
ABSTRACT
Although the proteasome inhibitor bortezomib has significantly improved the survival of patients with multiple myeloma (MM), the disease remains fatal as most patients eventually develop progressive disease. Recent data indicate that MM cells can evade bortezomib-induced cell death by undergoing autophagy as a consequence of endoplasmatic reticulum (ER)-stress induced by proteasome inhibition. Here we show that bortezomib sensitizes MM cells to NK cell killing via two distinct mechanisms: a) upregulation of the TRAIL death receptor DR5 on the surface of MM cells and b) ER-stress induced reduction of cell surface HLA-E. The latter mechanism is completely novel and was found to be exclusively controlled by the inhibitory receptor NKG2A, with NKG2A single-positive (NKG2ASP) NK cells developing a selective augmentation in tumor killing as a consequence of bortezomib-induced loss of HLA-E on the non-apoptotic MM cells. In contrast, the expression of classical HLA class I molecules remained unchanged following bortezomib exposure, diminishing the augmentation of MM killing by NK cells expressing KIR. Further, we found that feeder cell-based ex vivo expansion of NK cells increased both NK cell TRAIL surface expression and the percentage of NKG2ASP NK cells compared to unexpanded controls, substantially augmenting their capacity to kill bortezomib-treated MM cells. Based on these findings, we hypothesize that infusion of ex vivo expanded NK cells following treatment with bortezomib could eradicate MM cells that would normally evade killing through proteasome inhibition alone, potentially improving long-term survival among MM patients.
Introduction
Natural killer (NK) cells are immune cells that can kill tumor cells via death receptor pathways (TRAIL/TRAIL receptor and FasL/Fas) and the release of cytotoxic granules.Citation1 The latter is controlled by signals from activating and inhibitory cell surface receptors. While a large array of receptors mediate NK cell activation, inhibition is mainly controlled by HLA class I-binding receptors including leukocyte immunoglobulin-like receptor subfamily B member 1 (LILRB1; Lir-1), killer immunoglobulin-like receptors (KIRs), and CD94/NKG2A.Citation1 The KIRs and CD94/NKG2A receptor are also involved in NK cell education, a process that dynamically tunes the potency of an NK cell response to cells losing HLA class I expression.Citation2,Citation3
Bortezomib is a reversible proteasome inhibitor that upon binding to the 26S subunit of the proteasome causes cellular growth arrest, inhibition of migration and apoptosis.Citation4 Proteasome inhibition also induces endoplasmatic reticulum (ER)-stress due to accumulation of misfolded and unfolded proteins in the ER.Citation5 Cells respond to ER-stress by activating the unfolded protein response (UPR) which reduces unfolded protein burden via production of a set of pro-survival enzymes and chaperons that facilitate proper protein folding in the ER.Citation6 In addition to promoting cell survival via proper protein folding, UPR signaling also drives cells into autophagy,Citation7 a state where cells become quiescent while recycling damaged organelles and proteins before returning to a normal cellular state.Citation8 Cells that cannot resolve ER-stress undergo cell death via apoptosis.Citation6
Administration of bortezomib to patients with multiple myeloma (MM) has led to significantly improved survival.Citation9,Citation10 However, despite these positive results, disease recurrence inevitably occurs. Resistance to bortezomib has been reported to occur due to a variety of factors including the upregulation of the multidrug resistance protein,Citation11 heat-shock protein 70,Citation12 and the proteasome subunit beta-5 (PSMB5)Citation13 or from mutations in PSMB5,Citation13,Citation14 or from ER-stress-mediated quiescence.Citation15
In addition to its direct toxic effects, bortezomib also sensitizes tumor cells to TRAIL-mediated apoptosis.Citation16 We and others have reported that bortezomib augments TRAIL-mediated NK cell cytotoxicity against several different types of tumors in vitro by upregulating death receptor 5 (DR5) on the tumor cell surface.Citation17–Citation19 However, it remains to be determined whether bortezomib sensitizes MM cells to NK cells in vivo via this mechanism.
Here we describe a completely novel mechanism through which bortezomib sensitizes MM cells to NK cells. Following in vitro exposure to bortezomib at concentrations achieved pharmacologically in humans, we observed reduced cell surface expression of HLA-E on MM cells which increased their susceptibility to killing by NK cells that expressed CD94/NKG2A as their only inhibitory receptor (NKG2ASP). Remarkably, tumor sensitization to NK cells via the NKG2A/HLA-E axis occurred independent of sensitization that concomitantly occurred via the TRAIL pathway. Using a panel of drugs, we found bortezomib-induced upregulation of DR5 and downregulation of HLA-E on tumor cells was mediated through ER-stress that directed cells into autophagy. Finally, we observed that NK cells expanded ex vivo using irradiated EBV-LCL feeder cells increased both TRAIL surface expression and the percentage of NKG2ASP NK cells compared to unexpanded overnight IL-2 activated NK cells. Consistent with the above, we observed that overall killing of bortezomib-exposed MM cells by NK cells was greater with expanded NK cells compared to their unexpanded IL-2 activated counterparts. Based on these findings, we hypothesize that adoptive transfer of ex vivo expanded NK cells following treatment with bortezomib may contribute to eradication of MM cells that escape bortezomib-induced apoptosis, potentially improving disease free survival of patients treated with this agent.
Results
Bortezomib sensitizes multiple myeloma cells to NK cells via pathways additional to the TRAIL/DR5 pathway
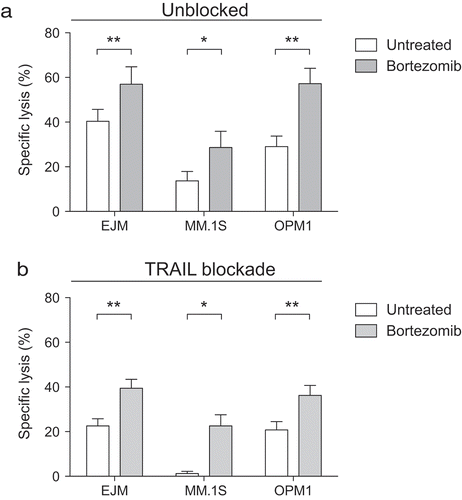
Previous studies have shown that bortezomib sensitizes various tumor cell types to TRAIL-expressing NK cells via upregulation of death receptor 5 (DR5) on the target cells.Citation17–Citation19 However, prior studies have not established that MM sensitization to NK cell killing following proteasome inhibition is exclusively TRAIL dependent. To address this, we treated three MM cell lines with bortezomib for 24 hours prior to co-culturing with NK cells. As MM cells are highly sensitive to bortezomib, our experiments were conducted with a 5 nM concentration of bortezomib, which represents the pharmacological levels achieved in vivo following treatment.Citation20 As shown in , pretreatment with bortezomib augmented NK cell-mediated killing of MM cells. However, antibody-mediated blockade of TRAIL on NK cells only partly reduced their capacity to kill MM cells and did not diminish the sensitizing effect of bortezomib to NK cell killing ( and Supplemental ). These data demonstrate that pathways other than the previously established TRAIL/DR5 pathway are involved in bortezomib-induced tumor sensitization to NK cells.
Figure 1. Bortezomib sensitizes multiple myeloma cells to NK cells, but only partially via the TRAIL/DR5 pathway.
Overnight IL-2 activated NK cells were co-cultured with the MM cell lines EJM (n = 8), MM.1S (n = 6), OPM1 (n = 8) either pre-exposed (grey bars) or not (white bars) to 5 nM bortezomib for 24 hours. (a) Lysis of MM cells by NK cells following a 4-hour co-culture (n = 10). (b) Lysis of MM cell lines following a 4-hour co-culture with NK cells pre-treated with a TRAIL blocking antibody. Bars, mean. Error bars, standard deviation. * p < 0.05, ** p < 0.01.
Multiple myeloma cells become susceptible to CD94/NKG2A+ NK cells following exposure to bortezomib
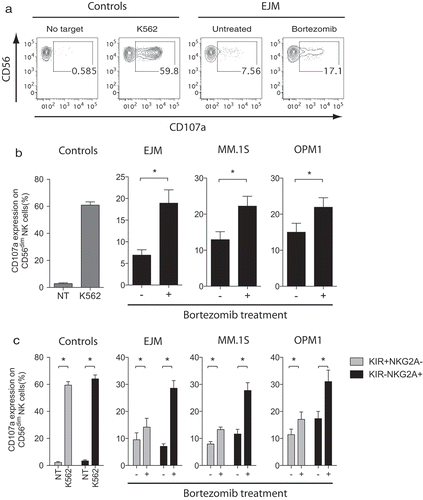
We next evaluated NK cell degranulation following co-cultures with MM cells to assess if NK cell activity was increased following bortezomib treatment. This analysis revealed that NK cell anti-tumor activity was significantly increased when MM cells were pretreated with bortezomib compared to untreated MM cells (–). Based on the report by Shi et al., describing loss of HLA class I on MM cells following treatment with bortezomib, we also analyzed if NK cells that are educated and inhibited by KIR and NKG2A had increased activity when co-cultured with bortezomib-treated MM cells. For this evaluation, we used NK cells from individuals with KIR haplotype A/A who only carry inhibitory but not activating KIRs. To remove any potential influence of the pan-HLA class I-binding receptor Lir-1, we excluded this receptor in the analysis. As shown in , NK cells expressing NKG2A but not KIR (and Lir-1) displayed a substantial and significantly higher reactivity to bortezomib-treated MM cells compared to untreated target cells. In contrast, there was a much smaller increase in the level of degranulation observed in NK cells controlled by inhibitory KIRs (). Taken together, these data establish that bortezomib sensitizes MM cells to NK cells via both a TRAIL/DR5 pathway and a previously uncharacterized selective sensitization to NKG2A+ NK cells.
Figure 2. Following exposure to bortezomib, MM cells become susceptible to NK cell killing via the degranulation pathway primarily triggered in CD94/NKG2A+ NK cells.
Overnight IL-2 activated NK cells (n = 7) were co-cultured without target (no target; NT) (negative control), with K562 cells (positive control), or MM cell lines (EJM, MM.1S, OPM1) either pre-exposed or not to 5 nM bortezomib for 24 hours. (a) Representative FACS plots showing degranulation levels by CD56dim NK cells using the CD107a marker following co-cultures with controls or the MM cell line EJM. (b) Degranulation levels by CD56dim NK cells following co-cultures with controls or MM cells. (c) Degranulation levels by KIR+NKG2A− (grey bars) or KIR−NKG2A+ (black bars) CD56dim NK cells following co-cultures with controls or MM cells. Bars, mean. Error bars, standard deviation. * p < 0.05.
Selective and transient reduction of cell surface HLA-E, but not classical HLA class I, on bortezomib treated multiple myeloma cells
The NKG2A receptor inhibits NK cell cytotoxicity following ligation to the non-classical HLA class I molecule HLA-E expressed on target cell.Citation21 Based on our functional data, and data on bortezomib-induced loss of HLA class I reported by Shi et al.,Citation22 we next assessed the impact of exposing MM cells to bortezomib in terms of HLA class I and HLA-E expression. Consistent with our finding that tumor sensitization occurred predominantly to NKG2A-expressing NK cells, we observed bortezomib induced a loss of HLA-E expression on MM cells while HLA class I levels remained unchanged or slightly increased (). In line with data for other tumor cell types, bortezomib also induced upregulation of DR5 on the surface of MM cells. These drug-induced phenotypic alterations observed in MM cells were dose-dependent (Supplemental Figure 2). Dose-dependent loss of cell surface HLA-E was also confirmed on bortezomib-exposed primary MM cells (Data not shown).
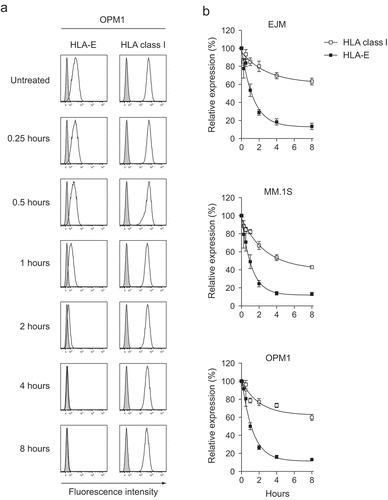
Figure 3. Bortezomib induces a selective and transient reduction of HLA-E, but not classical HLA class I, while transiently upregulating DR5 expression on MM cells.
MM cell lines were either exposed or not to 5 nM bortezomib prior to assessment of cell viability and cell surface expression of HLA-E, HLA class I and DR5. (a) Representative FACS plots showing the viability as measured by 7-AAD and Annexin V and the expression of HLA-E, HLA class I and DR5 on the cell surface of 7-AAD−Annexin V− live the OPM1 MM cell line. (b) The cell surface expression of HLA-E (black bars), HLA class I (grey bars) and DR5 (white bars) on live MM cells after 24 hours of exposure to bortezomib (n = 10). (c) The cell surface expression of HLA-E (circles), HLA class I (squares) and DR5 (triangles) on live MM cells before and after 24 hours of exposure to bortezomib, as well as up to 72 hours from wash-off (WO) of bortezomib (n = 5). (d) The viability of MM cells before and 24 hours after exposure to bortezomib, as well as up to 72 hours from WO of bortezomib (n = 5). Bars or symbols, mean. Error bars, standard deviation. * p < 0.05.
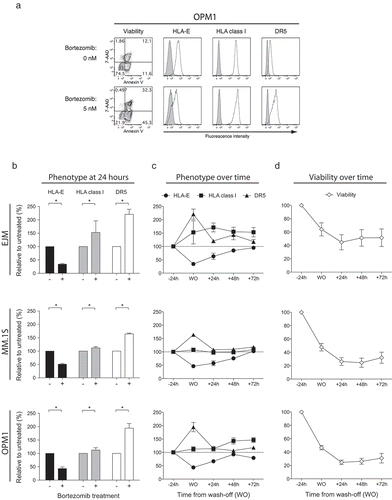
To further understand the kinetics of these phenotypic alterations, we next studied the expression of HLA class I, HLA-E and DR5 after wash-off of bortezomib from tumor cells in vitro. This experiment revealed that DR5 expression almost normalized 24 hours after withdrawal of the drug, in contrast to HLA-E expression which did not return to baseline levels on myeloma cells until approximately 48 hours after wash-off (). In contrast, HLA class I expression remained unchanged or was actually slightly increased over time after withdrawal of bortezomib. Importantly, bortezomib concentrations that are achieved pharmacologically in humans were utilized for all experiments,Citation20 which notably resulted in cell death in only a fraction of the MM cells (). Collectively, these data show for the first time that bortezomib upregulates DR5 expression on MM cells while simultaneously downregulating HLA-E without having any suppressive effect on HLA class I expression.
Bortezomib-induced reduction of HLA-E and upregulation of DR5 expression on multiple myeloma cells is a result of ER-stress and rapid turnover of cell surface HLA-E molecules
Since bortezomib is cytotoxic to MM cells, loss of HLA-E may simply be a consequence of cell death. To rule out this possibility, we evaluated HLA-E expression on MM cells following exposure to the chemotherapeutic agent cisplatin, which in contrast to the proteasome inhibitor bortezomib, induces cell death primarily via crosslinking of DNA. As shown in , although cisplatin induced MM cell death, HLA-E expression remained unchanged and no changes in cell surface expression of DR5 and HLA class I were observed. To completely exclude that HLA-E expression was reduced due to bortezomib-induced apoptosis, we treated MM cells with bortezomib for 24 hours and then sorted the cells by flow cytometry based on their relative expression of HLA-E. If the loss of HLA-E had been a consequence of cell death, one would predict that there would be a higher proportion of apoptosis among those cells that displayed the highest degree of HLA-E reduction. However, no such difference in viability could be observed between the HLA-Ehigh and HLA-Elow fractions after 24 hours of culture in bortezomib-free medium following flow cytometry sorting (Supplemental Figure 3).
Figure 4. ER-stress, and not cell death, prompts downregulation of HLA-E and upregulation of DR5 on the surface of MM cells.
MM cell lines were either exposed or not to cisplatin, bortezomib or tunicamycin prior to assessment of cell viability and cell surface expression of HLA-E, HLA class I and DR5. (a) Representative FACS plots showing the viability as measured by 7-AAD and Annexin V and the expression of HLA-E, HLA class I and DR5 on 7-AAD−Annexin V− live OPM1 MM cells following exposure to cisplatin (n = 10), bortezomib (n = 10) or tunicamycin (n = 8). (b) The relative viability of OPM1 MM cells exposed to either cisplatin, bortezomib or tunicamycin for 24 hours compared to untreated MM cells. (c) The relative cell surface expression of HLA class I (black bars), DR5 (grey bars) and HLA-E (white bars) on live OPM1 MM cells exposed to either cisplatin, bortezomib or tunicamycin for 24 hours compared to untreated MM cells. (d) The intracellular expression levels of the ER-stress related proteins CHOP and BiP at 12 and 24 hours, respectively. (e) The intracellular expression levels of the autophagy related proteins LC3 and LAMP-1 at 24 and 48 hours, respectively. D and E show representative data for the OPM1 MM cell line. Bars or symbols, mean. Error bars, standard deviation. * p < 0.05, ** p < 0.01.
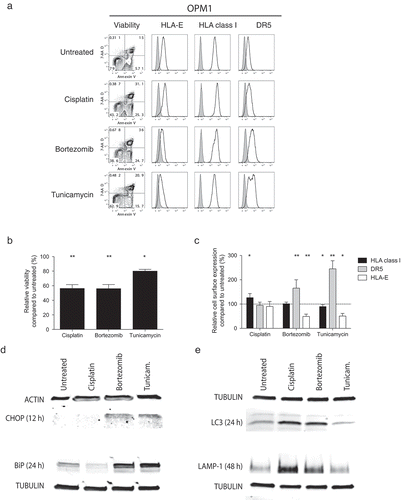
Bortezomib can alter tumor protein expression levels through a number of different mechanisms,Citation4 including as a consequence of ER-stress.Citation5 To further dissect if bortezomib-induced ER-stress was involved in reducing HLA-E expression, we exposed tumor cells to tunicamycin, a well-known ER-stressor that does not inhibit the proteasome. Treatment of MM cells for 24 hours with tunicamycin resulted in a simultaneous loss of HLA-E expression and upregulation of DR5, phenotypic changes that mirrored those that occurred when MM cells were treated with bortezomib (). Similar to bortezomib, tunicamycin also induced these changes without a major loss of viability in the MM cells (), confirming the phenotypic changes induced by bortezomib occur as a consequence of ER-stress rather than being caused nonspecifically as a consequence of drug-associated apoptosis. To definitively establish that bortezomib induces ER-stress and autophagy in our experimental model system like previously reported for several proteasome inhibitors,Citation5,Citation15 we confirmed the upregulation of CHOP and BiP as well as conversion of LC3-I to LC3-II and LAMP-1, respectively, using Western blots ().
HLA-E has been shown to have a relatively more unstable tertiary structure compared to classical HLA class I molecules with its expression being more dependent on TAP and tapasin in combination with ß2-microglobulin and a narrow set of peptides.Citation21 Based on this, we next evaluated if the turnover of cell surface HLA-E on MM cells was different from that of classical HLA class I molecules. Using brefeldin A (BFA) that inhibits fusion of late ER vesicles to Golgi, thereby preventing transportation to new molecules to the cell surface, we found that the cell surface half-life of HLA-E on MM cells was significantly shorter than for classical HLA class I molecules (). Because HLA-E expression appears to be more dependent on constant de novo synthesis than classical HLA class I molecules, these data provide the mechanism accounting for why HLA-E expression was significantly more affected by bortezomib-induced ER-stress compared to HLA class I expression.
Figure 5. Blockade of the delivery of de novo synthesized molecules from the ER reveals that HLA-E molecules have a shorter cell surface half-life on MM cells compared to classical HLA class I molecules.
HLA class I and HLA-E expression on MM cell lines following treatment with the ER to Golgi blocking agent brefeldin A (BFA). (a) Representative example of the HLA class I and HLA-E expression on the MM cell line OPM1 up to 8 hours after exposure to BFA. Histogram lines – HLA class I or HLA-E expression, tinted histograms – isotype controls. (b) Expression of HLA class I (open squares) and HLA-E (filled squares) on MM cells treated with BFA relative to unexposed MM cells up to 8 hours from start of exposure (n = 5). Symbols, mean. Error bars, standard deviation.
Taken altogether, these experiments establish that bortezomib reduces cell surface expression of HLA-E by triggering ER-stress and thereby interfering with de novo HLA-E protein synthesis.
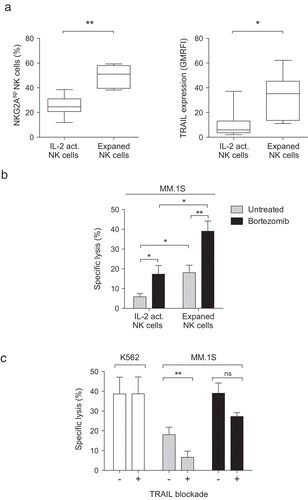
Ex vivo expanded NK cells with a large NKG2ASP subset and high level of cell surface TRAIL represents an optimal NK cell preparation for targeting bortezomib-exposed multiple myeloma cells
Based on data showing bortezomib enhances NK cell TRAIL-mediated tumor killing, we are currently conducting a phase I dose-escalating clinical trial to establish the safety of adoptive transfer of ex vivo expanded NK cells in patients with refractory malignancies pre-treated with bortezomib. The new discovery described in this report that MM cells downregulate HLA-E following bortezomib treatment, which selectively sensitizes tumors to NKG2ASP NK cells (only inhibited by the CD94/NKG2A receptor, while lacking KIR and Lir-1), led us to retrospectively investigate the effects of ex vivo NK cell expansion on NKG2ASP NK cells. Remarkably, we observed that ex vivo expansion using irradiated EBV-LCL feeders, the method currently being utilized to expand NK cells in our clinical trial,Citation23 not only upregulated NK cell TRAIL as described but also enriched for NKG2ASP NK cells ( and Supplemental Figure 4). In light of this, we next compared the MM targeting capacity of expanded NK cells to that of their unexpanded overnight IL-2 activated counterpart. Using the same NK cells to MM cells ratio, we observed that expanded NK cells were more potent at killing MM cells compared to overnight IL-2 activated NK cells (17% vs 5%, respectively) (). Although pretreatment with bortezomib sensitized MM cells to killing by both NK cell preparations, remarkably bortezomib pretreatment led to substantially higher overall killing by ex vivo expanded NK cells compared to overnight IL-2 activated NK cells (17% to 40% vs 5% to 15%, respectively). Blockade of TRAIL on expanded NK cells prior to co-culture with MM cells only slightly reduced their killing capacity, showing the sensitizing effect of bortezomib on MM cells is mediated to a lesser degree by the TRAIL/DR5 pathway compared to HLA-E downregulation sensitizing them to killing by NKG2ASP NK cells (). Finally, we also confirmed that killing by isolated NKG2ASP NK cells was potentiated when MM cells had been exposed to bortezomib as compared to unexposed cells (Data not shown). Collectively, these data show that ex vivo expanded NK cells represent an optimal NK cell preparation for future clinical trials utilizing bortezomib to sensitize MM cells to NK cell killing. Given the high probability that bortezomib is toxic to and impairs the function of circulating NK cells when administered to patients, as could be predicted based on our in vitro exposure of non-expanded NK cells to bortezomib (Supplemental Figure 5), the use of an adoptive transfer protocol to space out NK cell infusion from bortezomib administration may be key to obtain maximal clinical efficacy.
Figure 6. Ex vivo expanded NK cells with a large proportion of NKG2ASP cells and high levels of cell surface TRAIL are the optimal NK cell preparation for targeting bortezomib-exposed MM cells.
The proportion of NKG2ASP NK cells and level of TRAIL expression and MM lysis capacity of overnight IL-2 activated and ex vivo expanded NK cells were assessed. (a) The proportion of NKG2ASP NK cells among overnight IL-2 activated and ex vivo expanded NK cells as well as TRAIL expression on both NK cell preparations were measured (n = 9). (b) The capacity of overnight IL-2 activated and ex vivo expanded NK cells to lyse the MM cell line MM.1S either unexposed (grey bars) or pre-exposed (black bars) to 5 nM bortezomib (n = 8). (c) The capacity of unblocked or TRAIL blocked ex vivo expanded NK cells to lyse the MM cell line MM.1S either unexposed (grey bars) or pre-exposed (black bars) to 5 nM bortezomib (n = 8). The capacity of unblocked or TRAIL blocked ex vivo expanded NK cells to lyse K562 cells (white bars) was measured as a control (n = 8). Bars, mean. Error bars, standard deviation. ns, non-significant, * p < 0.05, ** p < 0.01.
Discussion
In this report, we identify a novel method through which bortezomib sensitizes MM cells to NK cell killing. Importantly, data from these experiments suggest this newly discovered sensitizing effect, which occurs as a consequence of bortezomib reducing tumor cell surface expression of HLA-E, is more potent than bortezomib-induced sensitization via the TRAIL pathway. By inducing ER-stress following proteasome inhibition with bortezomib, MM cells rapidly lose cell surface expression of HLA-E, but not classical HLA class I, rendering them susceptible to killing by NK cells that are exclusively regulated by the inhibitory receptor CD94/NKG2A (NKG2ASP NK cells). Remarkably, sensitization to NKG2ASP NK cells via this pathway was independent of tumor sensitization to TRAIL. As both pathways contributed to augmented overall tumor targeting by NK cells, we evaluated and subsequently established that ex vivo expanded NK cells maximize the sensitizing effects of bortezomib as they contain a higher percentage of NK cells that are NKG2ASP and express higher levels of TRAIL compared to short-term IL-2 activated NK cells. Given that bortezomib-induced ER-stress has been shown to drive MM cells into autophagy and that they thereby can evade bortezomib-induced cell death, we hypothesize that the addition of adoptive NK cell infusions following conventional bortezomib treatment might be a maneuver that could be used to eradicate the bortezomib-escaping quiescent MM cells that otherwise may cause disease relapse.
Since its FDA-approval in 2003, bortezomib has significantly improved the survival of patients with MM.Citation9,Citation10 However, despite these positive results, MM patients treated with bortezomib invariably relapse. Although drug resistance represents one potential mechanism of tumor escape,Citation11–Citation14 reports also indicate that MM cells can evade the apoptotic effects of bortezomib by undergoing ER-stress-mediated autophagy.Citation15 Data from studies conducted in this report show that bortezomib rapidly induces ER-stress in MM cells, which triggers tumor cell surface loss of HLA-E and upregulation of DR5 concomitant with the activation of autophagy pathways. Induction of ER-stress in MM cells exposed to sub-toxic doses of tunicamycin resulted in a marked reduction of HLA-E expression and a robust upregulation of cell surface DR5 expression on non-apoptotic cells. Although DR5 upregulation has previously been shown to be mediated via ER-stress-induced CHOP activation and ATF3 following proteasome inhibition,Citation24 our study is the first to show that HLA-E expression on tumor cells is substantially reduced as a consequence of ER-stress triggered by proteasome inhibition. We speculate that ER-stress leads to misfolding of and improper peptide loading of the HLA-E molecule in the ER, events which are critical for cell surface presentation of functional and stable HLA-E molecules.Citation25 However, additional mechanisms cannot be excluded, such as previously reported proteasome inhibition-mediated restriction of the peptide pool for HLA-E, leading to abrogation of the dissociation between HLA-E and TAP in the ER.Citation26,Citation27 Moreover, reduced translocation of HLA-E molecules from the ER to the cell surface, a phenomenon previously reported to occur following proteasome inhibition, can also prevent surface expression of new HLA-E molecules.Citation26 Further studies are needed to fully characterize the degree to which each of these mechanisms contributes to the reduction of HLA-E on the cell surface of MM cells following proteasome inhibition.
An important observation from our experiments showing bortezomib-induced suppression of HLA-E on the cell surface was that classical HLA class I expression remained intact. This contrasts the previously reported data by Shi et al. where bortezomib led to a reduction in pan-HLA class I expression on MM cells.Citation22 The discrepancies between that study and ours may be that substantially higher concentrations of bortezomib (10–50 nM vs 5 nM) were used in the study by Shi et al. In our study, we observed that bortezomib doses of 10 nM or higher induced significant cell death in all MM cell lines. As a consequence, we exposed the MM cells to lower concentrations of bortezomib and assessed HLA class I expression specifically on viable cells that were neither apoptotic nor dead (negative for both Annexin V and 7-AAD). In contrast, Shi et al. excluded dead cells by only using the late apoptosis marker 7-AAD, thereby potentially including pre-apoptotic cells in their analysis. Hence, in the study by Shi et al., it is possible their analysis identified cells having a reduction in HLA class I as a consequence of drug-induced early apoptosis. Loss of HLA class I in this context would be less relevant in terms of any drug-induced impact on NK cell tumor cytotoxicity as these MM cells would already be destined to die. Our analysis excluded this potential confounder by including Annexin V in addition to 7-AAD. Furthermore, by flow sorting Annexin V−7-AAD− bortezomib-exposed HLA-Elow MM cells, we confirmed reduction of HLA-E was not part of an apoptosis process. Together with our data demonstrating significant loss of HLA-E expression on MM cells exposed to sub-toxic doses of the ER-stressor tunicamycin, our report collectively supports HLA-E loss rather than HLA class I loss as a mechanism by which MM cells that evade bortezomib-induced cell death become sensitized to NK cell killing.
Several mechanisms may be involved in the selective loss of HLA-E on non-apoptotic MM cells exposed to bortezomib. Our data on BFA-mediated blockade of the transportation of de novo produced HLA molecules to the cell surface of MM cells demonstrate the cell surface half-life of HLA-E is significantly shorter than that of HLA class I. As mentioned above, the tertiary structure and stability of HLA-E is much more dependent on proper protein folding and peptide loading in the ER compared to that of classical HLA class I molecules.Citation21 We speculate that this may explain why HLA-E but not HLA class I expression declines in non-apoptotic cells following bortezomib-induced ER-stress. In this report we do not assess whether proteasome inhibition affects the transcription or translation of HLA-E or HLA class I molecules. However, previous studies showing rapid loss of HLA-E expression following induction of intracellular oxidative stress,Citation28 which also induces ER-stress, have established HLA-E loss occurs mechanistically at a post-translational level rather than at a transcriptional or translational level. Taken together, the vast literature on the synthesis and presentation of HLA-E on the cell surface, the established link between ER-stress and DR5 upregulation following bortezomib treatment of cancer cells and previous work on drug-induced rapid suppression of HLA-E on cancer cells, all strongly support bortezomib-induced loss of HLA-E on MM cells is caused by ER-stress-mediated misfolding of de novo synthesized HLA-E molecules.
A critical role for NKG2A – HLA-E interactions in NK cell-mediated killing of tumor cells has been pointed out in numerous in vitro studies.Citation29,Citation30 As observed in this report, the functional consequence of bortezomib-induced loss of HLA-E expression on MM cells is activation and augmentation of NK cell tumor killing that is controlled solely by the NKG2A receptor. Circumventing tumor escape by down-regulating HLA-E expression using this approach would theoretically be more effective than blockade of the NKG2A receptor on NK cells, as the latter strategy may result in detuned NK cell responsiveness, as was observed in a recent clinical trial exploring KIR blockade in patients with smoldering MM.Citation31 Moreover, employing adoptive transfer of NK cells to bortezomib pre-treated patients would also bypass the need to rely on utilizing endogenous patient NK cells that often are dysfunctional in cancer patients, including in patients with advanced MM.Citation32–Citation35 One such approach currently under investigation in the clinic utilizes a strategy of delaying adoptive transfer of ex vivo expanded NK cells following bortezomib treatment until the drug is cleared from the circulation.Citation36 Infusion of NK cells following administration of bortezomib also opens up the possibility of utilizing a number of different methods to manipulate NK cells ex vivo to optimize their persistence, homing and targeting of MM in vivo.Citation37 We observed that NK cells expanded ex vivo using EBV-LCL feeder cells had superior killing of bortezomib-treated MM cells compared to unexpanded overnight IL-2 activated NK cells. This enhanced killing by expanded NK cells was likely the consequence of them having higher TRAIL expression and containing a larger percentage of NKG2ASP NK cells, which would augment their cytotoxicity against tumors with upregulated DR5 and downregulated HLA-E expression following bortezomib exposure. These preclinical data suggest an expanded NK cell population would have superior clinical efficacy compared to unexpanded overnight IL-2 activated NK cells following adoptive NK cell transfer in myeloma patients pre-treated with bortezomib.
In addition to MM, many tumor types, including breast cancer,Citation38 ovarian carcinoma,Citation39 colon cancer,Citation40 cervical cancer,Citation41 melanoma,Citation42 glioma,Citation43 gastric cancer,Citation44 renal cell carcinoma,Citation45 acute myeloid leukemia,Citation29 over-express HLA-E. While bortezomib has been reported to have single-agent activity in vitro against several of these tumor types, no study to date has addressed whether this drug alters the expression of HLA-E on their cell surface. Based on our findings in MM, studies assessing whether bortezomib or other proteasome inhibitors would trigger suppression of HLA-E expression on non-myeloma tumor cells, potentially sensitizing them to killing by NKG2ASP NK cells, are warranted. In this context, it is important to consider that MM cells are highly sensitive to proteasome inhibition due to their high antibody production, therefore proteasome inhibitor induction of ER-stress may not occur equally amongst other tumor types. Drugs that more directly trigger ER-stress independently of proteasome inhibition could likewise be tested. Groups have established that resveratrol,Citation46 valproic acidCitation47 and tunicamycinCitation48 can be used to induce ER-stress in leukemic cells. Sodium selenite, a compound that is currently being evaluated in the clinic,Citation49 has recently also been shown to induce ER-stress in lymphoma cellsCitation50 and could represent another drug candidate to explore in the context of augmenting NK cell tumor killing, especially given the observation this agent suppresses HLA-E expression on tumor cells in vitro.Citation28 Further studies are clearly needed to address whether bortezomib-induced ER-stress or ER-stress induced by other means can trigger loss of cell surface HLA-E on tumors other than MM to sensitize them to killing by NK cells. Moreover, the potential of these drugs, including bortezomib, to also induce up-regulation of ligands for activation NK cell receptors has yet to be studied as additional mechanisms of tumor sensitization to NK cells.
In conclusion, we discovered that bortezomib triggers the loss of cell surface HLA-E on MM cells, resulting in a novel mechanism through which these tumor cells become selectively sensitized to killing by NKG2ASP NK cells that are only inhibited via ligation to HLA-E on the target cell. Further, we establish for the first time that tumor sensitization to NK cell killing via the TRAIL/DR5 pathway, previously observed on other tumors, also occurs on MM cells. Finally, we show that ex vivo expanded NK cells that have both higher TRAIL surface expression and a greater percentage of NKG2ASP NK cells compared to unexpanded overnight IL-2 activated NK cells mediate a stronger response to bortezomib-exposed MM cells compared to their unexpanded counterpart. Our findings showing ER-stress leads to the selective loss of HLA-E on MM cells suggests combined modality treatment utilizing the adoptive transfer of ex vivo expanded NK cells following treatment with bortezomib could potentially lead to the eradication of MM cells that escape bortezomib-induced apoptosis through ER-stress-induced autophagy. However, before this approach is explored in a clinical setting, additional studied are needed to confirm this mechanism in primary MM cells and establish proof-of-concept in an animal model.
Material and methods
Cells
The MM cell lines EJM, and OPM1 were obtained from Leslie Brents at Walter Reed National Military Medical Center (MD, USA) and the MM.1S was kindly provided by Dr. Irene Ghobrial at Dana-Farber Cancer Institute (MA, USA). The K562 cell line was from ATCC (VA, USA). All cells were propagated in complete media (RPMI 1640 with 2 mM Glutamine (Life Technologies, NY, USA) supplemented with 10% heat-inactivated FBS (Life Technologies). The NK cell isolation kit from Miltenyi (Bergisch Gladbach, Germany) was used to isolate NK cells from frozen peripheral blood mononuclear cells (PBMCs) of healthy donors collected and cryopreserved following informed consent (protocol 99-H-0050). Unexpanded NK cells were activated overnight in complete medium supplemented with 1000 IU/ml recombinant human IL-2 (Proleukin®; Chiron, USA).
NK cell expansion
Isolated from healthy donor NK cells were combined with irradiated EBV-SMI-LCL cells at a ratio of 1:20 in NK cell media (X-VIVO 20 (Lonza) supplemented with 10% heat-inactivated human AB plasma (Sigma Aldrich) and 500 IU/ml of recombinant human IL-2 (Chiron).Citation36 The cells were cultured at 37°C, 6.5% CO2. Half of the media was replaced with fresh NK cell media 5 days into the expansion. NK cells were thereafter counted and adjusted to 0.5–1.0 × 106 cells/ml every 48 hours from day 7 until utilized in experiments at day 14 of expansion.
KIR and HLA genotyping
Genomic DNA was isolated from peripheral blood using the DNeasy® Blood & Tissue Kit (Qiagen). The KIR genotyping kit from Olerup-SSP AB (Stockholm, Sweden) was used for KIR genotyping. KIR ligands were determined by using the KIR HLA ligand kit (Olerup-SSP AB) for detection of the -Bw4, -Cw3 (C1) and -Cw4 (C2) motifs.
TRAIL blockade
Cell surface TRAIL was blocked on NK cells using the unconjugated anti-TRAIL antibody clone RIK-2 (Biolegend, CA, USA) at a final concentration of 10 μg/ml for 30 minutes. Following pre-blockade, the antibody was present in any functional assay at 5 μg/ml.
Cytotoxicity assay
A classical 51chromium (51Cr) release assay (CRA) was used to assess target lysis by NK cells. In brief, NK cells were co-cultured at a ratio of 1:1 with 51Cr-labeled target cells in a final volume of 200 µl in 96-well plates at 37°C and 5% CO2. After 4 hours, supernatant was harvested onto a Luma plate. Counts were measured using a Perkin Elmer 1450 Microbeta Counter and specific target lysis was calculated ((NK cell-induce 51Cr-release – spontaneous 51Cr-release)/(maximum 51Cr-release – spontaneous 51Cr-release) x 100).
Antibodies and reagents
anti-CD56 (B159), anti-CD56 (NCAM-1), anti-CD3 (UCHT1), Brefeldin A, 7-AAD and Annexin V were from Becton Dickinson (BD) (CA, USA). Anti-KIR2DL1/DS1 (EB6), anti-KIR2DL2/3/DS2 (GL183) and anti-NKG2A (Z199) were from Beckman Coulter (CA, USA). The anti-CD107a (H4A3), KIR3DL1 (Dx9), HLA class I (W6/32), HLA-E (3D12), DR5 (MDS5-1) were from Biolegend. The anti-Lir-1 (HP-F1) was from eBioscience (CA, USA). The LIVE/DEAD Fixable Dead Cell Stain kit was from Invitrogen (CA, USA). Tunicamycin was from Sigma Aldrich. Bortezomib was from Millennium Pharmaceuticals (MA, USA). Cisplatin was from APP Pharmaceuticals, LLC (IL, USA).
Brefeldin a assay
MM cells were resuspended in complete media supplemented with 0.5 nM Brefeldin A (BFA). Cells were harvested at the denoted time points to assess HLA class I and HLA-E expression by flow cytometry.
Phenotyping of cells
NK cells or MM cell lines were incubated with appropriate antibodies diluted in FACS buffer (PBS supplemented with 2% FCS and 0.5 mM EDTA) on ice for 15 minutes. Cells were washed three times in FACS buffer prior to acquisition on the BD LSRFortessa. For samples stained with Annexin V, cells were washed in Annexin V-binding buffer (BD) followed by resuspension in Annexin V-containing Annexin V-binding buffer prior to acquisition. When used, 7-AAD was added prior to acquisition.
NK cell degranulation assay
NK cells and target cells were mixed at a ratio of 1:1 in a final volume of 200 µl in 96-well plates. After one hour of co-culture at 37°C and 5% CO2, cells were stained with cell surface antibodies and the LIVE/DEAD marker (Invitrogen) for 15 minutes on ice, followed by washing in FACS buffer and fixation in 1% PFA (MP Biomedicals, CA, USA). Cells were acquired on a BD LSRFortessa. In the analysis, NK cells were defined by CD56+CD3−LIVE/DEAD− lymphocytes.
Flow cytometry cell sorting
MM cells were exposed to 5 nM bortezomib for 24 hours prior to staining. A BDAria Flow Cytometer was used to sort 7-AAD− live HLA-Elow and HLA-Ehigh cells. The sorted cells were collected in medium and directly plated for culturing at 37ºC 5% CO2. The viability was assessed 24 hours later by flow cytometry using Annexin V and 7-AAD.
Er-stress and viability assay
MM cells were resuspended in complete media supplemented with either 1 μM Tunicamycin, 20 μM Cisplatin or 5 nM bortezomib. Cells were harvested after 24 hours to assess viability, HLA class I, HLA-E and DR5 expression by flow cytometry and intracellular protein expression.
Western blot
Cells were lysed in CHAPS buffer (Cell Signal Technology) according to manufacturer’s protocol. Purity and protein quantification was performed using a NanoDrop instrument (Thermo Fisher Scientific, CA, USA). Following pre-heating at 95°C, the isolated protein fractions were separated on a 4–20% Mini-PROTEAN® TGX™ Precast Gels (Bio-Rad) in Tris/Glycine/SDS Running Buffer (Bio-Rad). The Invitrogen™ Novex™ SeeBlue™ Plus2 Pre-stained Protein Standard Ladder (Invitrogen) ladder was used to identify the approximate protein sizes. Proteins were transferred to nitro-cellulose membranes using Trans-Blot® Turbo™ Mini Nitrocellulose Transfer Packs (Bio-Rad). The membrane was blocked with Odyssey® Blocking Buffer diluted in PBS before overnight incubation at + 4°C with either of the following mouse anti-human antibodies binding CHOP (clone L63F7, Cell Signaling Technology), BiP (clone C50B12, Cell Signaling Technology), LC3 (clone D3U4C, Cell Signaling Technology), LAMP-1 (clone D2D11, Cell Signaling Technology), β-Tubulin (Li-Core Biosciences) at 1:1000–1:10000 ratios. Antibodies were incubated in a solution of Odyssey® Blocking Buffer (Biosciences) in PBS with 0.1% Tween-20. After washing, the membrane was incubated with goat-anti mouse or goat-anti-rabbit fluorescent-conjugated antibodies (Li-Core Biosciences) at a 1:10,000 ratio. The blots were imaged with the Odyssey instrument (Li-Cor Biosciences).
Data and statistical analysis
Flow cytometry data was analyzed using the FlowJo software (Treestar Inc.). Western blot data was analyzed using ImageJ (http://rsbweb.nih.gov/ij/). CRA data was analyzed using Excel (Microsoft). Graphs and statistical analyses were performed with PRISM (GraphPad Software Inc.). Wilcoxon was used for paired, and Mann-Whitney was used for unpaired T-tests.
Authorship Contributions
M.C. has designed and conducted all experiments and outlined and written the manuscript. A.N. has performed some experiments. R.R., E.L. and M.B. have helped out with ex vivo expansion of NK cells. C.S.H. has helped out with performing Western blots. R.C. supported in the design of the experiments and writing of the manuscript.
Financial support
This work was supported by funding from the National Heart Lung and Blood Institute, Division of Intramural Research (DIR), NIH, the Dean O’Neill/Rancic Fellowship and the Swedish Research Council for funding of M.C.
Supplemental Material
Download PDF (2.4 MB)Acknowledgments
This work was supported by funding from the National Heart Lung and Blood Institute, Division of Intramural Research (DIR), National Institutes of Health. We would like to thank Leslie Brents and Irene Ghobrial for providing us with MM cell lines, and Keyvan Keyvanfar for assisting with FACS sortings. We would also like to acknowledge the Dean O’Neill/Rancic Fellowship for their kind support of the lab and the Swedish Research Council for funding of M.C.
Disclosure statement
The authors report no conflicts of interest.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510. doi:10.1038/ni1582.
- Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, Breso V, Frassati C, Reviron D, Middleton D, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25(2):331–342. doi:10.1016/j.immuni.2006.06.013.
- Brodin P, Karre K, Hoglund P. NK cell education: not an on-off switch but a tunable rheostat. Trends Immunol. 2009;30(4):143–149. doi:10.1016/j.it.2009.01.006.
- Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23(11):1964–1979. doi:10.1038/leu.2009.173.
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. doi:10.1182/blood-2005-08-3531.
- Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32(10):469–476. doi:10.1016/j.tibs.2007.09.003.
- Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14(9):1576–1582. doi:10.1038/sj.cdd.4402200.
- Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi:10.1038/nrm3735.
- Chauhan D, Hideshima T, Mitsiades C, Richardson P, Anderson KC. Proteasome inhibitor therapy in multiple myeloma. Mol Cancer Ther. 2005;4(4):686–692. doi:10.1158/1535-7163.MCT-04-0338.
- Mateos MV, Oriol A, Martinez-Lopez J, Gutierrez N, Teruel AI, de Paz R, García-Laraña J, Bengoechea E, Martín A, Mediavilla JD, et al. Bortezomib, melphalan, and prednisone versus bortezomib, thalidomide, and prednisone as induction therapy followed by maintenance treatment with bortezomib and thalidomide versus bortezomib and prednisone in elderly patients with untreated multiple myeloma: a randomised trial. Lancet Oncol. 2010;11(10):934–941. doi:10.1016/S1470-2045(10)70187-X.
- Gutman D, Morales AA, Boise LH. Acquisition of a multidrug-resistant phenotype with a proteasome inhibitor in multiple myeloma. Leukemia. 2009;23(11):2181–2183. doi:10.1038/leu.2009.123.
- Chauhan D, Li G, Shringarpure R, Podar K, Ohtake Y, Hideshima T, Anderson KC. Blockade of Hsp27 overcomes Bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Research. 2003;63(19):6174–6177.
- Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112(6):2489–2499. doi:10.1182/blood-2007-08-104950.
- Ri M, Iida S, Nakashima T, Miyazaki H, Mori F, Ito A, Inagaki A, Kusumoto S, Ishida T, Komatsu H, et al. Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia. 2010;24(8):1506–1512. doi:10.1038/leu.2010.137.
- Schewe DM, Aguirre-Ghiso JA. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Research. 2009;69(4):1545–1552. doi:10.1158/0008-5472.CAN-08-3858.
- de Wilt LH, Kroon J, Jansen G, de Jong S, Peters GJ, Kruyt FA. Bortezomib and TRAIL: a perfect match for apoptotic elimination of tumour cells? Crit Rev Oncol Hematol. 2013;85(3):363–372. doi:10.1016/j.critrevonc.2012.08.001.
- Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, Childs R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Research. 2006;66(14):7317–7325. doi:10.1158/0008-5472.CAN-06-0680.
- Ames E, Hallett WH, Murphy WJ. Sensitization of human breast cancer cells to natural killer cell-mediated cytotoxicity by proteasome inhibition. Clin Exp Immunol. 2009;155(3):504–513. doi:10.1111/j.1365-2249.2008.03818.x.
- Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, Sayers TJ, Hudig D, Murphy WJ. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunology. 2008;180(1):163–170. doi:10.4049/jimmunol.180.1.163.
- Moreau P, Coiteux V, Hulin C, Leleu X, van de Velde H, Acharya M, Harousseau J-L. Prospective comparison of subcutaneous versus intravenous administration of bortezomib in patients with multiple myeloma. Haematologica. 2008;93(12):1908–1911. doi:10.3324/haematol.13285.
- Braud VM, Allan DS, O’Callaghan CA, Soderstrom K, D’Andrea A, Ogg GS, Lazetic S, Young NT, Bell JI, Phillips JH, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391(6669):795–799. doi:10.1038/35869.
- Shi J, Tricot GJ, Garg TK, Malaviarachchi PA, Szmania SM, Kellum RE, Storrie B, Mulder A, Shaughnessy JD, Barlogie B, et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2008;111(3):1309–1317. doi:10.1182/blood-2007-03-078535.
- Reger R, Berg M, Lundqvist A, Donohue T, Carlsten M, Betters D, Cook L, Ramos C, Grasmeder S, Su S, et al. A Phase I trial of adoptively transferred Ex-vivo expanded autologous natural killer (NK) cells following treatment with bortezomib to sensitize tumors to NK cell cytotoxicity. American Society of Hematology. Volume 118: Blood; 2011. p Abstract 1001.
- Xu L, Su L, Liu X. PKCdelta regulates death receptor 5 expression induced by PS-341 through ATF4-ATF3/CHOP axis in human lung cancer cells. Mol Cancer Ther. 2012;11(10):2174–2182. doi:10.1158/1535-7163.MCT-12-0602.
- Ulbrecht M, Kellermann J, Johnson JP, Weiss EH. Impaired intracellular transport and cell surface expression of nonpolymorphic HLA-E: evidence for inefficient peptide binding. J Exp Med. 1992;176(4):1083–1090.
- Ulbrecht M, Modrow S, Srivastava R, Peterson PA, Weiss EH. Interaction of HLA-E with peptides and the peptide transporter in vitro: implications for its function in antigen presentation. J Immunology. 1998;160(9):4375–4385.
- Bland FA, Lemberg MK, McMichael AJ, Martoglio B, Braud VM. Requirement of the proteasome for the trimming of signal peptide-derived epitopes presented by the nonclassical major histocompatibility complex class I molecule HLA-E. J Biol Chem. 2003;278(36):33747–33752. doi:10.1074/jbc.M305593200.
- Enqvist M, Nilsonne G, Hammarfjord O, Wallin RP, Bjorkstrom NK, Bjornstedt M, Hjerpe A, Ljunggren H-G, Dobra K, Malmberg K-J, et al. Selenite induces posttranscriptional blockade of HLA-E expression and sensitizes tumor cells to CD94/NKG2A-positive NK cells. J Immunology. 2011;187(7):3546–3554. doi:10.4049/jimmunol.1100610.
- Godal R, Bachanova V, Gleason M, McCullar V, Yun GH, Cooley S, Verneris MR, McGlave PB, Miller JS. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol Blood Marrow Transplant. 2010;16(5):612–621. doi:10.1016/j.bbmt.2010.01.019.
- Nguyen S, Beziat V, Dhedin N, Kuentz M, Vernant JP, Debre P, Vieillard V. HLA-E upregulation on IFN-gamma-activated AML blasts impairs CD94/NKG2A-dependent NK cytolysis after haplo-mismatched hematopoietic SCT. Bone Marrow Transplant. 2009;43(9):693–699. doi:10.1038/bmt.2008.380.
- Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, Landgren O, Childs RW. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res. 2016;22(21):5211–5222. doi:10.1158/1078-0432.CCR-16-1108.
- Dosani T, Carlsten M, Maric I, Landgren O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of myeloma [corrected] and their uses in immunotherapies. Blood Cancer J. 2015;5:e306. doi:10.1038/bcj.2015.32.
- Carlsten M, Norell H, Bryceson YT, Poschke I, Schedvins K, Ljunggren HG, Kiessling R, Malmberg K-J. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J Immunology. 2009;183(8):4921–4930. doi:10.4049/jimmunol.0901226.
- Carlsten M, Baumann BC, Simonsson M, Jadersten M, Forsblom AM, Hammarstedt C, Bryceson YT, Ljunggren H-G, Hellström-Lindberg E, Malmberg K-J. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia. 2010;24(9):1607–1616. doi:10.1038/leu.2010.149.
- Fauriat C, Just-Landi S, Mallet F, Arnoulet C, Sainty D, Olive D, Costello RT. Deficient expression of NCR in NK cells from acute myeloid leukemia: evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood. 2007;109(1):323–330. doi:10.1182/blood-2005-08-027979.
- Childs RW, Berg M. Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology Am Soc Hematol Educ Program. 2013;2013:234–246. doi:10.1182/asheducation-2013.1.234.
- Carlsten M, Childs RW. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front Immunol. 2015;6:266. doi:10.3389/fimmu.2015.00266.
- de Kruijf EM, Sajet A, van Nes JG, Natanov R, Putter H, Smit VT, Liefers GJ, van Den Elsen PJ, van de Velde CJH, Kuppen PJK. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunology. 2010;185(12):7452–7459. doi:10.4049/jimmunol.1002629.
- Malmberg K-J, Levitsky V, Norell H, de Matos CT, Carlsten M, Schedvins K, Rabbani H, Moretta A, Söderström K, Levitskaya J, et al. IFN-gamma protects short-term ovarian carcinoma cell lines from CTL lysis via a CD94/NKG2A-dependent mechanism. J Clin Invest. 2002;110(10):1515–1523. doi:10.1172/JCI15564.
- Levy EM, Bianchini M, Von Euw EM, Barrio MM, Bravo AI, Furman D, Domenichini E, Macagno C, Pinsky V, Zucchini C, et al. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int J Oncol. 2008;32(3):633–641.
- Goncalves MA, Le Discorde M, Simoes RT, Rabreau M, Soares EG, Donadi EA, Carosella ED. Classical and non-classical HLA molecules and p16(INK4a) expression in precursors lesions and invasive cervical cancer. Eur J Obstet Gynecol Reprod Biol. 2008;141(1):70–74. doi:10.1016/j.ejogrb.2008.06.010.
- Derre L, Corvaisier M, Charreau B, Moreau A, Godefroy E, Moreau-Aubry A, Jotereau F, Gervois N. Expression and release of HLA-E by melanoma cells and melanocytes: potential impact on the response of cytotoxic effector cells. J Immunology. 2006;177(5):3100–3107. doi:10.4049/jimmunol.177.5.3100.
- Wischhusen J, Friese MA, Mittelbronn M, Meyermann R, Weller MHLA-E. protects glioma cells from NKG2D-mediated immune responses in vitro: implications for immune escape in vivo. J Neuropathol Exp Neurol. 2005;64(6):523–528.
- Dutta N, Majumder D, Gupta A, Mazumder DN, Banerjee S. Analysis of human lymphocyte antigen class I expression in gastric cancer by reverse transcriptase-polymerase chain reaction. Hum Immunol. 2005;66(2):164–169. doi:10.1016/j.humimm.2004.10.010.
- Seliger B, Jasinski-Bergner S, Quandt D, Stoehr C, Bukur J, Wach S, Legal W, Taubert H, Wullich B, Hartmann A. HLA-E expression and its clinical relevance in human renal cell carcinoma. Oncotarget. 2016;7(41):67360–67372. doi:10.18632/oncotarget.11744.
- Liu BQ, Gao YY, Niu XF, Xie JS, Meng X, Guan Y, Wang H-Q. Implication of unfolded protein response in resveratrol-induced inhibition of K562 cell proliferation. Biochem Biophys Res Commun. 2010;391(1):778–782. doi:10.1016/j.bbrc.2009.11.137.
- Segar KP, Chandrawanshi V, Mehra S. Activation of unfolded protein response pathway is important for valproic acid mediated increase in immunoglobulin G productivity in recombinant Chinese hamster ovary cells. J Biosci Bioeng. 2017. doi:10.1016/j.jbiosc.2017.05.005.
- Haefliger S, Klebig C, Schaubitzer K, Schardt J, Timchenko N, Mueller BU, Pabst T. Protein disulfide isomerase blocks CEBPA translation and is up-regulated during the unfolded protein response in AML. Blood. 2011;117(22):5931–5940. doi:10.1182/blood-2010-08-304485.
- Brodin O, Eksborg S, Wallenberg M, Asker-Hagelberg C, Larsen EH, Mohlkert D, Lenneby-Helleday C, Jacobsson H, Linder S, Misra S, et al. Pharmacokinetics and toxicity of sodium selenite in the treatment of patients with carcinoma in a Phase I clinical trial: the SECAR study. Nutrients. 2015;7(6):4978–4994. doi:10.3390/nu7064978.
- Shigemi Z, Manabe K, Hara N, Baba Y, Hosokawa K, Kagawa H, Watanabe T, Fujimuro M. Methylseleninic acid and sodium selenite induce severe ER stress and subsequent apoptosis through UPR activation in PEL cells. Chem Biol Interact. 2017;266:28–37. doi:10.1016/j.cbi.2017.01.027.