1.2; HMC JQ1: 5.1±2.1; epi MPM ctrl: 2.1±0.6; epi MPM JQ1: 18.9±1.9; bip MPM ctrl: 2.0±0.7; bip MPM JQ1: 25.7±2.5; sar MPM ctrl: 2.3±0.8; sar MPM JQ1: 21.3±2.1. Mean fluorescence intensity±SEM of pooled data for ERp57: HMC ctrl: 2.3±0.2; HMC JQ1: 3.3±1.0; epi MPM ctrl: 2.5±0.4; epi MPM JQ1: 15.9±2.9; bip MPM ctrl: 3.2±0.5; bip MPM JQ1: 21.4±2.8; sar MPM ctrl: 2.8±0.4; sar MPM JQ1: 18.9±3.4. Significance for both CRT and ERp57: p < 0.001: MPM cells vs HMC ctrl; p < 0.001: JQ1-treated cells vs respective untreated cells. Blank: non-immune isotypic antibody. (E) Cells were incubated as indicated in (A, B), then stained with PKH2-Green FITC, incubated at 1:1 ratio for 18 h at 37 °C with DC labelled with an anti-HLA-DR antibody (APC-conjugated). PKH2-Green FITC/HLA-DR APC-positive cells, i.e. DC that have phagocytized MPM cells, were counted by flow cytometry. Pooled data of MPM samples are means±SEM. *p < 0.05; **p < 0.01; ***p < 0.001: MPM cells vs HMC ctrl; °°°p < 0.001: JQ1-treated cells vs respective untreated cells. (F) The amount of proliferating T-cells co-cultured 6 days with HMC or MPM cells, treated as reported in (A, B), was measured by labelling them with [3H]-thymidine. As positive control of proliferation, PBMC were treated with the anti-CD3 and anti-CD28 antibodies; as negative control, the PBMC were grown in medium alone. Pooled data of MPM samples are means±SEM. *p < 0.05; **p < 0.01: MPM cells vs HMC ctrl; °°°p < 0.001: JQ1-treated cells vs respective untreated cells. (G) After 10 days of co-culture with DC that have phagocytized HMC or MPM cells, IFN-γ was measured by ELISA in the supernatant of CD3+ CD8+ T-cells. Pooled data of MPM samples are means±SEM. *p < 0.05; **p < 0.01; ***p < 0.001: MPM cells vs HMC ctrl; °°°p < 0.001: JQ1-treated cells vs respective untreated cells. (H) Representative dot plots of CD8+ CD107+ T-lymphocytes, isolated from T-cells co-cultured with DC as reported in (G), determined by flow cytometry. Mean fluorescence intensity±SEM of pooled data: HMC ctrl: 2.4±0.2; HMC JQ1: 3.0±0.3; epi MPM ctrl: 1.3±0.4; epi MPM JQ1: 6.3±0.5; bip MPM ctrl: 1.5±0.3; bip MPM JQ1: 6.2±0.8; sar MPM ctrl: 1.4±0.1; sar MPM JQ1: 8.2±1.2. Significance: p<0.001: MPM cells vs HMC ctrl; p < 0.001: JQ1-treated cells vs respective untreated cells.](/cms/asset/89ee5389-bc0e-4e0f-8109-e635edf57328/koni_a_1398874_f0003_oc.gif)
BBI treatment of patient-derived MPM cells promotes the expansion of CD8+ T-lymphocytes, the reduction of myeloid derived suppressor cells (MDSC) and modulates the expression of PD-1 and PD-L1 in lymphocytes and MPM cells
Comparison of the immune phenotype of peripheral blood mononuclear cells (PBMC) co-cultured with HMC or MPM cells revealed that MPM cells decreased the number of CD8+ T-lymphocytes, and increased the amount of T-regulatory cells (Treg), granulocytic-derived (Gr-MDSC) and monocytic-derived myeloid derived suppressor cells (Mo-MDSC) (, , Table S3). These data suggested that patient-derived MPM cells primed immune cell populations to an immune-suppressive rather than an immune-active environment. Interestingly, treatment with either JQ1 or OTX015 counteracted the immune-suppressive potential of MPM cells, increasing CD8+ T-lymphocytes (, , Table S3) and decreasing Gr-MDSC and Mo-MDSC ( and , , Table S3).
Figure 4. JQ1 prevents the decrease of CD8+ T-lymphocytes and the increase of granulocytic-/monocytic-derived myeloid derived suppressor cells induced by MPM cells. (A) Representative dot plots of CD3+ CD8+ T-lymphocytes, isolated from PBMC after 6-days co-culture with HMC or MPM cells (epi: epithelioid; bip: biphasic; sar: sarcomatoid), in medium containing DMSO (ctrl) or 250 nM JQ1, as per flow cytometry. (B) Representative dot plots of Gr-MDSC, isolated from HLA-DR− CD14− CD11b+ PBMC co-cultured as in (A). To identify Gr-MDSC, first HLA-DR− CD11b+ cells were gated (R1), then CD14− CD11b+ cells (R2 on R1, not shown). This cell population was used to identify CD11b+ CD15+ cells. Gating of CD11b+ CD15+ is shown. (C) Representative dot plots of Mo-MDSC, isolated from HLA-DR− CD15low CD11b+ PBMC co-cultured as in (A). Specifically to identify Mo-MDSC, first HLA-DR− CD11b+ cells (R1) were gated, then CD15low CD11b+ cells (R2 on R1, not shown). This cell population was used to identify CD11b+ CD14+ cells. Gating of CD11b+ CD14+ cells is shown.
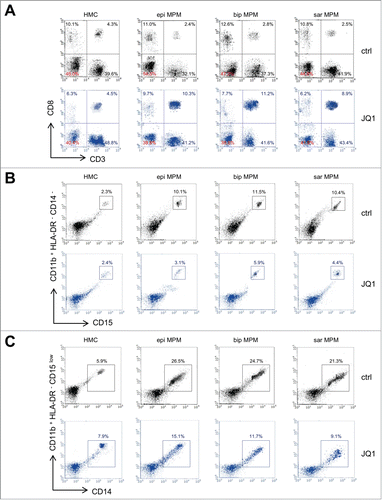
Table 1. Immune phenotype analysis of immune cells co-cultured with HMC and MPM cells treated with JQ1.
The expression of the immune checkpoints on CD8+ and CD4+ T-lymphocytes (e.g. PD-1, CTLA-4, TIM-3 and LAG-3), plays a key role in MPM-induced immune suppression.Citation4 Indeed, CD8+ T-lymphocytes co-cultured with patient-derived MPM cells showed an increased expression of PD-1, CTL-4 and LAG-3 ( and , ). Notably, JQ1-treated MPM cells reduced the proportion of PD-1and LAG-3 positive CD8+ ( and , ) and CD4+ (Table S4) T-lymphocytes. Although PD-L1 and LAG-3 were expressed at higher levels in MPM than in HMC, JQ1 reduced both markers at levels comparable to HMC cells (, ). Treatment of MPM cells with OTX015 induced the same modulation of immune checkpoints on CD8+ and CD4+ T-lymphocytes (Tables S5 and S6), as well as in tumor cells (Table S7).
Figure 5. JQ1 reduces the levels of PD-1/PD-L1 and LAG-3 in CD8+ T-lymphocytes and in MPM cells. (A, B) Representative dot plots of CD8+ PD-1+ and CD8+ LAG-3+ T-lymphocytes, isolated from PBMC after 6-days co-culture with HMC or MPM cells (epi: epithelioid; bip: biphasic; sar: sarcomatoid), in medium containing DMSO (ctrl) or 250 nM JQ1, determined by flow cytometry. (C) Representative dot plots of PD-L1+ and LAG-3+ HMC or MPM cells, incubated for 6 days in medium containing DMSO (ctrl) or 250 nM JQ1.
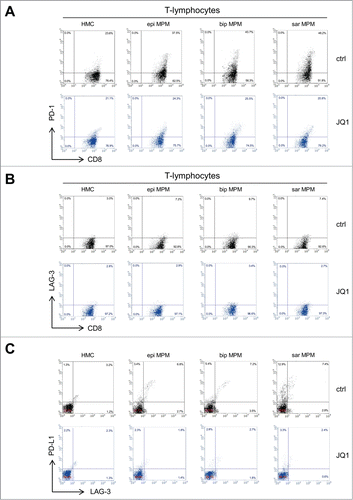
Table 2. Immune checkpoints expression on CD3+ CD8+ T-lymphocytes co-cultured with HMC and MPM cells treated with JQ1.
Table 3. Immune checkpoints expression on HMC and MPM cells treated with JQ1.
JQ1 reduces tumor growth and immunosuppressive tumor-infiltrating cells in vivo
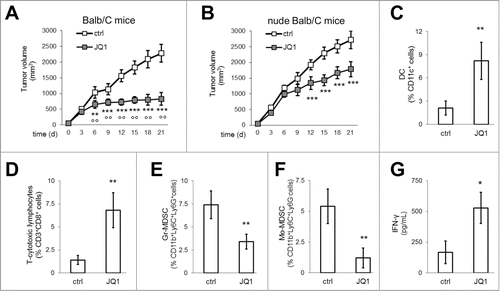
The efficacy of BBI was finally evaluated against murine MPM AB1 cells, implanted in syngeneic immunocompetent or immunodeficient Balb/C mice. AB1 derived tumors grew more rapidly in nude Balb/C mice than in immunocompetent hosts ( and ). JQ1 was particularly effective in restraining tumor growth in immunocompetent animals ( and ), suggesting that BBI activity could be partly ascribed to modulation of the immune system. Accordingly, flow cytometry analysis of intra-tumor immune infiltrate revealed that JQ1 increased the amount of DC (, Fig. S4A) and CD8+ T-lymphocytes (, Fig. S4B), and reduced the amount of Gr-MDSC (, Fig. S4C) and Mo-MDSC (, Fig. S4C). The production of IFN-γ from draining lymph nodes was also increased (). Notably, JQ1 did not elicit signs of myelosuppression or liver, kidney and heart toxicity (Table S8).
Figure 6. JQ1 delays MPM growth and reduces the immune-suppressive tumor-infiltrating cells. (A, B) Six week-old female immunocompetent or nude Balb/C mice were inoculated subcutaneously with 1 × 107 AB1 cells and treated as detailed under Materials and methods. Tumor growth data are means±SEM (n = 15/group). **p < 0.01; ***p < 0.001: JQ1-treated vs. untreated animals; °°p < 0.01: JQ1-treated immunocompetent animals vs JQ1-treated nude animals. (C-F) After digestion of tumors from immunocompetent mice, single cell suspension was analyzed by flow cytometry to measure the percentage of DC, CD3+ CD8+ T-lymphocytes, Gr-MDSC, Mo-MDSC. Data are means±SEM. **p < 0.01: JQ1-treated animals vs untreated animals. (G) IFN-γ was measured by ELISA in the supernatant of tumor-draining lymph nodes of immunocompetent mice. Data are means±SEM. *p < 0.05: JQ1-treated animals vs untreated animals.
Discussion
Resistance to conventional chemotherapy, lack of effective targeted therapies, low antigenicity of MPM and its ability to induce an immune-suppressive environment suggest that novel therapeutic strategies, including epigenetic drugs, should be explored to treat MPM patients. Histone deacetylase and DNA methyltransferase inhibitors have been evaluated in clinical trials in combination with chemotherapy, obtaining only a low rate of partial response associated to a high degree of toxicity.Citation21
Since BBIs have well documented activities on both tumor and immune system cells, we hypothesized that they could represent a novel potential therapeutic option in MPM. Data released from TCGA and analysis of our series of primary MPM samples indicate that several BRD members are overexpressed in MPM compared to HMC. In a screening of more than 650 cancer cell lines treated with JQ1, cells were classified as “JQ1-sensitive” if their IC50 was lower than 1 µM.Citation11 Since JQ1 reduced MPM cell proliferation and induced cell cycle arrest at nanomolar concentrations, MPM cells may be reasonably considered sensitive to the drug. Differently from previous reports that tested JQ1 in the range of 0.5–5 µM,Citation10,11 in this work the reduction of cell proliferation was not paralleled by increased apoptosis. Accordingly, also the treatment at nanomolar concentrations with OTX015 was associated to a very modest apoptotic index. It can be argued that the induction of apoptosis is a concentration-dependent event and that 250 nM of BBIs is below a putative “pro-apoptotic” threshold. Alternatively, it can be hypothesized that different mechanisms of cell death are involved. ICD is a process that makes dying tumor cells visible to the immune system, following stress events such as chemotherapy or radiotherapy that induce endoplasmic reticulum (ER) stress and/or alter autophagy mechanisms. This is associated to ATP and HMGB1 release in the extracellular environment, and to the exposure on the cell surface of ER-residing proteins, such as calreticulin and ERp57. All these signals contribute to recruitment and activation of local DC to remove dying cancer cells.Citation18 MPM cells are known to be refractory to chemotherapy-induced ICD.Citation19 Of note, both JQ1 and OTX015 BBIs overcame such refractoriness and induced a typical ICD signature in MPM cells, increasing DC-mediated phagocytosis and the subsequent expansion of anti-tumor CD8+ T-lymphocytes characterized by cytotoxic activity. It has already been reported that JQ1 activates antigen-presenting cells against melanoma,Citation22 a tumor with high immunogenicity. Our results are particularly relevant because MPM is a poorly immunogenic tumor.Citation2,3 Moreover, MPM-infiltrating DC are defective in presenting tumor antigens and inducing a CD8+-mediated anti-tumor response.Citation23 Interestingly, both BBIs spared not-transformed mesothelial cells from ICD. The differential expression of BRD between HMC and MPM cells may explain such selectivity, and may represent an advantage for using BBIs in MPM.
MPM is associated to an immune-suppressive rather than immune-active micro-environment, as documented by increased amount of anergic CD4+ and CD8+ T-lymphocytes, Treg and MDSC.Citation24–26 Our experimental data from MPM/PBMC co-cultures well fit with the findings from the analysis of the immune infiltrate in murine models and MPM patients. Indeed, compared to HMC, patient-derived MPM cells decreased the amount of CD8+ T-lymphocytes, and increased the amount of Treg, Gr-MDSC and Mo-MDSC. Importantly, BBIs modified two critical cell populations in the immune-environment associated to MPM. First, BBI-treated MPM cells showed an increase in CD8+ T-lymphocytes. Second, BBIs significantly reduced the percentage of Gr-MDSC and Mo-MDSC that are critical in sustaining MPM progression.Citation27 Since active anti-MPM CD8+ T-lymphocytes induce the apoptosis of MDSC,Citation28 BBIs likely induced a virtuous circle: by increasing MPM cell immunogenicity and priming it for ICD, the drugs activate anti-tumor CD8+ clones that eliminate MDSC; in turn, the reduction of MDSC rescues the cytotoxic functions of CD8+ T-lymphocytes and restores an anti-tumor immune-environment. Recently, the expression of immune checkpoints on T-lymphocytes and of their respective ligands on MPM cells emerged as a critical mechanism at the basis of MPM-induced immunesuppression.Citation4 Consistently, we found that both CD4+ and CD8+ T-lymphocytes co-cultured with patient-derived MPM cells had increased expression of PD-1, CTLA-4 and LAG-3 compared to lymphocytes co-cultured with HMC. PD-L1 and LAG-3 were also more expressed on MPM cells than on HMC. The prognostic implications of PD-1/PD-L1 axis in MPM are well characterized.Citation29 For instance, PD-L1 expressing MPM have a worst prognosisCitation29,30 and are characterized by an increased number of immune-suppressive Treg and anergic PD-1/TIM-3-positive CD4+ and CD8+ T-lymphocytes.Citation31 Interestingly, PD-L1 is a direct target of BRD4 and JQ1 has been identified as the most effective BBI in reducing PD-L1 transcription in ovarian cancer and increasing the anti-tumor activity of CD8+ T-lymphocytes.Citation32 Also in our experimental MPM models, JQ1 and OTX015 significantly reduced PD-L1 expression in MPM cells, as well as PD-1 expression in co-cultured CD8+ and CD4+ T-lymphocytes. The disruption of this biological circuit may further contribute to overcome the immune anergy induced by MPM cells.
The high expression of BRD members may underlie the efficacy of BBIs in our series of primary MPM samples. Our results indicate that the broad activity of BBIs seen in MPM models is not related to specific clinical or histological features of MPM patients from which they were derived. However, we acknowledge that our MPM series, although representative of the three histotypes and of the main clinical and pathological features of MPM, are rather limited and can potentially lead to data over interpretation. Expansion of this collection of MPM patient-derived models will help to identify potential unresponsive tumors and characterize the molecular bases of refractoriness to BBIs.
A limitation of this study may be related to the challenge of MPM cells with PBMC of healthy donors. On one hand, our results may provide useful indications about the alterations induced by MPM on a healthy immune system and about the rescuing activity of BBIs. However, our work cannot predict the effect of BBIs on the immune infiltrate of MPM patients that is known to change during MPM progression.Citation33 To partially overcome this limitation, we measured the effects of JQ1 on local immune system in a preclinical model of MPM. JQ1 resulted significantly more effective against MPM growing in immunocompetent rather than in immunodeficient animals. These results suggest that a significant fraction of JQ1 effect was due to the restoration of a proper anti-tumor immune activity. Murine MPM growth is characterized by a first phase of progressive increase of Treg cells that suppress T-lymphocytes functions, followed by a second phase of progressive increase of MDSC.Citation27 MDSC are well detectable within MPM of untreated Balb/C mice, suggesting that our model mirrors an advanced stage of MPM. The intratumor immune infiltrate profiling of JQ1-treated animals recapitulates the events induced by the drug in ex vivo assays, i.e. the increase of DC and CD8+ T-lymphocytes, and the reduction of Gr-MDSC and Mo-MDSC. The high ratio of CD8+ T-lymphocytes/MDSC observed in JQ1-treated animals indicates a clear shift from an immune-suppressive to an immune-active environment. Indeed, the higher production of IFN-γ from tumor-draining lymph nodes confirmed the presence of active cytotoxic CD8+ T-lymphocytes. Collectively, our results well reconcile with the experimental observation that JQ1, in combination with histone deacetylase inhibitors, fosters a T cell-mediated anti-tumor immunity against non-small cell lung cancer.Citation34
In summary, we demonstrated that BBIs induce the reduction of MPM cell proliferation and the reversion of the MPM-induced immune-suppression. Strategies combining epigenetic drugs and immunotherapyCitation35 are under investigations, and several immunotherapy-based phase II clinical trials for MPM treatment are open (https://clinicaltrials.gov). BBIs are relatively well-tolerated compared to other epigenetic drugsCitation36 and may be reasonably included among the novel agents to be further tested in combination therapy in MPM.
Materials and methods
Chemicals
Cell culture plasticware was obtained from Falcon. Electrophoresis reagents were obtained from Bio-Rad Laboratories. The protein content of cell lysates was assessed with the BCA kit from Sigma Chemicals Co. JQ1 and OTX015 were purchased from Tocris Bioscience and Selleckchem, respectively, and both drugs were dissolved in dimethyl sulfoxide (DMSO). Untreated cells were incubated with the same concentration of DMSO. Unless specified otherwise, all reagents were purchased from Sigma Chemicals Co.
Cells
HMC were isolated from two patients with pleural fluid secondary to congestive heart failure, with no history of malignant disease. MPM cells were obtained after written informed consent from the Biologic Bank of Malignant Mesothelioma, S. Antonio e Biagio Hospital (Alessandria, Italy). MPM samples, identified with an Unknown Patient Number (UPN), were used within passage 6. The Ethical Committee of the S. Antonio e Biagio Hospital in Alessandria approved the study (#9/11/2011). Murine AB1 cells were purchased from Sigma Chemicals Co. (#10092305), and authenticated by microsatellite analysis using the PowerPlex kit (Promega Corporation; last authentication: January 2017). Cells were grown in Ham's F12 nutrient mixture medium (primary MPM cells) or DMEM (AB1 cells), supplemented with 10% v/v fetal bovine serum (FBS), 1% v/v penicillin-streptomycin. Cells were checked for Mycoplasma spp. contamination by PCR every three weeks; contaminated cells were discharged.
Immune-phenotype of primary MPM cells
The mesothelial origin of the isolated cells was confirmed by positive immune-staining, after cell centrifugation (1 200 × g for 5 min) and fixation in 4% v/v formalin at 4 °C overnight, using the following antibodies: calretinin (1:100, rabbit polyclonal #RB-9002-R7, Thermo Fisher Scientific), Wilms tumor-1 antigen (WT1, 1:10, mouse clone 6FH2, Thermo Fisher Scientific), cytokeratin 5 (1:100, mouse clone D5, ImPath, Menarini Diagnostics), podoplanin (1:150, mouse clone D2-40, Dako), pancytokeratin (1:500, mouse clone AE1/AE3, Dako), epithelial membrane antigen (EMA, 1:6000, mouse clone E29, Dako), carcino-embryonic antigen (CEA, 1:15000, rabbit polyclonal #IR52661-2, Dako). Only cells positive for at least one mesothelial antigen among calretinin, WT1, podoplanin and cytokeratin 5 or positive for pancytokeratin, were considered for subsequent experiments and included in the study.
BRD expression
RNA was extracted from cells using TRIzol (Invitrogen). 1 µg of total RNA was used for reverse transcription with iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed with iQ SYBR Green (Bio-Rad) using the following primers: BRD2: for:5′-GGAAGATGAGGAGGACGAGG-3’; rev:5′-TGGGCTTGGATATTGGACCC-3′; BRD3: for:5′-GAGAGTACCCAGACGCACAG-3′; rev:5′-TCTCAAACACGTCCTGGAGC-3′; BRD4: for:5′-ATACCTGCTCAGAGTGGTGC-3′; rev:5′-TGTTCCCATATCCATAGGCGT-3′; BRD9: for:5′-GACGCTGATGAGGAGGAGAC-3′; rev: 5′-GCAGTAGTGGCACTGGAGAG-3′; HuPO: for:5′-GCTTCCTGGAGGGTGTCC-3′; rev:5′-GGACTCGTTTGTACCCGTTG-3′. Real-time PCR parameters were as follows: cycle 1: 95 °C for 3 minutes; cycle 2: 95 °C for 15 seconds, 60 °C for 30 seconds (40 cycles). The 2-DDCT method was employed to analyze the data. HuPO expression was used to normalize the results.
Cell proliferation analysis
For long-term cell proliferation assays, 2 × 103 cells were seeded in each well of a 24-well plate in complete growth media containing the indicated JQ1 or OTX015 concentrations. After 10 days, medium was aspirated, cells were fixed and stained with 5% w/v crystal violet solution in 66% v/v methanol, washed and analyzed under bright field Olympus IX73 microscope (Olympus Corporation), equipped with the CellSense Dimension imaging system (10 × objective; 10 × ocular lens). For short-term proliferation assay, cells were plated in 96-well plates at a density of 2 × 103 per well. Proliferation was evaluated by CellTiter-Glo (Promega). Proliferation at day 0 was considered as 100%; the results were expressed as percentage of proliferation vs day 0.
Cell cycle analysis
Cells were plated at a density of 6 × 104 in 6-well plates. Cells were harvested, washed with PBS, treated with RNAse (0.25 mg/ml) and stained for 15 min with propidium iodide (50 μg/ml). The cell-cycle distribution G0/G1, S, and G2/M was analyzed by FACScan cell sorter (Becton Dickinson) equipped with CellQuest software (Becton Dickinson).
Apoptosis
Cells were plated at a density of 6 × 104 in 6-well plates. After 3 days, floating and harvested cells were washed with PBS and stained for 15 min with 200 nM tetramethylrhodamine methyl ester perchlorate (TMRM). The percentage of apoptotic cells was analyzed by FACScan using the CellQuest Software.
ATP and HMGB1 release
The amount of extracellular ATP was measured with the ATP Bioluminescent Assay Kit (FL-AA, Sigma Chemicals Co.), using a Synergy HT Multi-Detection Microplate Reader (Bio-Tek Instruments). ATP was quantified as arbitrary light units; data were converted into nmoles/mg proteins. The extracellular release of the HMGB1 was measured with the High Mobility Group Protein 1 ELISA kit (Cloud-Clone Corp.). Results were expressed in pg/mg total cellular proteins.
Flow cytometry analysis
Cells were washed with PBS, detached with Cell Dissociation Solution (Sigma Chemicals Co.) and re-suspended at 5 × 105 cells/ml in culture medium containing 5% v/v FBS. Cells were incubated for 45 min at 4oC with anti-calreticulin (1:100, rabbit polyclonal, #PA3-900, Affinity Bioreagents) or anti-ERp57 (1:100, rabbit polyclonal, #PA3-009, Thermo Fisher Scientific) in 0.25% w/v bovine serum albumin (BSA)-PBS, washed, incubated with the secondary fluorescein isothiocyanate (FITC)-conjugated antibody (1:50, Sigma Chemicals Co.) for 30 min at 4oC, washed and fixed with 2.5% v/v paraformaldehyde. 1 × 105 cells were analyzed by a Guava® easyCyte flow cytometer (Millipore), using the InCyte software (Millipore). Control experiments included incubation of cells with non-immune isotypic antibody, followed by secondary antibody.
Phagocytosis
DC were generated from peripheral blood samples obtained from healthy donors provided by Blood Bank of AOU Città della Salute e della Scienza, Torino, Italy (#DG-767/2015), as previously reported.Citation19 The percentage of phagocytized cells at 4 °C was less than 5% than the phagocytized cells at 37 °C (not shown). Phagocytosis rate was expressed as phagocytic index, calculated as previously reported.Citation37
T-lymphocyte proliferation in HMC/MPM co-cultures
1 × 106/ml human PBMC, isolated from buffy coats of healthy donors (Blood Bank, Città della Salute e della Scienza di Torino Hospital, Torino, Italy) by centrifugation on Ficoll-Hypaque density gradient, were treated with anti-CD3 (1:2 000, mouse clone OKT3, BioLegend) and anti-CD28 (1:500, mouse clone 37.51, BioLegend) antibodies, to induce the specific proliferation of T-lymphocytes. PBMC were co-cultured at an effector/target (HMC or MPM cells) ratio of 10:1 for 6 days. The proliferation of T-lymphocytes was assessed by adding 1 μCi of [3H]thymidine (PerkinElmer) 18 h before the end of the co-cultures, then harvesting the plates and counting the radioactivity.
T-lymphocyte activation
After MPM cell phagocytosis, DC were washed and co-cultured 10 days at a 1:5 ratio with autologous T-lymphocytes, isolated by immuno-magnetic sorting with the Pan T Cell Isolation Kit (Miltenyi Biotec.). The percentage of CD8+CD107+T-lymphocytes, indicative of active anti-tumor cytotoxic T-lymphocytes, was determined by flow cytometry.Citation19 IFN-γ in the culture supernatant of CD8+T-cells co-cultured with DC or in the supernatant of tumor-draining lymph nodes was measured with the Human IFN-γ DuoSet Development Kit (R&D Systems). Results were expressed as ng/mg cell proteins or pg/ml, according to the respective calibration curves.
Immune phenotyping
PBMC isolated from buffy coats as indicated above, were incubated for 6 days with HMC or MPM cells, then harvested, washed and re-suspended in PBS containing 5% v/v FBS. A three- and four-color flow cytometry was performed on 1 × 106 cells, with the appropriate combinations of antibodies (all diluted 1:10, Miltenyi Biotec.) for: CD3 (mouse clone REA613), CD4 (mouse clone M-T466), CD8 (mouse clone BW135/80) for T-lymphocytes; CD56 (mouse clone AF127H3), CD335/NKp46 (mouse clone 9E2) for Natural Killer (NK) cells; CD4 (mouse clone M-T466), CD25 (mouse clone 4E3), CD127 (mouse clone MB1518C9) for Treg cells; CD11b (rat clone M1/70.15.11.5), CD14 (mouse clone TÜK4), CD15 (mouse clone VIMC6), HLADR (mouse clone AC122) for Gr-MSDC and Mo-MDSC; CD14 (mouse clone TÜK4) and CD68 (mouse clone Y1/82A) for monocytes and macrophages. The analysis of immune infiltrate of murine MPM was performed by using antibodies recognizing: CD11c (hamster clone N418, Miltenyi Biotec.) for DC; CD3 (mouse clone REA641, Miltenyi Biotec.) and CD8 (rat clone 53–6.7, Miltenyi Biotec.) for T-lymphocytes; CD11b (rat clone M1/70.15.11.5, Miltenyi Biotec.), Ly6C (rat clone AL-21, BD Biosciences), Ly6G (rat clone 1A8, BD Biosciences) for Gr-MDSC and Mo-MDSC. Cells were analyzed using a Guava® easyCyte flow cytometer (Millipore), equipped with the InCyte software.
Immune check-point detection
CD3+ cells were isolated from 1 × 106 PBMC co-cultured with HMC or MPM cells for 6 days, with the Pan T Cell Isolation Kit (Miltenyi Biotec.), washed and re-suspended in PBS containing 5% v/v FBS. The detection of immune checkpoints on T-lymphocytes and/or immune checkpoint ligands on MPM cells were performed using antibodies for CD279/PD-1 (mouse clone PD1.3.1.3), CD223/LAG-3 (mouse clone REA351), CD366/TIM-3 (mouse clone F38-2E2), CD152/CTLA-4 (mouse clone BNI3; all diluted 1:10, Miltenyi Biotec.), CD274/PD-L1 (1:1 000, mouse clone 29E.2A3, BioLegend). 1 × 105 cells were analyzed by as reported above.
In vivo tumor growth, immune-environment analysis and hematochemical parameters
1 × 107 AB1 cells, mixed with 100 µL Matrigel, were injected subcutaneously in 6 weeks-old female immunocompetent or nude Balb/C mice (Charles River Laboratories), housed (5 per cage) under 12 h light/dark cycle, with food and drinking provided ad libitum. Tumor growth was measured daily by caliper and was calculated according to the equation (LxWCitation2)/2, where L = tumor length; W = tumor width. When the tumor reached the volume of 50 mm3 (day 15 after injection), mice were randomized into 2 groups, treated intraperitoneally twice a week for 3 consecutive weeks, as follows: 1) control group, treated with 0.1 ml saline solution; 2) JQ1 group, treated with 0.1 ml JQ1 (in 1:10 sterile saline solution/DMSO solution; final dosage: 50 mg/kg). Tumor volumes were monitored daily by caliper and animals were euthanized with zolazepam (0.2 ml/kg) and xylazine (16 mg/kg) at the end of treatment. Tumor were excised, cut into 1 mm3-pieces and digested (in DMEM medium containing 1 mg/ml collagenase and 0.2 mg/ml hyaluronidase) for 1 h at 37 °C. The material was filtered in a syringe using a 70 µm-cell strainer to obtain a single cell suspension, and washed in DMEM. Infiltrating immune cells were collected by centrifugation on Ficoll-Hypaque density gradient and subjected to immune phenotyping by flow cytometry. Draining lymph nodes were collected, homogenized for 30s at 15 Hz, using a TissueLyser II device (Qiagen) and centrifuged at 12 000 × g for 5 minutes. The supernatant was used to measure the amount of IFN-γ. The hemocromocytometric analyses were performed with a UniCel DxH 800 Coulter Cellular Analysis System (Beckman Coulter Inc.) on 0.5 ml of blood collected immediately after mice sacrifice; lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, creatinine, creatine phosphokinase were measured using kits from Beckman Coulter Inc.
Animal care and experimental procedures were approved by the Bio-Ethical Committee of the Italian Ministry of Health (#122/2015-PR).
Statistical analysis
All data in the text and figures are provided as means±SEM. The results were analyzed by a one-way analysis of variance (ANOVA), using Statistical Package for Social Science (SPSS) software (IBM SPSS Statistics v.19). p < 0.05 was considered significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2017ONCOIMM0553R-s02.docx
Download MS Word (27.7 MB)Acknowledgment
We are grateful to Mr. Costanzo Costamagna, Dept. of Oncology, for technical assistance.
Additional information
Funding
References
- Remon J, Reguart N, Corral J, Lianes P. Malignant pleural mesothelioma: new hope in the horizon with novel therapeutic strategies. Cancer Treat Rev. 2015;41:27–34. doi:10.1016/j.ctrv.2014.10.007. PMID:25467107
- Izzi V, Masuelli L, Tresoldi I, Foti C, Modesti A, Bei R. Immunity and malignant mesothelioma: from mesothelial cell damage to tumor development and immune response-based therapies. Cancer Lett. 2012;322:18–34. doi:10.1016/j.canlet.2012.02.034. PMID:22394996
- Aerts JG, Lievense LA, Hoogsteden HC, Hegmans JP. Immunotherapy prospects in the treatment of lung cancer and mesothelioma. Transl Lung Cancer Res. 2014;3:34–45. doi:10.3978/j.issn.2218-6751.2013.11.04. PMID:25806279.
- Marcq E, Pauwels P, van Meerbeeck JP, Smits EL. Targeting immune checkpoints: New opportunity for mesothelioma treatment? Cancer Treat Rev. 2015;41:914–924. doi:10.1016/j.ctrv.2015.09.006. PMID:26433514
- Guo G, Chmielecki J, Goparaju C, Heguy A, Dolgalev I, Carbone M, Seepo S, Meyerson M, Pass HI. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015;75:264–269. doi:10.1158/0008-5472.CAN-14-1008. PMID:25488749
- Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, Gnad F, Nguyen TT, Jaiswal BS, Chirieac LR, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48:407–416. doi:10.1038/ng.3520. PMID:26928227
- Stahel RA, Weder W, Felley-Bosco E, Petrausch U, Curioni-Fontecedro A, Schmitt-Opitz I, Peters S. Searching for targets for the systemic therapy of mesothelioma. Ann Oncol. 2015;26:1649–1660. doi:10.1093/annonc/mdv101. PMID:25722383
- de Reyniès A, Jaurand MC, Renier A, Couchy G, Hysi I, Elarouci N, Galateau-Sallé F, Copin MC, Hofman P, Cazes A, et al. Molecular classification of malignant pleural mesothelioma: identification of a poor prognosis.subgroup linked to the epithelial-to-mesenchymal transition. Clin Cancer Res. 2014;20:1323–1334. doi:10.1158/1078-0432.CCR-13-2429. PMID:24443521
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung C, Hopf C, Savitski MM, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi:10.1038/nature10509. PMID:21964340
- Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, and Weinstock DM. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120:2843–2852. doi:10.1182/blood-2012-02-413021. PMID:22904298
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi:10.1158/2159-8290.CD-12-0418. PMID:23430699
- Vázquez R, Licandro SA, Astorgues-Xerri L, Lettera E, Panini N, Romano M, Erba E, Ubezio P, Bello E, Libener R, et al. Promising in vivo efficacy of the BET bromodomain inhibitor OTX015/MK-8628 in malignant pleural mesothelioma xenografts. Int J Cancer. 2017;140:197–207. doi:10.1002/ijc.30412. PMID:27594045
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi:10.1158/2159-8290.CD-12-0095. PMID:22588877
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi:10.1126/scisignal.2004088. PMID:23550210
- Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, Jin L, Pupo G, Cullinane C, McArthur GA, et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res. 2014;27:1126–1137. doi:10.1111/pcmr.12282. PMID:24924589
- Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, Beavis PA, Darcy PK, Martin BP, Spencer A, et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017;18:2162–74. doi:10.1016/j.celrep.2017.02.011. PMID:28249162
- Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691. doi:10.4161/21624011.2014.955691. PMID:25941621
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi:10.1038/nri.2016.107. PMID:27748397
- Riganti C, Castella B, Kopecka J, Campia I, Coscia M, Pescarmona G, Bosia A, Ghigo D, and Massaia M. Zoledronic acid restores doxorubicin chemosensitivity and immunogenic cell death in multidrug-resistant human cancer cells. PLoS One. 2013;8:e60975. doi:10.1371/journal.pone.0060975. PMID:23593363
- Salaroglio IC, Campia I, Kopecka J, Gazzano E, Orecchia S, Ghigo D, et al. Zoledronic acid overcomes chemoresistance and immunosuppression of malignant mesothelioma. Oncotarget. 2015;6:1128–1142. doi:10.18632/oncotarget.2731. PMID:25544757
- Vandermeers F, Neelature Sriramareddy S, Costa C, Hubaux R, Cosse JP, Willems L. The role of epigenetics in malignant pleural mesothelioma. Lung Cancer. 2013;81:311–318. doi:10.1016/j.lungcan.2013.05.014. PMID:23790315
- Gallagher SJ, Tiffen JC, Hersey P. Histone Modifications, Modifiers and Readers in Melanoma Resistance to Targeted and Immune Therapy. Cancers (Basel). 2015;7:1959–1982. doi:10.3390/cancers7040870. PMID:26426052
- McDonnell AM, Lesterhuis WJ, Khong A, Nowak AK, Lake RA, Currie AJ, Robinson BWS. Tumor-infiltrating dendritic cells exhibit defective cross-presentation of tumor antigens, but is reversed by chemotherapy. Eur J Immunol. 2015;45:49–59. doi:10.1002/eji.201444722. PMID:25316312
- Hegmans JP, Hemmes A, Hammad H, Boon L, Hoogsteden HC, Lambrecht BN. Mesothelioma environment comprises cytokines and T-regulatory cells that suppress immune responses. Eur Respir J. 2006;27:1086–1095. doi:10.1183/09031936.06.00135305. PMID:16540497
- Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, Hegmans JP. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. 2010;10:e464. doi:10.1186/1471-2407-10-464. PMID:20804550
- Ujiie H, Kadota K, Nitadori JI, Aerts JG, Woo KM, Sima CS, Travis WD, Jones DR, Krug LM, and Adusumilli PS. The tumoral and stromal immune microenvironment in malignant pleural mesothelioma: A comprehensive analysis reveals prognostic immune markers. Oncoimmunology. 2015;4:e1009285. doi:10.1080/2162402X.2015.1009285. PMID:26155428
- Miselis NR, Lau BW, Wu Z, Kane AB. Kinetics of host cell recruitment during dissemination of diffuse malignant peritoneal mesothelioma. Cancer Microenviron. 2010;4:39–50. doi:10.1007/s12307-010-0048-1. PMID:21505561
- Yu Z, Tan Z, Lee BK, Tang J, Wu X, Cheung KW, Lo NTL, Man K, Liu L, Chen Z. Antigen spreading-induced CD8+T cells confer protection against the lethal challenge of wild-type malignant mesothelioma by eliminating myeloid-derived suppressor cells. Oncotarget. 2015;6:32426–32438. doi:10.18632/oncotarget.5856. PMID:26431275
- Khanna S, Thomas A, Abate-Daga D, Zhang J, Morrow B, Steinberg SM, Orlandi A, Ferroni P, Schlom J, Guadagni F, et al. Malignant Mesothelioma Effusions Are Infiltrated by CD3(+) T Cells Highly Expressing PD-L1 and the PD-L1(+) Tumor Cells within These Effusions Are Susceptible to ADCC by the Anti-PD-L1 Antibody Avelumab. J Thorac Oncol. 2016;11:1993–2005. doi:10.1016/j.jtho.2016.07.033. PMID:27544053
- Cedrés S, Ponce-Aix S, Zugazagoitia J, Sansano I, Enguita A, Navarro-Mendivil A, Martinez-Marti A, Martinez P, Felip E. Analysis of expression of programmed cell death 1 ligand 1 (PD-L1) in malignant pleural mesothelioma (MPM). PLoS One. 2015;10:e0121071. doi:10.1371/journal.pone.0121071. PMID:25774992
- Awad MM, Jones RE, Liu H, Lizotte PH, Ivanova EV, Kulkarni M, Herter-Sprie GS, Liao X, Santos AA, Bittinger MA, et al. Cytotoxic T Cells in PD-L1-Positive Malignant Pleural Mesotheliomas Are Counterbalanced by Distinct Immunosuppressive Factors. Cancer Immunol Res. 2016;4:1038–1048. doi:10.1158/2326-6066.CIR-16-0171. PMID:27856426
- Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, Yokoyama Y, Kossenkov AV, Bradner JE, Conejo-Garcia JR, et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016;16:2829–2837. doi:10.1016/j.celrep.2016.08.032. PMID:27626654
- Lievense LA, Bezemer K, Cornelissen R, Kaijen-Lambers ME, Hegmans JP, Aerts JG. Precision immunotherapy; dynamics in the cellular profile of pleural effusions in malignant mesothelioma patients. Lung Cancer. 2017;107:36–40. doi:10.1016/j.lungcan.2016.04.015. PMID:27168021
- Adeegbe D, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, Dries R, Li Y, Liu S, Wang X, et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non-small Cell Lung Cancer. Cancer Discov. 2017;7:852–867. doi:10.1158/2159-8290.CD-16-1020. PMID:28408401
- Hamaidia M, Staumont B, Duysinx B, Louis R, Willems L. Improvement of malignant pleural mesothelioma immunotherapy by epigenetic modulators. Curr Top Med Chem. 2016;16:777–787. doi:10.2174/1568026615666150825141152. PMID:26303419
- Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, Morschhauser F, Karlin L, Broussais F, Rezai K, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3:196–204. doi:10.1016/S2352-3026(16)00021-1. PMID:27063978
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi:10.1038/nm1523. PMID:17187072