
ABSTRACT
Despite approval for the treatment of various malignancies, clinical application of cytokines such as type I interferon (IFN) is severely impeded by their systemic toxicity. AcTakines (Activity-on-Target cytokines) are optimized immunocytokines that, when injected in mice, only reveal their activity upon cell-specific impact. We here show that type I IFN-derived AcTaferon targeted to the tumor displays strong antitumor activity without any associated toxicity, in contrast with wild type IFN. Treatment with CD20-targeted AcTaferon of CD20+ lymphoma tumors or melanoma tumors engineered to be CD20+, drastically reduced tumor growth. This antitumor effect was completely lost in IFNAR- or Batf3-deficient mice, and depended on IFN signaling in conventional dendritic cells. Also the presence of, but not the IFN signaling in, CD8+ T lymphocytes was critical for proficient antitumor effects. When combined with immunogenic chemotherapy, low-dose TNF, or immune checkpoint blockade strategies such as anti-PDL1, anti-CTLA4 or anti-LAG3, complete tumor regressions and subsequent immunity (memory) were observed, still without any concomitant morbidity, again in sharp contrast with wild type IFN. Interestingly, the combination therapy of tumor-targeted AcTaferon with checkpoint inhibiting antibodies indicated its ability to convert nonresponding tumors into responders. Collectively, our findings demonstrate that AcTaferon targeted to tumor-specific surface markers may provide a safe and generic addition to cancer (immuno)therapies.
Introduction
Interferon-alpha (IFNα) is a type I IFN (IFN), approved for the treatment of some neoplasms, including hematological (hairy cell leukemia and other lympho- and myeloproliferative neoplasms) and solid cancers (melanoma, renal cell carcinoma, Kaposi's sarcoma).Citation1 Unfortunately, IFN therapy experienced variable and unpredictable success in the clinic, and is severely limited due to side effects such as flu-like symptoms, leukopenia, anemia, hepatotoxicity, cognitive dysfunction, neurologic toxicity and depressionCitation2.
It was initially assumed that the direct inhibitory effect of IFN on tumor cells was essential. Indeed, IFNs regulate the expression of genes affecting tumor cell growth, proliferation, differentiation, survival, migration and other functions,Citation3 but it is known since a long time that the key mechanism of IFN antitumor activity is more likely indirect, via immune activation.Citation4 Several host immune cells, including dendritic cells (DC), T and B lymphocytes, Natural Killer (NK) cells and macrophages, respond to IFN and may be involved in antitumor activity.Citation3,5 Furthermore, endogenous IFN is essential for many anti-cancer therapies, including chemotherapy, radiotherapy, immunotherapies and checkpoint inhibition.Citation5
Safe exploitation of the clinical potential of IFN, and many other cytokines, requires strategies to target their activity to selected target cells only, thus avoiding systemic toxicity. One strategy to accomplish this is by developing immunocytokines, fusions of wild type (WT) cytokines coupled to antibodies recognizing cell-specific surface-expressed markers. For most immunocytokines in development, an approximately 10-fold increase in targeted activity is achieved, increasing the therapeutic index modestly.Citation6,7 Indeed, even if coupled to a targeting moiety, WT cytokines still exert unwanted effects on untargeted cells while in passage to their target, due to the ubiquitous expression of their cognate receptors. Related to the latter, WT (immuno)cytokines may also disappear from the circulation before reaching their target cells (the so-called “sink effect”).Citation8 To improve the therapeutic index of toxic cytokines, we developed AcTakines (Activity-on-Target Cytokines), optimized immunocytokines, using mutated cytokines with strongly reduced affinity for their receptor complex instead of WT cytokines. Fusing the mutated cytokine to cell-specific targeting domains selectively restores AcTakine activity on the selected cell population only. Consequently, AcTakines are much less potent to signal while traveling through the body. We have proven the basic AcTakine concept of IFN using human IFNα2 (which is not active on mouse cells) with a Q124R mutation rendering it about 100-fold less active on mouse cells than murine (m) IFNα.Citation9 We coined this mutated and targetedCitation10 IFN ‘AcTaferon’ (AFN), and evaluated its potential as a safe and generic cancer treatment in preclinical mouse models by targeting its activity to the tumor.
Results
Tumor-targeted delivery of IFN activity: mCD20–AcTaferon proof-of-principle
To study the antitumor potential of targeting type I IFN activity specifically to tumor cells, we coupled hIFNα2-Q124RCitation9 (from now on designated as ‘AcTaferon’ or AFN) to a VHH single domain antibody (sdAb) targeting mCD20 (). As control (untargeted) constructs, we used AFN coupled to sdAb targeting either hCD20, GFP or BcII10, epitopes absent in the mouse (confirmed by imaging).Citation11 CD20 is a B lymphocyte-specific antigen present on all B cell stages except early pro-B lymphocytes and plasma cells. We preferred mCD20 targeting in view of the success of CD20 antibodies (rituximab, obinutuzumab) in patients with B cell malignancies, which clearly proves the efficacy of CD20 as a tumor-associated antigen (TAA) useful for therapy. Since the frequently used CD20+ mouse B cell lymphoma A20 is highly sensitive to IFN, we chose this model to obtain initial in vivo antitumor proof-of-concept.
Figure 1. mCD20-AcTaferon proof-of-principle. (A) General lay-out of an AcTaferon (AFN). The hIFNa2-Q124R, human IFNa2 with a Q124R point mutation, active on mouse cells but at a 100-fold lower level than murine IFN, is coupled to a sdAb module recognizing mCD20, hCD20, GFP or BcII10 via a GGS linker molecule. (B) Phospho-STAT1 as a read-out for IFN signaling in spleen CD19+ B lymphocytes isolated from mice treated 45 min earlier with i.v. PBS, serial dilutions of B cell targeted mCD20-AFN or untargeted GFP-AFN. (C) Proliferation of A20 cells treated in vitro for 72 hours with serial dilutions of mCD20-AFN or GFP-AFN. Results are expressed as percentage of cells versus untreated culture. (D) Growth of s.c. inoculated A20 tumors in syngeneic Balb/c mice after treatments with PBS, mCD20-mIFN (immunocytokine) or hCD20-mIFN (untargeted mIFN), mCD20-AFN ( = targeted), hCD20-AFN (untargeted control) or mCD20-hIFN (“sdAb-only” control). Arrows indicate treatment days, starting at day 6 after tumor inoculation. Shown is a representative experiment. Error bars represent mean ± s.e.m.; ***P < 0.001 and ****P < 0.0001 compared with PBS treated animals by two-way ANOVA with Dunnett's multiple comparison test (n = 5 mice per group).
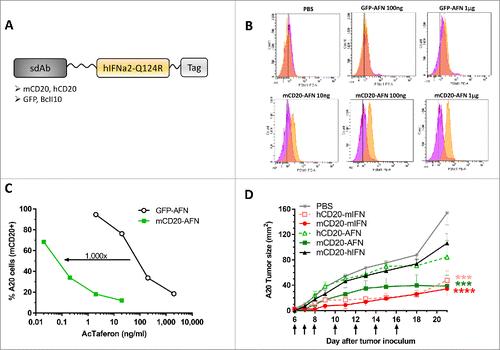
CD20-targeting of IFN activity was demonstrated in both primary CD19+ B cells, activated either in vitro (not shown) or in vivo (), as well as using CD20+ IFN-sensitive A20 cells (). For primary B cells, IFN signaling was evaluated via intracellular phospho-STAT1 determination (). Importantly, mCD20-targeting of wild type (WT) hIFN (which is not active on mouse cells) did not induce any phospho-STAT1 signal in murine B cells (Supplementary ). For A20, the anti-proliferative efficacy in vitro was determined, indicating a 1,000-fold increased activity of AFN due to targeting (). Because of this remarkable targeting efficacy, we decided to use A20 to evaluate whether mCD20-targeted AFN can be efficiently delivered to CD20+ tumors in vivo. In A20-bearing mice, treatment with mCD20-AFN prevented tumor growth () comparable to WT mIFN targeted to the tumors or not (the latter using sdAb recognizing hCD20 instead of mCD20). Untargeted hCD20-AFN or WT hIFN-coupled mCD20 sdAb (which does not have detectable IFN signaling capacity in mice) did not have a significant antitumor effect.
Targeted delivery of AcTaferon activity to the tumor host-dependently controls tumor growth without systemic toxicity
In some cancers, such as B cell malignancies, IFN exerts direct antitumor effects. Although combination therapies with anti-CD20 and IFN showed improved efficiency in B cell lymphomas, including in targeting strategies,Citation12 we envisaged the development of a non-toxic safe antitumor therapy applicable to various tumor types, including non-B cell and IFN-insensitive malignancies. Central to this concept is the observation that the antitumor efficacy of IFN may also depend on indirect effects via activation of immune cells.Citation4 Recombinant IFNα2 was in 1995 the first cytokine to be approved for the treatment of cancer, i.e. malignant melanoma. Until the approval of checkpoint inhibitors, IFN was actually the only effective adjuvant therapy for melanoma patients at high risk for recurrence and death.Citation2 Since 2011, several immunotherapies have been approved, primarily for the treatment of advanced metastatic melanoma. In view of this, we decided to evaluate the use of tumor-targeted AFNs by using the B16 melanoma model, which is not sensitive to the anti-proliferative activity of IFN, and is considered a non- or low-immunogenic tumor, reflecting the poor immunogenicity of metastatic tumors in humans, thus representing a “tougher test” for immunotherapy.Citation13,14 To enable the use of our proof-of-principle mCD20-AFN, we engineered B16-mCD20+ and B16-hCD20+ clones using CD20 as a surrogate tumor-specific surface marker.
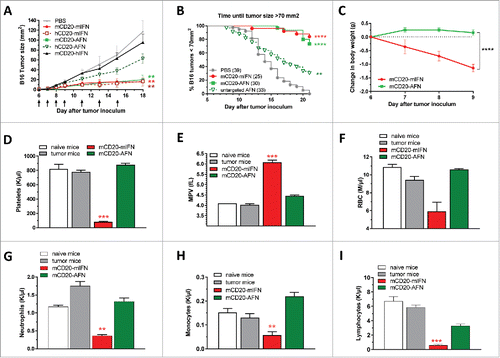
In mice bearing a B16-mCD20 tumor, mCD20-AFN treatment inhibited tumor growth comparable to WT mIFN, while untargeted hCD20-AFN only exhibited some minor, mostly non-significant, antitumor effect starting after 3–4 treatments (). Targeting WT hIFN (mCD20-hIFN) to the mCD20+ tumors did not have any effect, excluding antitumor activities induced by the anti-mCD20 sdAb on its own. Although mCD20-AFN and mIFN had comparable antitumor effects when used at the same protein concentration (), there was a dramatic difference in systemic toxicity. While mIFN caused body weight loss, severe thrombocytopenia, anemia and leukopenia, AFN therapy did not (). Reduced platelet numbers combined with increased platelet sizes, as seen after mIFN (), indicate platelet destruction.
Figure 2. Targeted AcTaferon delivery to B16 tumors controls tumor growth without toxicity. (A) Growth of s.c. inoculated B16-mCD20+ tumors in syngeneic C57BL/6J mice after treatments with PBS, mCD20-mIFN (immunocytokine) or hCD20-mIFN (untargeted mIFN), mCD20-AFN (targeted) or hCD20-AFN (untargeted), or mCD20-hIFN as a negative control for “sdAb-only” effects. Shown is a representative experiment of 7 independent repeats (n = 5 mice per group), arrows indicate treatment days. (B) Seven independent experiments were pooled to plot the time necessary for each mouse to reach a tumor of 70 mm2 (total n = indicated in the legend). (C) Body weight changes of tumor-bearing mice treated with mCD20-targeted mIFN or AFN (n = 5). (D-I) Hematological analyses (platelet counts, mean platelet volume, red blood cell, neutrophil, monocyte and lymphocyte counts) in fresh EDTA-blood collected 1 day after the last treatment. ‘Naive mice’ are tumor-free, ‘tumor mice’ are tumor-bearing treated with PBS. All values depicted are mean ± s.e.m.; *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared with PBS treated animals unless otherwise indicated; by two-way ANOVA with Dunnett's multiple comparison test (A, C), one-way ANOVA with Dunnett's multiple comparison test (D-I) or log-rank test (B). Shown are representative results of 7 independent repeats (C-I).
Bioactivity measurements of mIFN and mCD20-AFN, on murine cells which do not express mCD20, revealed that the AFN dose used for therapy was at least 1,000-fold lower than mIFN. For the representative experiment (), the doses injected were 6,000,000 and 5,500 IU for mIFN and mCD20-AFN, respectively. Injection of lower doses of WT mIFN reduced systemic toxicity concomitantly with the antitumor potential (). In contrast with lower doses (5,500 or 1,100 IU) of mCD20-AFN, lower doses of mIFN did not inhibit tumor growth, not even when targeted to the tumor as immunocytokine ().
Figure 3. Partial lymphopenia due to mCD20-AcTaferon therapy is not required for antitumor efficacy. (A) Growth of s.c. inoculated B16-mCD20+ tumors in C57BL/6J mice after treatments with PBS, or different doses of mCD20-mIFN or mCD20-AFN. Shown is a representative experiment of 2 independent repeats (n = 5 mice per group). (B-D) Lymphocyte, neutrophil and platelet counts in fresh EDTA-blood collected 1 day after the last treatment. ‘Naive mice’ are tumor-free. All values depicted are mean ± s.e.m.; *P < 0.05, **P < 0.01 compared with PBS treated animals, by two-way ANOVA with Dunnett's multiple comparison test (A), or one-way ANOVA with Dunnett's multiple comparison test (B-D).
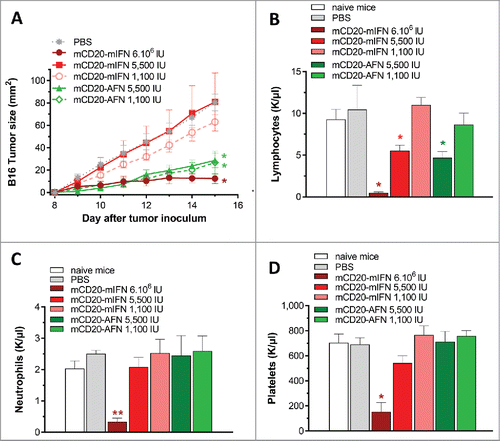
As already mentioned, AFN therapy did not cause hematological deficits. However, although not significant, lymphocyte counts were consistently lower after mCD20-AFN (). Flow cytometry revealed partial B cell depletion from circulation, and normal CD8+ and CD4+ populations, in line with specific AFN targeting to CD20+ cells (Supplementary ). Lowering the mCD20-AFN dose to 1,100 IU resulted in efficient tumor inhibition () without the partial lymphocyte depletion (), indicating that B-lymphopenia is not required for antitumor responses. To unequivocally evaluate whether B cells are involved, we employed B16-hCD20+ tumors. Treatment with hCD20-AFN was equally potent as hCD20-mIFN, without lymphopenia (Supplementary ).
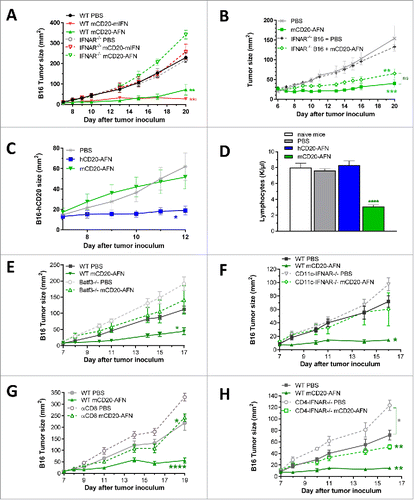
The B16 melanoma model was chosen partly because there is no anti-proliferative effect of mCD20-AFN on B16-mCD20 cells in vitro (not shown). To confirm that the in vivo antitumor effects depend on host cells, we used mice lacking functional IFNAR1 (IFNAR1−/−). As shown in , mCD20-targeted mIFN or AFN were indeed ineffective in IFNAR1−/−, in contrast to WT mice. Although these experiments indicate the crucial involvement of IFN signaling in host cells, direct effects of IFN on tumor cells that may contribute to antitumor efficacy (e.g. by releasing chemokines attracting the immune cells necessary for tumor cell eradicationCitation15) could also be involved. To evaluate this, we genetically engineered B16-mCD20+ tumor cells that lack IFNAR1. As shown in , the absence of IFNAR1 on the tumor cells did not significantly affect the antitumor effectiveness of mCD20-AFN, indicating that IFN signaling in the tumor cells is not crucial. However, mCD20-targeted AFN did not prevent tumor growth of B16-hCD20+ tumor cells, in contrast to hCD20-targeted AFN (), demonstrating that targeting to the tumor cells (and thus the tumor microenvironment) is critical. Moreover, since our hCD20 sdAb is not cross-reactive with mCD20 (not shown), the antitumor effectiveness of hCD20-AFN in the B16-hCD20 model is not accompanied by partial lymphocyte depletion in mice (). These results imply that the lymphopenia that can be observed is entirely due to anti-CD20 effects, and that the lymphopenia and IFN effects on B cells are not critically involved in the antitumor response.
Figure 4. Antitumor efficacy of tumor-targeted AcTaferon depends on cDC1 and CTL. (A) Growth of B16-mCD20+ tumors in IFNAR1-deficient versus WT mice after 7 treatments with PBS or tumor-targeted mIFN or AFN (n = 5 mice per group). (B) Growth of B16-mCD20+ or B16-mCD20+-IFNAR−/− tumors in WT mice after 10 treatments with PBS or tumor-targeted AFN (n = 12 mice per group, pooled results of 2 different IFNAR−/− B16-mCD20+ clones). (C) Growth of s.c. inoculated B16-hCD20+ tumors in C57BL/6J mice after treatments with PBS, hCD20-AFN or mCD20-AFN. Shown is a representative experiment (n = 5 mice per group). (D) Lymphocyte counts in fresh EDTA-blood collected 1 day after the last treatment of mice represented in C. ‘Naive mice’ are tumor-free. (E) Growth of B16-mCD20+ tumors in Batf3−/− mice (lacking cDC1) and WT littermates after 6 treatments with PBS or mCD20-AFN (n = 7 mice per group). (F) Growth of B16-mCD20+ tumors in CD11c-IFNAR-deficient mice (lacking IFNAR in cDC1 and cDC2) and WT littermates after 5 treatments (n = 4 mice per group). (G) Growth of B16-mCD20+ tumors in CD8-depleted mice and controls after 6 treatments (n = 5 mice per group). (H) Growth of B16-mCD20+ tumors in CD4-IFNAR-deficient mice (lacking IFNAR in T lymphocytes) and WT littermates after 5 treatments (n = 4 mice per group). All results shown are a representative of two independent repeats. Shown are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared with PBS treated animals unless otherwise indicated; determined by two-way ANOVA with Dunnett's multiple comparison test.
Tumor-targeted AcTaferon effects critically depend on DC and CTL
In the cancer-immunity cycle described by Chen and Mellman,Citation16 priming and activation of tumor-killing cytotoxic T lymphocytes (CTLs) represents a crucial step, for which activation and maturation of antigen-presenting DCs is key. A specific DC subset is essential for CTL responses in mice and men. This XCR1+ Clec9A+ conventional (c) DC1 subset, also known as CD8+ DC in mice and CD141+ or BDCA3+ DC in humans, displays superior cross-presentation capacities and requires type I IFN signaling for efficient tumor rejection.Citation17−23 As cDC1 require the Batf3 transcription factor for their differentiation, deletion of Batf3 ablates their development.Citation24 Experiments in cDC1-deficient Batf3−/− mice indicated the absolute requirement for cDC1 for the antitumor efficacy of tumor-targeted AFN (). Also in mice where type I IFN signaling is absent in cDC only (CD11c-IFNAR−/−),Citation19 mCD20-AFN could not prevent tumor growth (). The most important cells to destroy cancer cells and control tumor growth are believed to be the CD8+ CTL. They get selectively activated to recognize tumor cells by cDC1 cross-presenting tumor antigen. Indeed, depletion of CD8+ cells largely reduced mCD20-AFN antitumor efficacy (). Still, a minor but significant antitumor effect was seen in CD8-depleted conditions, suggesting the involvement of other immune cells as well. CD4 depletion, however, did not affect antitumor efficacy of mCD20-AFN (not shown). In contrast to CD11c-IFNAR−/- (), mCD20-AFN could still prevent tumor growth in mice lacking IFN signaling in T cells (CD4-IFNAR−/−), demonstrating the requirement for IFN signaling in DC rather than T lymphocytes ().Citation19
DC activation and T cell proliferation induced by targeted AcTaferon delivery
To evaluate DC activation, we analyzed different populations isolated from tumor-draining lymph nodes after treatment with mCD20-targeted or untargeted AFN. While untargeted (BcII10-coupled) AFN had a moderate effect on the expression of PDL1, MHCII, CD80, CD86 and CD40 in XCR1+ cDC1, mCD20-AFN treatment was clearly much better ( upper row). Of note, mCD20-AFN also significantly increased the activation of CD11b+ cDC2 in comparison with untargeted AFN ( lower row). Similar effects were also seen in non-tumor-draining lymph nodes (not shown).
Figure 5. DC and CTL responses during AcTaferon treatments. (A) Flow cytometric profiling of the DC activation status in the tumor draining lymph node in response to AFN treatment. DCs were identified as CD3− CD19− Ly6C− CD11cint-hi MHCIIint-hi cells and subdivided into XCR1+ cDC1 and CD11b+ cDC2. Expression levels of PDL1, MHCII, CD80, CD86 and CD40 are displayed as MFI in the respective fluorescence channels. Results shown are a representative of two independent repeats (n = 5). (B) Flow cytometric analysis of CD3+ CD8+ T cell phenotype based on the expression of CD44 and CD62L was performed on tumor-draining lymph nodes of mice bearing B16 tumors, five days after perilesional delivery of the AFNs indicated in the figure legend (n = 3). Effector T cells were identified as CD44 high and CD62L low. (C) Flow cytometric analysis of Pmel-1 T cell proliferation in the tumor-draining lymph node in response to perilesional AFN treatment of B16-mCD20 tumor-bearing mice. Data show the percentage of T cells having undergone at least one division. (D-E) Flow cytometric analysis of CD3+ CD8+ T cell phenotype based on the expression of CD44 and CD62L was performed on B16 tumors, five days after perilesional delivery of the AFNs indicated in the figure legend (n = 3). Naive cells (D) were identified as CD44 low and CD62L high, effector T cells (E) as CD44 high and CD62L low. Shown are individual values (A) and mean ± s.e.m. of a representative experiment of at least 2 independent repeats (A-E); *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared with PBS treated animals unless otherwise indicated; by one-way ANOVA with Tukey's multiple comparison test.
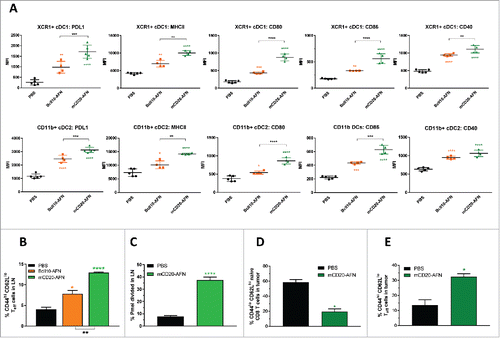
CD8+ CTLs are considered the most important cells to control tumor growth by killing cancer cells. Treatment with mCD20-tumor-targeted AFN was much more efficient than PBS or untargeted AFN to induce tumor-specific activated CD8+ effector T cells (expressing high levels of CD44 and low levels of CD62L) in lymph nodes and in the tumors themselves (). In addition, mCD20-AFN also significantly increased tumor-antigen-specific CTL proliferation in the lymph nodes ().
Complete and safe tumor eradication by tumor-targeted AcTaferon in combination treatments
The cancer-immunity cycle indicates the sequential involvement of several steps necessary for tumor eradication, requiring various immune cells and signals, and possibilities to interact or influenceCitation16. First of all, we examined whether chemotherapy causing immunogenic cell death could enhance tumor-targeted AFN therapy. Doxorubicin is such an anthracycline routinely used in the clinic. Used in a non-curative dose, doxorubicin synergized with mIFN or mCD20-AFN to completely eradicate B16-mCD20 tumors (). Combined with mIFN, doxorubicin dramatically amplified toxicity resulting in exaggerated weight loss, hematological deficiency and even 100% mortality (). In stark contrast, mCD20-AFN plus doxorubicin completely destroyed tumors without any detectable toxicity or mortality ().
Figure 6. Tumor-targeted AcTaferon combination treatments eradicate tumors without toxicity. (A) Growth of s.c. inoculated B16-mCD20+ tumors in C57BL/6J mice, body weight loss (B), neutrophil counts (C) and mortality (D) after 8 treatments with PBS, tumor-targeted mCD20-mIFN or mCD20-AFN (n = 5 mice per group, shown is a representative experiment, arrows indicate treatment days). When indicated in the legends, tumors were also treated with dox(orubicine) every second day. (E) Growth of s.c. inoculated B16-mCD20+ tumors in C57BL/6J mice, body weight loss (F), platelet counts (G) and mortality (H) after 8 treatments with PBS, tumor-targeted mCD20-mIFN or mCD20-AFN (n = 5 mice per group, shown is a representative experiment, arrows indicate treatment days). When indicated in the legends, tumors were also treated with low-dose (0.6 μg/mouse) TNF every second day. (I-K) Growth of s.c. inoculated B16-mCD20+ tumors in C57BL/6J mice treated with PBS or mCD20-AFN. When indicated, treatment was combined with anti-PDL1 sdAb or a combination of Treg-depleting (TregΔ) anti-CTLA4 + anti-OX40 antibodies. Dividend/divisor in the figures indicates the number of tumor-free mice over the number of total mice at the day the experiment was ended, indicated in the X axis. For all figures, a representative experiment is shown (n = 4-6 per group), repeated at least twice. All values are mean ± s.e.m.; *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared with PBS treated animals unless otherwise indicated; by two-way ANOVA with Dunnett's multiple comparison test (A-B, E-F, I-K), one-way ANOVA with Dunnett's multiple comparison test (C, G) or log-rank test (D, H).
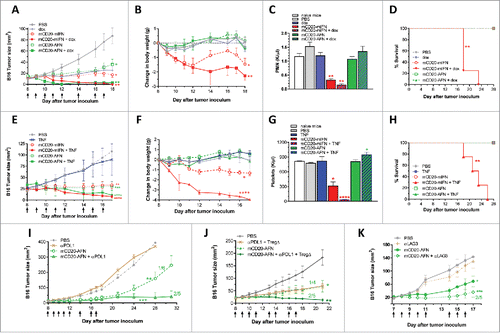
To facilitate tumor penetration of activated CTL and other immune cells involved in tumor eradication, we next combined IFN or mCD20-AFN with Tumor Necrosis Factor (TNF), known to activate and permeabilize endothelium in preclinical models and isolated limb perfusion in patients.Citation25,26 Low-dose TNF did not have an antitumor effect as such, but strongly synergized with mIFN or mCD20-AFN to fully destroy the B16-mCD20 tumor (). Comparable to doxorubicine, low-dose TNF also fatally worsened the toxicity of mIFN, but not of mCD20-AFN ().
Recently, immune checkpoint blockade strategies have become very efficient therapeutic options for many different malignancies. Both anti-CTLA4 and anti-PD1 treatments were first approved for advanced metastatic melanoma and show long-term cure in up to 40% of patients.Citation27 Unfortunately, most patients suffer from severe adverse effects, especially when treatments are combined.Citation28 In addition, the majority of patients are still either resistant to immunotherapy, or they relapse.Citation18 Recently, endogenous IFN was shown to be involved not only in conventional anti-cancer therapies such as chemo- and radiotherapy,Citation5,29,30 but also in immune checkpoint blockade efficacy.Citation18,31,32 Considering this, we combined mCD20-AFN with a neutralizing anti-PDL1 sdAb. The rationale for this was further boosted by PDL1 analysis; mIFN and mCD20-AFN increased PDL1 expression on B16 cells in vitro and in vivo (Supplementary ). Anti-PDL1 sdAb therapy significantly prolonged tumor stasis caused by mCD20-AFN and increased the number of tumor-free mice (). To escape CTL-mediated tumor killing following anti-PD1 therapy, upregulation of CTLA4 expression on tumor-infiltrating or –resident lymphocytes, and vice versa, has been described.Citation33,34 Therefore, we decided to add anti-CTLA4 and anti-OX40 antibodies to deplete intratumoral regulatory T cellsCitation35 to our anti-PDL1 treatment regime. This resulted in AFN-induced tumor shrinkage in all mice (). In control mice, anti-PDL1 as such had no effect (), while anti-CTLA4 + anti-OX40 slowed down tumor growth (). Although targeting the PDL1 and CTLA4 checkpoints has shown promising efficacy in several cancer types, several problems and questions still need to be resolved, such as the cause of resistance in many patients, their inefficacy in certain tumor types, as well as the development of severe adverse side effects. Lymphocyte‐activation gene‐3 (LAG3) is another vital checkpoint implicated in immunotherapy escape.Citation36 As shown in , tumor-targeted AFN was capable of converting the nonresponsive B16 tumors into anti-LAG3 therapy responders, resulting in 40% tumor-free mice. Importantly, adding anti-PDL1, anti-CTLA4, anti-OX40, or anti-LAG3 to AFN therapy did not cause extra toxicity (Supplementary –).
AcTaferon treatment provides tumor immunity
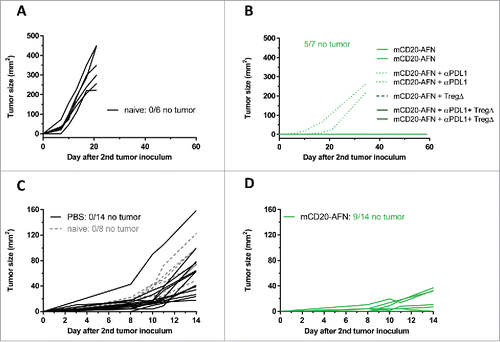
As certain combinations completely eradicated tumors (), we evaluated whether therapy induced immunity. Usually, AFN treatment was given until 16–17 days after initial tumor inoculation. If the successfully treated mice were still tumor-free on day 35, they were re-challenged with tumor cells on the contralateral flank. While all control (naive) mice developed a tumor within 7 days (), 70% of AFN-treated tumor-free mice did not develop a new tumor in the next 2 months (). Mice that developed a tumor did so later than naive mice. In addition, mice treated on days 5–10 following inoculation of their first tumor were challenged contralaterally on day 12. While all naive or PBS-treated mice developed a second tumor during the next 2 weeks (), 65% of the AFN-treated animals did not ().
Figure 7. Tumor-targeted AcTaferon therapy provides immunity. (A) Growth of s.c. inoculated B16-mCD20+ tumors in naive C57BL/6J mice. (B) Growth of B16-mCD20+ tumors inoculated on the contralateral flank on day 35 in mice where complete eradication of the primary tumor was achieved thanks to specific treatments (day 7–17) indicated in the figure legend. Tumor growth was evaluated for 60 days after the second tumor inoculation. (C) Growth of s.c. inoculated B16-mCD20+ tumors in naive C57BL/6J mice, or in mice where the primary tumor was treated with PBS (day 5–10). (D) Growth of B16-mCD20+ tumors inoculated on the contralateral flank on day 12 in mice that received p.l. treatment for their primary tumor with mCD20-AFN on days 5–10. The experiment was ended 14 days after the second tumor inoculation (26 days after the primary tumor inoculation). Tumor growth of individual mice are plotted, the number of mice that remained tumor-free for the duration of the experiment (60 days for A-B and 14 days for C-D) is indicated in each figure.
Discussion
Type I IFN has long been approved for the treatment of several malignancies. The leading indications for its use in oncology include melanoma, renal cell carcinoma, AIDS-related Kaposi's sarcoma, follicular lymphoma, hairy cell leukemia, and chronic myelogenous leukemia.Citation1 In addition, endogenous IFN is critical for cancer immunosurveillanceCitation37 as well as for many different antitumor strategies including chemotherapy, radiotherapy and immunotherapy.Citation5 Unfortunately, IFN-based cancer therapy is associated with severe dose-limiting side effects.Citation1,2 More recently, activation of endogenous IFN production has been suggested and applied, by means of treatment with Toll-like Receptor (TLR) ligands such as poly(I:C) or CpG oligodeoxynucleotides, or Stimulator of IFN Genes (STING) agonists, either as a primary treatment or to overcome resistance to targeted therapy or immunotherapy.Citation18,38–41 However, also these IFN-inducing strategies are likely to be limited by potential unacceptable toxic side effects.Citation41–44 In addition, IFN may not only activate but also suppress anti-cancer immunity.Citation1,18 Therefore, we developed AcTaferons (AFNs), targeting type I IFN activity to selected cell types only, in an attempt to segregate the beneficial from detrimental qualities and side effects of IFN. As targeting modules, we opted for small camelid single domain antibodies, which have superior penetration potential and stability, and are more easily conjugated than conventional antibodies.Citation45 In contrast to standard targeted immunocytokines that have been developed over the last decades,Citation6 AFNs do not consist of WT cytokines, but instead of a mutated cytokine (type I IFN in this case) with a substantially decreased receptor affinity (IFNAR1 in case of AFNs).Citation10 As such, AFNs are less active while traveling through the body, and restore their activity at the targeted cell type only. Furthermore, by not interacting with their cognate receptors ubiquitously expressed throughout the body, they will not get trapped or cleared before reaching their target cells,Citation8 nor can any interference by soluble receptors occur.
In view of the oncological successes of anti-CD20 targeted therapy, we decided to target AFN to CD20+ tumors to obtain proof of concept. Treatment with tumor-targeted AFN drastically reduced lymphoma and melanoma tumor growth without any sign of systemic toxicity, which was evaluated by monitoring body weight and temperature, as well as several blood parameters. Antitumor efficacy depended on the presence of XCR1+ cDC1 and CD8+ T lymphocytes, and on IFN signaling in conventional DC. Tumor-targeted AFN very efficiently activated both XCR1+ and CD11b+ conventional DC, and significantly increased effector CTL numbers in lymph nodes and in the tumor, as well as their proliferation in lymph nodes. Importantly, not only mouse CD8+ cDC1, but also their CD141+ human counterparts, both of which are XCR1+, have already been shown to be superior antigen presenting DC, efficiently stimulating both naïve and activated CTL.Citation20,21,23 In addition, TCGA data analysis has clearly indicated that a high cDC1 signature in the tumor provides the strongest pro-immune survival value across multiple human cancer types, as well as a robust CTL recruiting chemokine profile.Citation46,47 In addition, targeting IFNα as an adjuvant to human DC was recently shown to increase their capacity for antigen presentation and antigen-specific CTL responses to human cancer epitopes.Citation48 The antitumor potential of targeting AcTaferons specifically to DC instead of to the tumor microenvironment is currently being explored.
Recently, CD38-targeted attenuated IFN (referred to as an ‘Attenukine’) was reported to reduce tumor progression in a human multiple myeloma xenograft model in mice.Citation49 Importantly, however, this was entirely due to the direct anti-proliferative effect of hIFN on the human tumor cells, as hIFN is not active on mouse cells at all. For the same reason, this study did not allow appropriate assessment of toxic systemic side effects, in contrast to our study.
Tumor-targeted AFN was also very effective to efficiently eradicate tumors in combination treatments with immunogenic chemotherapy or low-dose TNF, again without any toxic adverse effects. This was in sharp contrast with WT mIFN, where combination with chemotherapy or TNF dramatically enhanced toxicity, resulting in mortality due to the treatment. After decades of fruitless immunotherapy attempts, recent years have shown impressive results of checkpoint inhibition therapy for melanoma, lung cancer and several other tumor types. Nevertheless, many non-immunogenic tumors are still resistant to immunotherapy, and even in the melanoma population less than half of the patients are responsive. On top, about a quarter of the responsive patients develop resistance.Citation50 It is generally accepted that modulation of the tumor microenvironment to convert non-immunogenic tumors into immunogenic responders will be key to the further optimization and success rate of checkpoint inhibition therapy.Citation15,18,32,51 Interestingly, tumor-targeted AFN was much more effective when combined with PDL1, CTLA4 or LAG3 blockade to shrink and even completely eradicate tumors, again without causing any detectable adverse effects, in contrast with WT IFN or immunocytokine treatments. While B16 tumor growth was slowed down by Treg-depleting anti-CTLA4 plus anti-OX40 treatment, B16 tumors were completely insensitive for the anti-PDL1 or anti-LAG3 monotherapy regime that we applied. Combination of the latter with tumor-targeted AFN therapy, however, efficiently converted the non-responding B16 tumors into responders with full tumor eradication as a result. Therefore, our experiments indicate that targeting type I IFN activity to the tumor, when possible, may represent a very potent and completely safe alternative for systemic treatment with WT IFN or IFN inducers, such as TLR or STING agonists, to convert non-immunogenic neoplasms. In contrast to WT IFN, TLR ligands or STING agonists, AFNs do not provoke any systemic side effects, not even when injected daily in a 4-fold higher dose than presented in this manuscript, either subcutaneously or intravenously (data not shown). In addition, direct effects on tumor cells may also potentially contribute to antitumor efficacy by the release of chemokines.Citation15 A possible drawback of targeting tumor-associated antigens (TAA) in cancer patients may, however, be on-target off-tumor effects with concomitant toxicity, as is the case for chimeric antigen receptor (CAR) T cell therapy.Citation52 Tumors that present tumor-specific antigens (TSA) on their surface, however, could be safely treated without any chance of adverse effects.
Although targeting AFN specifically to the tumor cells proved effective to stall tumor growth, the effect of the tumor-targeted AFN did not depend on IFN signaling in the tumor cells themselves. This may indicate that enhanced localization of AFN in the tumor niche suffices to mount an efficient antitumor immune response, which may involve signaling in the DCs that we identified using the CD11c-IFNAR-deficient mice. Furthermore, although we chose CD20 as a targeting strategy, the antitumor efficacy of CD20-targeted AFN did not depend on B lymphocytes either. In contrast, the antitumor effect of tumor-targeted AFN clearly relied on the presence of XCR1+ conventional DC as well as on IFN signaling in conventional DC. Hence, we conclude that the completely safe and highly efficient antitumor effect of tumor-targeted type I IFN activity critically requires IFN signaling in the XCR1+ conventional DC locally present in the tumor microenvironment. Of interest, these conventional DC have already been described to require type I IFN signaling to efficiently present tumor antigen to tumor-killing CTL.Citation19,22
Experimental procedures
Construction and production of AcTaferons and immunocytokines
The mutation Q124R was introduced into the IFNa2 sequence by site-directed mutagenesis using the QuikChange II-E Site-Directed Mutagenesis Kit (Agilent Technologies) and single domain llama VHH antibodies (sdAb) were generated at the VIB Protein Service Facility, as described previouslyCitation10. Mouse AcTaferons are composed of hIFNa2Q124RCitation9 coupled via a 20xGGS-linker to an N-terminal targeting sdAb. A C-terminal tag is added for easy purification. AcTaferons and immunocytokines (WT mIFNa11 coupled to sdAb) were constructed in pHen6 vectors, large scale productions of His-tagged AcTaferons were performed in E. coli. The bacteria were cultured till stationary phase (OD600 of 0.7–0.8) whereupon IPTG (BioScientific) was added to activate the LacZ promoter. Cell supernatant was collected after overnight culture. The proteins in the periplasmic fraction were released by osmotic shock using a sucrose solution and were purified by immobilized metal ion chromatography (IMAC) on a HiTrap Sepharose resin loaded with Kobalt ions (Clontech, Takara Biotechnology). After binding of the protein, columns were washed with 0.5% EMPIGEN (Calbiochem, Millipore), 0.5% CHAPS (Sigma-Aldrich) and PBS. Imidazole (Merck) was used for elution and removed using PD-10 gel filtration columns (GE Healthcare). Protein concentration was determined using the absorbance at 280 nm and purity was assessed via SDS-PAGE. LPS levels were quantified using Limulus Amebocyte Lysate (LAL) QCL-1000 (Lonza). If still present, LPS was removed using Endotoxin Removal Resin (Thermo Scientific). Biological activities of all products were assessed by a functional assay using the mouse luciferase reporter cell line LL171 against the WHO International mouse IFNα standard Ga02-901-511 as described previously.Citation10
Mice, cells and tumor models
Mice were maintained in pathogen–free conditions in a temperature-controlled environment with 12/12 hour light/dark cycles and received food and water ad libitum. Animal experiments followed the Federation of European Laboratory Animal Science Association (FELASA) guidelines and were approved by the Ethical Committee of Ghent University. Female C57BL/6J and Balb/c mice (Charles River Laboratories, Saint-Germain sur l'Arbresle, France) were inoculated at the age of 7–10 weeks. For experiments using knock-out mice (IFNAR1, Batf3), mice were bred in our own facilities and WT littermates were used as controls. For s.c. tumor models, cells were injected using a 30G insulin syringe, in 50 μl suspension, on the shaved flank of briefly sedated mice (using 4% isoflurane). For the s.c. A20 lymphoma model, 5.106 cells were inoculated; for the B16-mCD20 and B16-hCD20 clones 6.105 cells. The B16-mCD20 and B16-hCD20 cell lines were generated as follows: B16Bl6 cells were stably co-transfected with a plasmid containing the expression cassettes for mCD20 or hCD20, and with a plasmid containing the neomycin resistance gene. Stable transfected cells were selected with G418 (2 mg/ml)-containing medium, followed by FACS sorting of mCD20- or hCD20-expressing cells. From the pool of hCD20 and mCD20 expressing B16-Bl6 cells, single clones were selected by limited dilution. The B16-mCD20-IFNAR1−/− cell lines were generated via the CRISPR-Cas9 editing system, using 2 different gRNA sequences targeting exon 2 of IFNAR1, 5’-AGCAGCCACGGAGAGTCAAT-3’ and 5’-ATGTAGACGTCTATATTCTC-3’ (determined via http://crispr.mit.edu). The gRNA were cloned in the pSpCas9(BB)-2 A-Puro vector (PX459)Citation53 and transfected into B16-mCD20 cells via Jetprime. After 4 weeks of selection with 1 µg/ml puromycine, negative selection of the 2 different cell pools was performed using MACS with anti-IFNAR1-PE (eBioScience) and anti-PE microbeads (Miltenyi Biotec). The absence of IFNAR1 was verified with flow cytometry (evaluating mIFNAR1 presence using anti-IFNAR1-PE) as well as functionally determining P-STAT1 signaling 15 and 30 min after mIFN treatment. Cell lines were purchased from American Type Culture Collection and cultured in conditions specified by the manufacturer. All cells used for inoculation were free of mycoplasma. Tumor diameters were measured using a caliper. To analyze tumor immunity, mice were re-challenged on the contralateral flank with a new dose of tumor cells. Re-challenge was done either on tumor-free mice (after successful therapy), or on tumor-bearing mice after a short 6 day treatment schedule (day 5–10). Before the start of the treatments, mice were randomly and blindly allocated to a therapy group, the size of the groups was determined by the number of mice available with an appropriate tumor size; we strived to have at least 5 animals per experimental group. To determine clear-cut unambiguous antitumor effect, we know from experience that 5 animals suffice to obtain statistical significance. Monotherapy experiments were performed in at least 7 individual experiments, combination therapies in at least 2. Data were normally distributed, variance between groups was not significantly different. Differences in measured variables between the experimental and control group were assessed by using one-way or two-way ANOVA followed by Dunnett's or Tukey's multiple-comparison test. Survival curves were compared using the log-rank test. GraphPad Prism software was used for statistical analysis.
Tumor treatments
Unless otherwise indicated, tumor treatments were done perilesionally (p.l.), which is s.c. at the tumor border. As a control, mice were always treated with PBS. AcTaferons were given at 5,500 IU per treatment, WT mIFN or immunocytokine at ∼5-9.106 unless noted otherwise in the figure legend. These treatment doses corresponded to ∼30 μg protein (1.4 mg/kg). For combination therapies, we injected doxorubicine (3 mg/kg), rmTNF (28 μg/kg), anti-PDL1 sdAb (5.5 mg/kg), anti-CTLA4 Ab (450 μg/kg), anti-OX40 Ab (1.8 mg/kg), anti-LAG3 Ab (9 mg/kg). These were not injected daily, but every 2–3 days.
In vitro proliferation assays
A20 cells were cultured at 100,000 cells/ml in 25 cm2 cell culture flasks. Serial dilutions of mCD20-AcTaferon or GFP-AcTaferon were added and 72 hours later cells were counted by using a Scepter cell counter (Millipore). Results are expressed as percentage of A20 cells versus untreated culture (considered as the 100%). B16-mCD20 cells were cultured at 1,000 cells/well in a 96 well plate, and incubated with medium, mCD20-mIFN or mCD20-targeted or hCD20-untargeted AcTaferon for 24 hours. Cell proliferation was evaluated using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega).
Inhibitors, antibodies and checkpoint blockades
To inhibit the immune modulating PD1-PDL1 pathway, mice were treated with a neutralizing anti-PDL1 sdAb (120 μg/mouse), given i.p. every second day. To block CTLA4 signaling and deplete intratumoral regulatory T cellsCitation54, we used anti-CTLA4 (10 μg/mouse, BioXCell clone 9H10) and anti-OX40 (40 μg/mouse, BioXCell clone OX-86) given 3x/week. Anti-LAG3 (200 μg/mouse, BioXCell clone C9B7 W) was given in the same schedule. Depletion of CD8+ cells was performed by i.p. administration of 200 μg rat-anti-mouse CD8 Ab (BioXCell clone YTS169.4) one day prior to the first AcTaferon treatment. Additional depletion rounds were performed 4 and 10 days after the first. Control (non-depleted) mice were treated with 200 μg rat IgG2b Isotype Control Ab (BioXCell clone LTF-2). CD8+ cell depletion was evaluated with flow cytometry on blood, spleen, lymph nodes and tumor, as well as via IHC on spleen and tumor. Depletion was observed as soon as 4 hours after Ab injection and lasted at least 4 days. Thanks to additional depletion round CD8+ cells were absent during the entire AFN treatment period.
Flow cytometry analysis and sorting
For ex vivo P-STAT1 signaling analysis, different amounts of mCD20-AcTaferon or GFP-AcTaferon or mCD20-hIFN-R149A were injected intravenously through the retro-orbital vein in Balb/c mice (female, 14 weeks) and spleens were recovered 45 minutes later. Splenocytes were isolated, fixed, permeabilized and labelled with anti-CD19-APC and anti-phospho-Stat1-PE antibodies (BD Biosciences)Citation10. Samples were acquired on a FACS Canto (BD Biosciences) and data were analyzed using DIVA software (BD Biosciences). For analysis of CD19+ B, CD4+ and CD8+ T cell populations in circulation, blood was collected from the tail vein with a heparinized capillary and stained for flow cytometric analysis using CD19, CD4 or CD8 antibodies (CD19 FITC, BD; CD4 APC, Immunotools; CD8 a PE, eBioScience). PDL1 expression was analyzed on B16-mCD20 cells in vitro after 48 h, and in vivo (spleen + tumor) after 24 h.
Analysis of the DC activation status
To address the impact of perilesional AFN treatment on the DC activation status in the tumor draining lymph node, B16-mCD20 melanoma bearing mice were injected with BCII10-AFN, mCD20-AFN (5000 IU) or PBS. 24 hours post injection, tumor-draining lymph nodes were dissected and processed for flow cytometry. Cell suspensions were stained with CD16/CD32 to block Fc receptors, followed by CD3-AlexaFluor-700, CD19-AlexaFluor-700, Ly6C-PECy7, CD11b-APCCy7, CD86-eFluor450, PDL1-PE, CD40-APC, CD80-APC, CD11c-PEeFluor610, MHCI-FITC (all eBioscience), XCR1-BV650 (BioLegend). After exclusion of T and B cells and Ly6Chi monocytes, DCs were identified based on their expression of CD11c and MHCII. XCR1+ cDC1s were identified based on their XCR1+ CD11b− MHCIIint-hi CD11cint-hi phenotype, whereas CD11b+ cDC2s were identified based on their XCR1− CD11b+ MHCIIint-hi CD11cint-hi phenotype. Samples were acquired on a BD LSR Fortessa (5-laser) and analyzed using FlowJo software.
Analysis of CTL proliferation and activation
To analyze activated T cell phenotype, tumor draining lymph nodes were dissected at different time points after single perilesional delivery of AcTaferons and processed for flow cytometry. Fc receptors were blocked using CD16/CD32, whereupon single cell suspensions were stained with CD3-PeCy7 (clone 145-2C11), CD4-PE (clone RMA-5), CD8-APC (clone 53–6.7) (all BD Pharmingen), CD44-PercP-Cy5.5 (clone IMF7) and CD62 L-APC-Cy7 (clone MEL-14) (both BioLegend). Effector T cells were identified based on their CD44hiCD62Llow phenotype. Samples were acquired on an Attune NxT Acoustic Focusing Cytometer (Life Technologies) and analyzed using FlowJo software. To evaluate CTL proliferation, we used T cell receptor transgenic CD8+ T cells specifically recognizing the melanocyte differentiation antigen gp100 (Pmel-1) present on B16 tumor cells. Gp100-specific CD8 Pmel-1 T cells were isolated from the spleens of C57BL/6J Pmel-1–Thy1.1 mice, using the CD8α+ T Cell Isolation Kit (Miltenyi Biotec) and labeled with 5 µM of CFSE (Thermo Fisher). One million of CFSE-labeled T cells were adoptively transferred to C57BL/6J mice inoculated with 6.105 B16 melanoma cells. Subsequently, mice were treated with the indicated AcTakines. At least five days post adoptive T cell transfer, tumor-draining lymph nodes and spleen were dissected and specific T cell proliferation was assessed by Flow Cytometry. Samples were acquired on a BD LSR Fortessa (5-laser) or on an Attune Nxt Acoustic Focusing Cytometer (Life Technologies) and analyzed using FlowJo software.
Hematological analysis
One day after the last tumor treatment, blood was collected from the tail vein in EDTA-coated microvette tubes (Sarstedt), and analyzed in a Hemavet 950FS (Drew Scientific, Waterbury, USA) whole blood counter.
Disclosure of potential conflict of interest
J.T. and G.U. are scientific co-founders, and J.T. is CTO of Orionis Biosciences and holds stock-options in the company.
Author contributions
A.C. and J.T. designed experiments and analyzed and interpreted the data. A.C., S.V.L., G.G., J.v.D.H. conducted experiments. D.C., E.R. and A.V. provided technical support, including construction and purification and help with animal work. J.B. made the stable mCD20- and hCD20-B16 clones, L.D.C. the B16-mCD20-IFNAR1−/− clones, and S.G. helped with AcTaferon designs. G.U. designed and supervised the work proving the cell-targeting efficacy, performed by F.P. and Y.B. A.C., G.U. and J.T. wrote the manuscript.
2017ONCOIMM0699R-f02-z-4c.pptx
Download MS Power Point (1.2 MB)Acknowledgments
We thank Johan Grooten for the anti-PDL1 sdAb, Reza Hassanzadeh Ghassabeh (VIB Nanobody Core) for the selection of the anti-CD20 sdAbs, Claude Libert for the IFNAR1−/− mice, Veronique Flamand for the Batf3−/− mice, Ulrich Kalinke for CD11c-IFNAR- and CD4-IFNAR-deficient mice, Feng Zhang for the PX459 vector.
Additional information
Funding
References
- Jonasch E, Haluska FG. Interferon in oncological practice: review of interferon biology, clinical applications, and toxicities. Oncologist. 2001;6:34–55. doi:10.1634/theoncologist.6-1-34.
- Kirkwood JM, Bender C, Agarwala S, Tarhini A, Shipe-Spotloe J, Smelko B, Donnelly S, Stover L. Mechanisms and management of toxicities associated with high-dose interferon alfa-2b therapy. J Clin Oncol. 2002;20:3703–18. doi:10.1200/JCO.2002.03.052.
- Parker BS, Rautela J and Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16:131–44. doi:10.1038/nrc.2016.14.
- Belardelli F, Gresser I. The neglected role of type I interferon in the T-cell response: implications for its clinical use. Immunol Today. 1996;17:369–72. doi:10.1016/0167-5699(96)10027-X.
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–14. doi:10.1038/nri3845.
- List T, Neri D. Immunocytokines: a review of molecules in clinical development for cancer therapy. Clin Pharmacol. 2013;5:29–45.
- Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Chang CH. CD20-targeted tetrameric interferon-alpha, a novel and potent immunocytokine for the therapy of B-cell lymphomas. Blood. 2009;114:3864–71. doi:10.1182/blood-2009-06-228890.
- Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci U S A. 2015;112:3320–5. doi:10.1073/pnas.1416159112.
- Weber H, Valenzuela D, Lujber G, Gubler M, Weissmann C. Single amino acid changes that render human IFN-alpha 2 biologically active on mouse cells. EMBO J. 1987;6:591–8.
- Garcin G, Paul F, Staufenbiel M, Bordat Y, Van der Heyden J, Wilmes S, Cartron G, Apparailly F, De Koker S, Piehler J. High efficiency cell-specific targeting of cytokine activity. Nat Commun. 2014;5:3016. doi:10.1038/ncomms4016. PMID:24398568
- De Groeve K, Deschacht N, De Koninck C, Caveliers V, Lahoutte T, Devoogdt N, Muyldermans S, De Baetselier P, Raes G. Nanobodies as tools for in vivo imaging of specific immune cell types. J Nucl Med. 2010;51:782–9. doi:10.2967/jnumed.109.070078.
- Trinh KR, Vasuthasawat A, Steward KK, Yamada RE, Timmerman JM, Morrison SL. Anti-CD20-interferon-beta fusion protein therapy of murine B-cell lymphomas. J Immunother. 2013;36:305–18. doi:10.1097/CJI.0b013e3182993eb9.
- Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, Ohlfest JR, Hu P, Epstein AL. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477–89. doi:10.1097/01.cji.0000436722.46675.4a.
- Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol. 2001;Chapter 20:Unit 20 1. PMID:18432774
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–9. doi:10.1038/nm.3708.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi:10.1016/j.immuni.2013.07.012.
- Schlitzer A, Ginhoux F. Organization of the mouse and human DC network. Curr Opin Immunol. 2014;26:90–9. doi:10.1016/j.coi.2013.11.002.
- Minn AJ, Wherry EJ. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell. 2016;165:272–5. doi:10.1016/j.cell.2016.03.031.
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi:10.1084/jem.20101158.
- Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. 2010;207:1273–81. doi:10.1084/jem.20100348.
- Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med. 2010;207:1283–92. doi:10.1084/jem.20100223.
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–16. doi:10.1084/jem.20101159.
- Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207:1247–60. doi:10.1084/jem.20092140.
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–100. doi:10.1126/science.1164206.
- Lejeune FJ. Clinical use of TNF revisited: improving penetration of anti-cancer agents by increasing vascular permeability. J Clin Invest. 2002;110:433–5. doi:10.1172/JCI0216493.
- van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11:397–408. doi:10.1634/theoncologist.11-4-397.
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61. doi:10.1016/j.ccell.2015.03.001.
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi:10.1056/NEJMoa1504030. PMID:26027431
- Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71:2488–96. doi:10.1158/0008-5472.CAN-10-2820.
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–52. doi:10.1016/j.immuni.2014.10.019.
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42. doi:10.1016/j.immuni.2014.10.017.
- Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, Gaffal E, Steitz J, Tolba R, Kalinke U. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014;4:674–87. doi:10.1158/2159-8290.CD-13-0458.
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi:10.1038/ncomms10501. PMID:26883990
- Twyman-Saint\sVictor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7.
- Marabelle A, Kohrt H, Levy R. New insights into the mechanism of action of immune checkpoint antibodies. Oncoimmunology. 2014;3:e954869. doi:10.4161/21624011.2014.954869. PMID:25610751
- He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, Zhou C, Hirsch FR. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. 2016;107:1193–7. doi:10.1111/cas.12986.
- Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6:722–9.
- Dahal LN, Dou L, Hussain K, Liu R, Earley A, Cox KL, Murinello S, Tracy I, Forconi F, Steele AJ. STING Activation Reverses Lymphoma-Mediated Resistance to Antibody Immunotherapy. Cancer Res. 2017;77:3619–3631. doi:10.1158/0008-5472.CAN-16-2784.
- Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52. doi:10.1126/scitranslmed.aaa4306. PMID:25877890
- Li K, Qu S, Chen X, Wu Q, Shi M. Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways. Int J Mol Sci. 2017;18:pii: E404. doi: 10.3390/ijms18020404.
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–30. doi:10.1016/j.celrep.2015.04.031.
- Sparwasser T, Miethke T, Lipford G, Borschert K, Hacker H, Heeg K, Wagner H. Bacterial DNA causes septic shock. Nature. 1997;386:336–7. doi:10.1038/386336a0.
- Stevenson HC, Abrams PG, Schoenberger CS, Smalley RB, Herberman RB, Foon KA. A phase I evaluation of poly(I,C)-LC in cancer patients. J Biol Response Mod. 1985;4:650–5.
- Sparwasser T, Koch ES, Vabulas RM, Heeg K, Lipford GB, Ellwart JW, Wagner H. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol. 1998;28:2045–54. doi:10.1002/(SICI)1521-4141(199806)28:06%3c2045::AID-IMMU2045%3e3.0.CO;2-8.
- Wesolowski J, Alzogaray V, Reyelt J, Unger M, Juarez K, Urrutia M, Cauerhff A, Danquah W, Rissiek B, Scheuplein F. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol. 2009;198:157–74. doi:10.1007/s00430-009-0116-7.
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26:638–52. doi:10.1016/j.ccell.2014.11.010.
- Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31:711−723 e4. doi:10.1016/j.ccell.2017.04.003.
- Graham JP, Authie P, Karolina Palucka A, Zurawski G. Targeting interferon-alpha to dendritic cells enhances a CD8+ T cell response to a human CD40-targeted cancer vaccine. Vaccine. 2017;35:4532–4539. doi:10.1016/j.vaccine.2017.07.032.
- Pogue SL, Taura T, Bi M, Yun Y, Sho A, Mikesell G, Behrens C, Sokolovsky M, Hallak H, Rosenstock M. Targeting Attenuated Interferon-alpha to Myeloma Cells with a CD38 Antibody Induces Potent Tumor Regression with Reduced Off-Target Activity. PLoS One. 2016;11:e0162472. doi:10.1371/journal.pone.0162472. PMID:27611189
- Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA. 2016;315:1600–9. doi:10.1001/jama.2016.4059.
- Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187. PMID:25964783
- Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. doi:10.1038/mto.2016.11. PMID:27626062
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi:10.1038/nprot.2013.143.
- Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123:2447–63. doi:10.1172/JCI64859.