
ABSTRACT
Cancer-associated mutations, mostly single nucleotide variations, can act as neoepitopes and prime targets for effective anti-cancer T-cell immunity. T cells recognizing cancer mutations are critical for the clinical activity of immune checkpoint blockade (ICB) and they are potent vaccine antigens. High frequencies of mutation-specific T cells are rarely spontaneously induced. Hence, therapies that broaden the tumor specific T-cell response are of interest. Here, we analyzed neoepitope-specific CD8+ T-cell responses mounted either spontaneously or after immunotherapy regimens, which induce local tumor inflammation and cell death, in mice bearing tumors of the widely used colon carcinoma cell line CT26. A comprehensive immune reactivity screening of 2474 peptides covering 628 transcribed CT26 point mutations was conducted. All tested treatment regimens were found to induce a single significant CD8+ T-cell response against a non-synonymous D733A point mutation in the Smc3 gene. Surprisingly, even though Smc3 D733A turned out to be the immune-dominant neoepitope in CT26 tumor bearing mice, neither T cells specific for this neoepitope nor their T cell receptors (TCRs) were able to recognize or lyse tumor cells. Moreover, vaccination with the D733A neoepitope did not result in anti-tumoral activity despite induction of specific T cells. This is to our knowledge the first report that neoepitope specific CD8+ T cells primed by tumor-released antigen exposure in vivo can be functionally irrelevant.
Introduction
T cells are key effectors in cancer immunity. A preexisting T-cell response against tumor antigens, in particular mutated ones which may act as neoepitopes, is a prerequisite for PD-1/PD-L1 checkpoint inhibition to work.Citation1,Citation2 A high mutational burden is associated with higher numbers of neoepitope specific T cells which are believed to play a crucial role in tumor immunityCitation3 and responses to ICB.Citation4–Citation6,Citation7–Citation9 Thus, patients with immunogenic tumors such as melanoma or microsatellite instable tumors are more likely to benefit from ICB.Citation10 In this regard, it was demonstrated that tumor infiltration by CD8+ T cellsCitation9,Citation11 as well as a high mutational burdenCitation6,Citation7,Citation9 correlates with clinical response to ICB.
Neoepitope specific T-cell vaccinesCitation12–Citation14 have the potential to broaden tumor specific T-cell responses and to synergize with ICB. Vaccination requires selection of candidates from dozens to thousands of mutated genes identified by computational pipelines analyzing data from comparative next generation sequencing of tumor versus healthy tissue.Citation15 It is still unclear how to select the most relevant vaccine targets. Expression of mutations on the RNA level and their predicted capability of binding to the patient`s major histocompatibility complex (MHC) are the most basic selection criteria which are used. This, however, may identify false positives which are not expressed on the protein level in the cancer cells or not presented on MHC molecules, respectively. As a result, T cells elicited by vaccination against such candidate neoepitopes would not be able to recognize and lyse tumor cells.
An alternative to administering a vaccine for inducing tumor-specific T-cell responses may be to “vaccinate from within” by promoting inflammation and cancer cell death at the tumor site. Endogenous proteins released from tumor cells may be taken up and presented by Dendritic cells (DCs), which, if the right immunomodulatory context were provided, could result in priming of a tumor-specific T-cell response. A variety of treatment modalities are known to cause tumor cell death, antigen release and DC maturation, including Toll like receptor (TLR) agonists,Citation16 chemotherapy and radiation therapy.Citation17
Here we investigated the magnitude, frequency and therapeutic relevance of CD8+ T-cell responses to neoepitopes induced by such treatment regimens in the widely used murine colon carcinoma model CT26.
Results
We employed an unbiased forward immunology screening approach to systematically test point mutations identified by whole exome sequencing of CT26 cells for recognition by CD8+ T cells obtained from tumor-bearing mice. To this aim, we created a matrix of 2474 peptides covering all 628 cancer-associated point-mutated sequences for which transcription had been confirmed in RNA sequencing data of CT26 tumor cells. Each mutated candidate epitope was represented by four 15-mer peptides overlapping by 11 amino acids. For mutations at the 5ʹ or 3ʹ end of a gene fewer peptides were required to cover the relevant sequence. The peptide matrix featured 51 pools each with 96–105 peptides and each individual peptide was represented in 2 pools. This matrix was used to test T cells in splenocytes of tumor-bearing mice ex vivo by IFNγ ELISpot and determine against which naturally processed and presented point-mutated antigens T cells were prevalent ()).
Figure 1. Treatment of CT26 bearing mice with a TLR7 agonist induces a discrete neoepitope – specific T-cell response against mutated Smc3. A: Design of the peptide matrix encoding all 628 transcribed non synonymous single nucleotide variants (nsSNVs) of CT26. B: Splenocytes were isolated from CT26-WT tumor bearing mice (n = 3, 28 days after tumor inoculation, mean tumor size ~800mm3). 5 × 105 CD4 depleted cells per well were tested for recognition of matrix peptides or 5 × 104 CT26-WT cells in an IFNγ ELISpot. C-D: CT26-WT (C) or CT26-gp70KO (D) tumor bearing mice (n = 5) were treated repetitively with SC1, an TLR7 agonist injected into the tumor starting at day 14 (tumor size ~50 mm3). T-cell responses were analyzed by ELISpot on day 31 as described above. E-F: Splenocytes from TLR7 treated CT26-WT (E) or CT26-gp70KO (F) tumor bearing mice were tested for recognition of Smc3 and gp70 AH1 peptides at 0.4 µg/ml (same concentration as compared to the peptide matrix) or 2 µg/ml as well as CT26-WT or CT26-gp70KO cells by IFNγ ELISpot. Mean + s.e.m. of duplicates is shown.
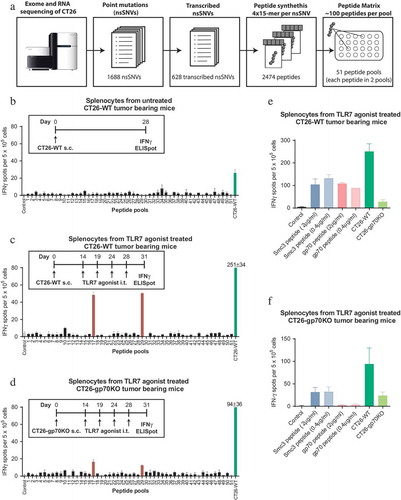
First, we tested for spontaneously occuring point-mutation specific CD8+ T cells in untreated mice bearing subcutaneous (s.c.) CT26 tumors. Splenocytes were harvested 28 days after the mice were inoculated with CT26 tumor cells (CT26-WT) and tumors had reached a mean size of ~800 mm3. CD4+ T cell depleted splenocytes were tested in IFNγ ELISpot for recognition of the peptide matrix pools. IFNγ secretion by CD8+ T cells co-cultured with CT26-WT cells was very low and none of the point mutations was specifically recognized ()). We only detected a T-cell response against the H2-Ld restricted epitope SPSYVYHQF (also called ‘AH1ʹ) of gp70, a well-known non-mutated immunodominant epitope derived from an endogenous retrovirus (Supplementary Figure 1),Citation18 which is the highest expressed gene in CT26.Citation19
Having shown the lack of spontaneously occurring neoepitope specific T cells in this mouse tumor model, we hypothesized that we could broaden the repertoire of tumor-directed T-cell responses by increasing tumor cell death and thereby antigen release in the context of immunomodulation. To this end we conducted three series of experiments in which tumor-bearing mice were treated with either a TLR7 agonist, were vaccinated in combination with local irradiation or were treated with an anti-PD-L1 antibody for immune checkpoint blockade.
SC1, a novel TLR7 agonistic small molecule, is reported to induce potent and durable T cell-mediated tumor control and inflammatory change of the tumor microenvironment,Citation20,Citation21manuscript in preparation. To ensure sufficient antigen exposure and time for priming of T cells, we treated mice 14 days after tumor inoculation when tumors reached a size of 50mm3 with intratumoral (i.t.) injection of SC1. On day 31 after tumor inoculation, about two weeks after starting treatment, CD4+ T cell-depleted splenocytes were tested for recognition of the peptide matrix pools. Two peptide pools, 17 and 29, were shown to induce IFNγ secretion above background levels ()). Both pools contained altered peptides derived from a mutant Smc3 (Structural maintenance of chromosomes 3) D733A neoepitope. Smc3 encodes a nuclear protein involved in mitosis that can be secreted after post-translational modification. OverexpressedCitation22 but not mutated Smc3 was described to be involved in tumorigenesis suggesting that the detected D733A alteration is a passenger mutation.
To exclude that the highly abundant gp70 epitope would inhibit the induction of diversified CD8+ T-cell responsesCitation23 we used the CRISPR/Cas9 system to generate a gp70 deficient variant (CT26-gp70KO), which was no longer recognized by gp70 AH1-specific CD8+ T cells (Supplementary Figure 2). T cells from CT26-gp70KO tumor bearing mice treated with the TLR7 agonist on day 14 (tumor size ~50mm3) still only recognized the two pools containing the Smc3 peptides ()). Single peptide testing confirmed that the induced T cells were specific for mutated Smc3 ()). As expected, T cell reactivity against gp70 was only observed in CT26-WT but not CT26-gp70KO mice ()). Consistently, reactivity against CT26-WT but not CT26-gp70KO cells was diminished in CT26-gp70KO compared to CT26-WT tumor bearing mice ()).
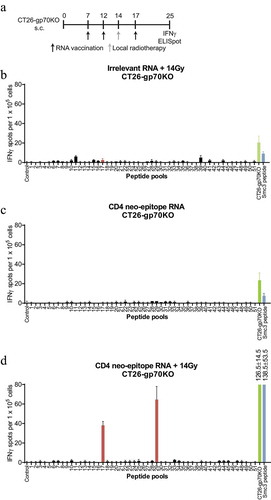
Previously, we have shown that vaccine-induced CD4+ T cells recognizing CT26-derived neoepitopes can license DCs for priming of tumor specific CD8+ T cellsCitation5 and thus promote antigen spread. We therefore applied vaccination with MHC class II neoepitopes and combined it with local tumor irradiation to further increase release and uptake of tumor antigens by intratumoral DCs. CT26-gp70KO tumor bearing mice were repetitively vaccinated on day 7, 12 and 17 with RNA lipoplexes encoding multiple MHC class II restricted neoepitopes (PMECitation5) and locally irradiated with 14 Gy on day 14. This combined regimen resulted in the induction of Smc3 neoantigen-specific CD8+ T cells. RNA vaccination or radiotherapy alone did not result in Smc3-specific T cells although reactivity towards CT26-WT and CT26-gp70KO cells could be measured ().
Figure 2. Combination of CD4 neoepitope vaccination and local radiotherapy induces a discrete neoepitope – specific T-cell response only against mutated Smc3. CT26-gp70KO tumor bearing mice (n = 2 per group) were treated with 14 Gy local radiotherapy, a 40 µg of a RNA lipoplex based MHC class II neoepitope vaccine or both as (schedule depicted). 1 × 105 isolated CD8+ splenocytes together with 5 × 104 syngeneic bone-marrow-derived DCs were tested for recognition of the matrix peptide pools, 5 × 104 CT26-gp70KO cells or 2 µg/ml Smc3 peptide by IFNγ ELISpot. Mean + s.e.m. of duplicates is shown.
In the CT26 model tumor, infiltrating CD8+ T cells express PD-1 and tumor cells express PD-L1 (data not shown). Consequently, we tested reinvigoration of these suppressed tumor-specific T cells by PD-L1 blockade as a possible means to mobilize a broader repertoire of T-cell responses. CT26 tumor bearing mice were treated on day 16, 19, 23 and 26 with an anti-PD-L1 antibody and mutation-specific T-cell responses in the spleen were analyzed on day 28. Again only a Smc3-specific response was observed in CT26-WT tumor bearing mice, which was weaker than responses induced by the other regimens (Supplementary Figure 3).
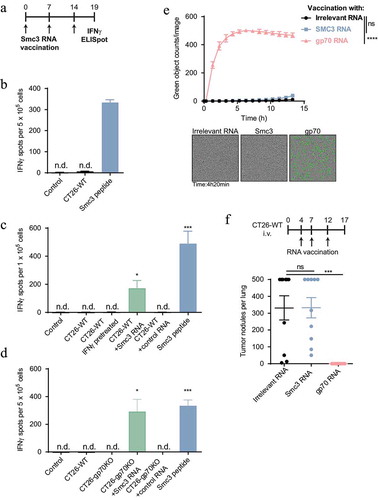
Having shown that three different treatment regimens result in induction of T-cell responses against Smc3, we set out to analyze T-cell responses against this antigen in more detail. We vaccinated mice with Smc3-encoding RNA lipoplexes and tested splenocytes for the recognition of CT26-WT cells ex vivo. Smc3 peptide loaded cells but not CT26-WT cells were recognized ()). As processing of the mutated Smc3 could depend on the immunoproteasome we pretreated CT26-WT cells with IFNγ, an inducer of immunoproteasomal genes. However, IFNγ pretreated CT26 cells were also not recognized by Smc3-specific T cells ()) despite their elevated MHC class I surface expression (Supplementary Figure 4). Only electroporation of CT26-WT cells with mutated Smc3 encoding RNA (27 amino acids of Smc3 flanked by the signal domain and transmembrane domain of MHC class I to deliver the antigen into the ER for improved immunogenicityCitation24) resulted in recognition by splenocytes indicating that in principle the epitope can be processed and presented by tumor cells ()). Similarly, CT26-gp70KO cells were only recognized by Smc3-specific T cells when electroporated with Smc3 RNA ()). Smc3-specific T cells did not mediate CT26-WT cell lysis in vitro ()) and failed to control tumor growth in vivo ()).
Figure 3. Smc3 neoantigen-specific T cells do not recognize and kill CT26 tumor cells ex vivo or in vivo. A: Schematic overview of Smc3 RNA lipoplex vaccination and testing of tumor cell recognition ex vivo. B-D: splenocytes of Smc3 RNA vaccinated mice (schedule see A, n = 1 for B, n = 3 for C and D) were tested ex vivo for recognition of 5 × 104 CT26 cells, Smc3 or control RNA electroporated CT26-WT (C) or CT26-gp70KO cells (D) as well as IFNγ stimulated CT26-WT cells (C). Smc3 peptide stimulation severed as control for successful vaccination. Mean + s.e.m. is shown. E: Cytotoxicity towards CT26-WT cells of CD8+ T cells isolated after vaccination with Smc3 RNA, gp70 RNA or control RNA lipoplexes induced CD8+ T cells towards CT26-WT cells (n = 3 per group, vaccination schedule as in A). Caspase-3/7 positive dying CT26-WT cells are stained green by IncuCyte® live-cell analysis. Quantification of green signals over time (mean ± s.e.m.) and exemplary cell images are shown. F: BALB/c mice (n = 10) were inoculated i.v. with CT26-WT tumors and vaccinated with Smc3, gp70 or irrelevant RNA lipoplexes. 17 days after tumor inoculation, lung tumor nodules were counted. Mean ± s.e.m. is shown. n.d.; not detected.
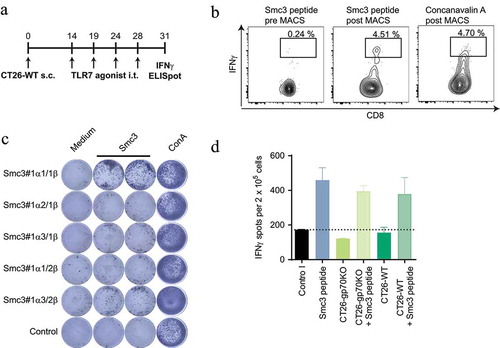
T cells primed upon tumor antigen release under inflammatory conditions in vivo may have a higher T cell receptor (TCR) avidity as compared to T cells induced by vaccination.Citation25 For testing this, we retrieved TCRs from Smc3-specific T cells induced in CT26-WT tumor bearing mice by treatment with TLR7 agonist therapy. Splenocytes were restimulated with Smc3 peptide pulsed BMDCs and IFNγ positive cells were sorted via magnetic cell separation followed by fluorescence-activated cell sorting ()). TCR sequences of sorted T cells were cloned and in vitro transcribed to RNA. RNA-encoded TCR alpha/beta pairs were electroporated into murine T cells for specificity testing. One of the five tested TCR alpha beta pairs induced strong IFNγ secretion upon stimulation with BMDCs, CT26-WT and CT26-gp70KO, which were loaded with Smc3 peptide ()). Unpulsed CT26 cells with endogenous Smc3 expression, however, were not recognized by these inflammation-induced T cells.
Figure 4. T cells engineered with Smc3-specific TCRs do not mediate recognition of CT26 tumor cells. Splenocytes of CT26-WT tumor bearing mice (n = 3) treated with TLR7 therapy i.t. (as summarized in A) were restimulated with Smc3 peptide pulsed BMDC or Concanavalin A. IFNγ positive cells were subsequently isolated via an IFNγ secretion assay followed by MACS enrichment and fluorescence-activated cell sorting of single labeled cells. Exemplary flow cytometry plots of IFNγ secreting CD8+ positive T cells pre and post enrichment are shown (B). Single Smc3 stimulated IFNγ s cells were subsequently isolated, TCRs cloned and RNA encoding TCR α and β chains electroporated into naïve T cells. C: Electroporated T cells were then tested for recognition of Smc3 peptide via IFNγ ELISpot. D: T cells expressing the Smc3 reactive TCR Smc3#1α1/1β were afterwards tested by ELISpot for recognition of CT26-WT or CT26-gp70KO cells with or without addition of Smc3 peptide. Mean + s.e.m. of duplicates is shown.
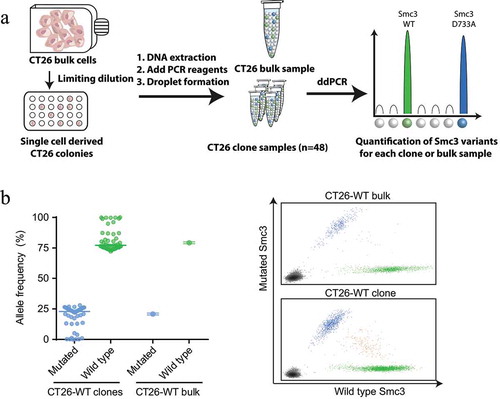
Analysis of the exome and RNA sequencing data of CT26 cells revealed that the fraction of reads covering the Smc3 mutation was as low as 10–16%, respectively (Supplementary Table 1). A low variant allele fraction may indicate subclonality of the mutation. Subclonal antigens have been shown to be associated with failure of anti-PD-1 checkpoint blockade.Citation26 Representation of the Smc3 mutation in a small fraction of CT26 tumor cells may explain the lack of tumor cell recognition and killing in vitro ()) and tumor control in vivo ()). To determine the degree of subclonality in CT26 cells we analyzed populations of single cell clones by droplet digital PCR using mutation- and wild type-specific TaqMan™ probes ()). The vast majority of clones had a similar variant allele frequency as the CT26 bulk population ()). One out of four gene copies contained the D733A alteration which suggests that the Smc3 gene was duplicated with one out of four alleles being mutated. Only 6 of 48 analyzed clones had a mutated variant allele frequency below 1%. Thus, this data does not support that lack of recognition of CT26 cells by Smc3-specific T cells is a matter of subclonality of Smc3.
Figure 5. Mutated Smc3 is detectable in the majority of CT26 tumor cells. A: Experiment outline. ddPCR: droplet digital PCR, WT: wild type. B: Depicted is the allele frequency of mutated and wild type Smc3 in a population of CT26-WT clones (mean, each dot represents one clone) or CT26-WT bulk cells (mean ± s.d. of four technical replicates) determined via droplet digital PCR (left). Exemplary graphs for CT26-WT bulk cells and a CT26-WT clone is depicted (right).
The finding that Smc3-specific T cells are capable of recognizing CT26-WT electroporated with Smc3 ()) may indicate that CT26 cells are able to process the mutated Smc3 epitope but do not produce sufficient amounts of the mutated epitope/MHC complexes from endogenous Smc3. To probe this hypothesis, we analyzed the presence of 20 predicted H2-Kd ligands of CT26-WT cells, including Smc3, (Supplementary Table 1) by a targeted ligandomics approach,Citation27 which provides higher specificity and increased sensitivity compared to the usually performed untargeted liquid chromatography–tandem mass spectrometry (LC-MS2). Using an H2-Kd specific antibody, MHC I binders were enriched by immunoprecipitation (IP) and separated by Ultra-Performance Liquid Chromatography (UPLC). Subsequently, isolated H2-Kd ligands were analyzed by the liquid chromatography–multiple reaction monitoring cubed (LC-MRM3) scanning modeCitation28 which resulted in MS3 spectra for pre-selected and pre-optimized peptide fragment ions. The non-immunogenic peptide derived from the mutated Nav2 gene (immunogenicity was tested by IFNγ ELISpot of splenocytes from RNA vaccinated mice, data not shown) was robustly detected in three independent replicates. The chromatographic profile for this peptide in the IP samples was measured at the same time as for the synthetic peptide and with matching relative intensities for all four measured fragments (), left). MS3 spectra of the fragment b9+ H2O+2 detected in the IP sample had the same MS3 fingerprint with characteristic peaks and their relative ratios as those obtained for the synthetic peptide (), right). The same was true for the other three measured fragments (not shown) confirming the presences of the mutated Nav2 peptide in the IP sample.
Figure 6. Direct identification of neoepitopes in immune precipitates from CT26-WT cells by targeted LC-MRM3 mass spectrometry. The presence of candidate neoepitopes (Supplementary Table 1) in CT26 H2-Kd immune precipitates was assessed by monitoring chromatographic profiles of four fragments of the peptide (A-B, left), and ascertaining the identity of these fragments by MS3 (A-B, right). Only one fragment MS3 spectrum per peptide is shown for clarity reasons. In all panels, profiles/spectra of immune precipitates are shown above the x-axis, those of the synthetic reference peptides below the x-axis. A: Detection of the mutated, non-immunogenic control peptide derived from the Nav2 protein. B: Analysis of the mutated Smc3 peptide. Experiments were conducted in three replicates. Results of a representative experiment are shown.
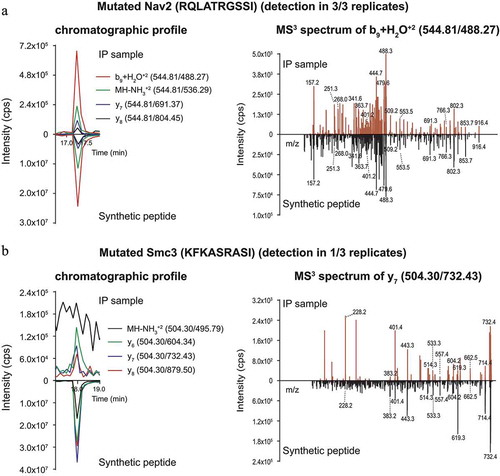
In contrast, the neoepitope derived from the mutated Smc3 protein was detected reliably in only one replicate, whereas signal intensities were very low (at the limit of detection (LOD)) in two other replicates ()). In these two IP samples, the peptide did elute at the same time as the synthetic counterpart. However, chromatographic profiles and MS3 spectra could only be measured for three out of four fragments. The chromatographic profiles of the detected fragments in the IP samples were measured at the same time as those of the synthetic peptide, but the intensity ratios did not match (), left). The MS3 fingerprints contained the characteristic peaks for all three detected fragments, but the ratios of peak intensities again did not match those obtained for the synthetic peptide (), right). This was also true for the other two detected fragments (data not shown). As the targeted LC-MRM3 method is extremely specific, the detection and identity confirmation by the main characteristic peaks of three out of four measured fragments should be sufficient to confirm the presence of mutated Smc3. However, the discrepancies in ratios of peak intensities and the problematic detection in 2 out of 3 replicates suggest that the Smc3 neoepitope is, if at all, presented only at minute amounts on MHC class I of CT26 cells. Thus, although mutated Smc3 protein was sufficiently expressed to induce priming after non-antigen-directed immunotherapy, our data suggests that the amount of MHC ligands on tumor cells most likely did not suffice to allow the recognition by Smc3-specific T cells.
Discussion
In this study we used a comprehensive forward immunology approach to test all 628 cancer-associated point mutations in CT26 colon cancer cells for recognition by T cells induced by three different cell death inducing immune modulatory regimens. The only mutation-specific immune response induced by all three treatments was directed against mutant Smc3. In theory, it is possible that screening of T cells derived from tumors or draining lymph nodes would have yielded additional responses. However, low cell numbers available from these organs prevented a through screening with our platform. In this regard, a study analyzing neoepitope specific CD8+ T cells in peripheral blood and tumor infiltrating lymphocytes (TILs) in melanoma patients showed that 8 out of 9 responses identified in TILs were also present in the bloodCitation29 indicating that neoepitope specific T cells can usually be found systemically. Similar to the study presented here, only a very low fraction of neoepitope specific T cells could be identified. From thousands of point mutations, 459 predicted MHC class I-binding peptides were selected but only 9 were found to be recognized by T cells. Moreover, a study performed in melanoma patients treated with adoptive T-cell or anti-CTLA-4 therapy found only roughly 0.5% of tested candidate neoepitopes to be recognized by CD4+ T cells.Citation30 In contrast, the fraction of immunogenic point mutations can be strongly increased when a neoepitope vaccine is administered.Citation12–Citation14 Along this line, 105 out of the 628 expressed point mutations in CT26 were predicted to bind MHC class I (IEDB V2.13, percentile rank ≤1). In a prior study, a fraction of these predicted MHC binders was tested for immunogenicity after RNA vaccination, with four of them being recognized by CD8+ T cells.Citation12 None of these four targets was found in the screens presented here hypothetically because their expression levels were too low to allow priming by tumor derived antigen exposure.
As our data also indicates that CT26 cells deficient for the immune dominant gp70 antigen are recognized by CD8+ T-cell responses, there are most likely additional immunogenic antigens beyond Smc3 and gp70 in CT26 cells, which are, however, most likely not derived from point mutations.
The Smc3 D733A specific T cells, which were induced under conditions of increased antigen release from CT26 tumors, were not able to recognize tumor cells. To the best of our knowledge this is the first reported example of a therapeutically induced neoepitope-specific CD8+ T-cell response, which remains without functional relevance due to lack of tumor cell recognition. This is an important finding as many studies assume that preexisting, tumor antigen-specific T cells are necessarily involved in tumor control without testing for direct tumor cell recognition. For personalized neoantigen vaccination, it is debated whether preexisting or de novo induced T-cell responses against neoantigens are to be preferred. Preexisting immunity is considered to assure the expression of the neoantigen in tumor cells and processing and presentation by professional antigen-presenting cells (APCs). However, our data exemplifies that this may not always be the case. The exact reason for lack of recognition of CT26 by Smc3 specific T cells remains ambiguous. We infer from the the ligandome analysis () and Smc3 overexpression experiments ()) that Smc3 neoepitopes are not sufficiently presented to allow T-cell recognition. The lack of sufficient presentation of Smc3 neoepitopes on tumor cells cannot be directly deduced from RNA sequencing data. For example, mutated Nav2 which was robustly detected on MHC I of CT26 is transcribed slightly lower than mutated Smc3 (4.16 compared to 4.52 RPKM). Most likely additional parameters such as RNA translation efficiency and protein half-life,Citation31 processing efficiency, as well as stability of MHC-peptide bindingCitation32 play a role. Using MS-based MHC class I ligandome analysis has been proposed to filter out neoepitopes not sufficiently presented by tumor cells.Citation33 However, sensitive neoepitope detection by standard MS techniques is challenging as neoantigens are in general not highly expressed.Citation34 The MS3 approach used here is of high sensitivityCitation27 but its labor intensiveness, long processing times and high sample requirements limits its use for individualized cancer immunotherapy. Ultimately, our finding supports the targeting of a multitude of neoantigens (both for expanding preexisting and inducing de novo T-cell responses) identified by next generation sequencing in parallel by vaccination to increase the probability to have therapeutically relevant ones among them.Citation12,Citation14,Citation35
It has been shown that high antigen amounts are required for cross-priming and activation of naïve T cells whereas primed CD8+ T cells need fewer peptides presented on target cells to be activated for killing.Citation36,Citation37 In contrast, we observed efficient priming of Smc3 specific T cells but lack of recognition of tumor cells. One explanation would be the ability of APCs to enrich tumor antigens. Enhanced macropinocytosis induced via TLR signaling,Citation38 or receptor-mediated uptake of immune complexes via Fc-receptorsCitation39 as well as actin binding proteins via Clec9A/DNGR-1Citation40,Citation41 may result in strong enrichment of certain antigens reaching MHC-peptide complex densities sufficient for priming of T cells.
Tumor specific CD4+ T cells preferentially recognize their cognate antigen on APCs in the tumor or draining lymph node but not on tumor cells, which are usually MHC II negative. We and others have previously shown that CD4+ neoepitope specific T-cell responses frequently mediate tumor control in mice.Citation12,Citation42 If tumor antigens are commonly enriched by APCs as hypothesized for Smc3, neoepitope specific CD4+ T-cell responses may benefit from this mechanism preferentially in comparison to neoepitope specific CD8+ T cells as they do not require the direct recognition of the antigen presented on tumor cells in order to exert anti-tumoral effects. Additional studies are required that particularly investigate whether the therapeutic relevance of MHC class I restricted neoepitopes can be predicted to enable the provision of neoepitope vaccines to cancer patients with a strong clinical impact.
Materials and methods
Cell lines and mice
Female 8–12 week old BALB/c mice (Janvier Labs) were kept and treated as approved by the Ethics Committee for animal research of Rhineland-Palatinate, Germany. CT26 colon carcinoma cells (CT26-WT) were purchased in 2011 (ATCC CRL-2638 lot no. 58494154). CT26-gp70KO cells were generated via CRISPR/Cas9 mediated introduction of indels into the gp70 locus. In brief, CT26-WT cells were lipofected daily for three days with Cas9 RNA and a one of five selected gp70 sgRNA. Colonies of single cell clones (~5x104 cells) were tested for recognition by gp70 AH1 specific splenocytes (5x105 cells) in an IFNγ ELISpot (Supplementary Figure 2). Clone 3–8 (sgRNA target sequence: 5ʹ-TTCCAGGCCGTATTGCACCG-3ʹ) was selected for subsequent studies based on lack of recognition by gp70-specific T cells and confirmation of MHC class I expression. Master and working cell banks were generated immediately upon generation/receipt, of which early passages were used for experiments. Cells were tested for mycoplasma contamination every 3 months.
Mutation identification and prioritization
Non-synonymous single nucleotide variations (nsSNVs) were identified via exome and RNA sequencing as described earlier.Citation5 nsSNVs with at least 1 read covering the mutation in the transcriptome were subjected to further analysis. Potential MHC ligands shown in Supplementary Table 1 were selected based on variant expression and MHC class I binding affinity prediction. The variant allele frequency (VAF) of nsSNVs multiplied with the mRNA expression level of the respective gene in RPKM (reads which map per kilobase of transcript length per million mapped reads) was computed as a surrogate for variant expression. MHC class I binding prediction was performed using IEDB consensus (V2.13) and NetMHCpan (V2.8). MHC ligand candidates were selected based on coinciding favorable H2-Kd binding and a matching predicted minimal epitope sequence of nsSNVs with a variant expression > 1 RPKM.
Peptides and RNA
Peptide synthesis was performed by JPT Peptide Technologies GmbH via fully automated SPOT-synthesis approach (PepTrack™ Fast Track specification). A cellulose membrane was functionalized with the individual C-terminal amino acid for each peptide. After Fmoc- deprotection and washing, the activated amino acids were spotted to the membrane in a computer controlled and spatially addressed fashion. Deprotection, washing and amino acid coupling cycles were repeated until the complete peptide sequences were assembled. Following side-chain deprotection peptides were individually cleaved into microtiter-plate wells, analyzed by high-throughput HPLC-MS and dried. Finally, peptides were pooled and aliquoted in antigen representing matrix pools using an automated liquid handling system. RNAs were synthesized by BioNTech RNA Pharmaceuticals GmbH.Citation12 Smc3 RNA encodes 27 amino acids with the mutated amino acid in the center (position 14). PME1 RNA represents five neoepitopes, CT26-ME1 to CT26-ME5 (27 amino acids per epitope) as described earlier.Citation12 gp70 RNA codes for the H-2Ld-restricted epitope AH1423-431 derived from the murine leukemia virus envelope glycoprotein 70 (gp70) with single amino acid substitution at position five (V427A).Citation18 All epitope sequences were embedded in a backbone described by Kreiter and colleagues.Citation24
Tumor models
Mice were injected subcutaneously into the flank with 5 × 105 CT26-WT or CT26-gp70KO cells in 100 µl PBS and tumor growth was measured with a caliper using the formula (A x B2)/2 (A as the largest and B the smallest diameter of the tumor). 3 × 105 CT26-gp70KO cells were injected for the experiment shown in . Lipoplex-formulated RNA was injected i.v. as indicated in the figure legends (20 µg per RNA in 200 µl if not otherwise stated).Citation43 The TLR7 agonist SC1 was injected directly into the tumor (80.5 µg in 30 µl) at day 14, 19, 24 and 28 after tumor inoculation. Local radiation (Precision X-Ray Inc., X-RAD 320, 0.47 Gy/min, 14 Gy in total) was performed under ketamine/xylazine narcosis on day 14 after tumor inoculation. Non-tumor tissue was protected by a custom made lead shield. PD-L1 antibody treatment (clone 10F.9G2 from BioXcell, 200 µg, i.p. in 200 µl PBS) was performed on day 16, 19, 23 and 26 after tumor inoculation. For induction of lung tumors, 4 × 105 CT26-WT were injected i.v. and mice were treated at day 4, 7 and 12 with 20 µg RNA lipoplexes encoding either Smc3 or the vaccine backbone alone (irrelevant RNA).
IFN-γ ELISpot
Enzyme-linked ImmunoSpot assays detecting IFNγ release of T cells were performed as previously described.Citation5 All samples were tested in duplicates or triplicates. 5 × 105 splenocytes, CD4+ T cell depleted splenocytes or (1–1.5x105) purified CD4+ or CD8+ T cells (MACS®, Miltenyi Biotec) were restimulated using either 5 × 104 tumor cells, 0.4 or 2 µg/ml peptide or peptide matrix pool (0.4 µg/ml per individual peptide) with or without 5 × 104 syngeneic bone-marrow-derived dendritic cells (BMDC).
Mutated allele frequency determined by droplet digital PCR
TaqMan™ assays
To discriminate wild type and mutated Smc3 genomic DNA using ddPCR™ analysis TaqMan™ assays were designed and ordered at Eurofins Genomics. The following probes were used for detection of Smc3 DNA:
wild type: 5ʹ-[HEX] CAT CCA GAG ATA GCA TAT TAT C [MGBEQ]-3ʹ
D733A variant: 5ʹ-[FAM] CAT CCA GAG CTA GCA TAT TAT C [MGBEQ]-3ʹ
with HEX being 6-carboxy-2ʹ,4,4ʹ,5ʹ,7,7ʹ-hexachlorofluorescein and FAM being 6-Carboxyfluorescin. Both probes were labeled at the 3ʹ-end with MGBEQ (Minor Groove Binder-Eclipse Quencher) to enhance binding specificity and reduce background signaling. Both TaqMan™ assays used the shared forward primer 5ʹ-GAT AGA GAC CCA ACA AAG-3ʹ and the reverse primer 5ʹ-TCT TTT AGC ATC TTC ATC TC-3ʹ.
Extraction of genomic DNA
Genomic DNA from CT26 bulk cells was extracted with the QIAGEN DNeasy® Blood & Tissue Kit according to the manufacturer’s protocol and eluted in nuclease-free, PCR-grade water. For the extraction of genomic DNA from CT26 single cell clones Invitrogen™’s PureLink™ Pro 96 Genomic DNA Kit was used. Preparation of cell pellets, clean-up and elution was performed according to the manufacturer’s manual using 50 µl nuclease-free, PCR-grade water for elution.
ddPCR™
Droplet Digital™ PCR (ddPCR™) was performed on a Bio-Rad QX200™ system. 1 µl of a mixture of 7 µM HEX-labeled probe or 5 µM FAM-labeled probe with 13.5 µM forward primer and 13.5 µM reverse primer was added to 11 µl ddPCR™ Supermix as well as 2.5 µl gDNA. Droplet generation was performed in the QX200™ Droplet Generator according to the manufacturer’s protocol. For the PCR a Bio-Rad C1000 Touch™ Thermal Cycler with 45 cycles and 58.0°C annealing temperature was used. Droplet readout was executed in the QX200™ Droplet Reader calling Smc3 wild type allelic gDNA positive droplets in the HEX-channel, Smc3 D733A variant DNA carrying droplets in the FAM-channel, and negative droplets, without any gDNA copy, in neither one of both channels. From the overall count of FAM vs. HEX positive droplets the distribution of Smc3 alleles was determined.
TCR sequencing
Cloning of Smc3-specific TCRs
Splenocytes of CT26-WT tumor bearing BALB/c mice were cultured 14 h in the presence of 4 µg/ml peptide or with 2 µg/ml Concanavalin A (ConA, Sigma Aldrich), respectively. Activated cells were labeled using IFNγ-secretion assay-APC kit (Miltenyi Biotec) and enriched via anti-APC-microbeads (Miltenyi Biotec) using MACS technique. CD4negCD8+IFNghi -single T cells (αCD8α-V500, BD Horizon, clone 53.6–7; αCD4-eFluor450, eBioscience, clone GK1.5) were sorted (BD FACSAria) into wells of 96 well V-bottom plates (Greiner Bio-One) containing CCD-1079Sk (ATCC® CRL-2097™) carrier cells.
Cloning of paired a/b TCRs of these single cells was performed as previously described.Citation44 TCR chains in pST1 expression vectorsCitation45 were used for in vitro transcription.
RNA transfer into cells
BALB/c splenocytes were pre-activated in the presence of 2 µg/ml ConA, rhIL-7/IL-15 (5 ng/ml each, Miltenyi Biotec) 72 h and rested for 3 days in the presence of IL-7/IL-15. Viable cells were separated from debris prior electroporation using 1.084 Ficoll®-Paque PREMIUM (GE Healthcare) density-gradient centrifugation. 10 µg IVT RNA of each TCR chain were electroporated (BTX ECM 630, 500 V, 3 ms, 1 pulse) into cells as previously described.Citation6 eGFP RNA electroporated T cells served as mock control. Freshly electroporated cells were functionally validated via IFNγ ELISpot.
Cytotoxicity assay
Antigen-specific T cells were activated for 24 h at 37 °C and 5% CO2 by culturing splenocytes from immunized mice (n = 3, 5 × 106 cells/ml) in the presence of 100 U/mL IL-2 (Proleukin S, Novartis) and 0.1 µg/ml Smc3 or gp70 AH1 peptide. CD8+ T cells were then isolated using CD8 MicroBeads (Miltenyi Biotec). 3 × 104 CT26-WT cells were seeded as targets into a transparent 96-well plate (TPP) and cultured at 37 °C and 5% CO2. After 24 h, 3 × 105 activated CD8+ T cells were plated and Caspase 3/7 reagent (Essen Bioscience) was added at a 1:2,000 dilution from a 5 mM solution. Cells were co-cultured at 37 °C and 5% CO2 and four images per well were acquired at tenfold magnification every hour for 24 h using an IncuCyte® Zoom Live Cell Analysis system (Essen Bioscience). The number of apoptotic cells per image as indicated by green fluorescence was determined using IncuCyte® analysis software.
Targeted MHC ligandome analysis
As described earlier,Citation27 CT26-WT cells were grown to 90% confluence, washed with 1x PBS and harvested with lysis buffer containing 1% IGEPAL® Ca-630 (Sigma-Aldrich), 2x protease inhibitor cocktail mix (Roche Diagnostics), and 1 mM PMSF (Roth) in 1x PBS. After removing cell debris (20,000x g, 30 min, 4 °C), the clear cell lysate of 1.7 × 108 cells was incubated with 45 µl anti-H-2Kd antibody (HB159/SF1.1.10, 100 µg, BioXCell)-coupled protein G coated GammaBind™ Plus Sepharose™ beads (GE Healthcare) for 4 h at 4 °C while rotating. Beads were washed extensively with 1x PBS and epitopes were eluted by acidic treatment (0.3% TFA in H2O, 15 min, RT).
Samples were purified using Sep-Pak® C18 extraction cartridges (Waters). In brief, cartridges were wetted with 100% ACN and washed with 29% ACN in 1x PBS, before samples in 29% ACN in 1x PBS were processed. The flow through was collected, dried, and desalted by reverse phase (RP) enrichment with C18 micro columns (self-packedCitation46). Peptide binding was performed in 3% ACN/0.1% TFA in H2O solution, peptides were washed with 0.1% TFA in H2O, and eluted with 27% ACN/0.1% TFA in H2O. The eluate was dried by vacuum centrifugation and dissolved in 3% ACN/0.1% FA in H2O for subsequent LC-MRM3 analysis with a nanoAcquity UPLC® (Waters)-QTrap6500 (ABSciex) system.
Chromatographic separation was performed on a 25 cm long C18 RP nanoAcquity BEH130 column (Waters) with an inner diameter of 75 µm. Separation was performed with a flow rate of 300 nL/min with a linear gradient increasing from 3% to 10% ACN/0.1% FA/0.01% TFA in H2O within 1 min. Elution from the column was performed with a gradient increasing from 10% to a final 40% ACN/0.1% FA/0.01% TFA in H2O at 50 min. The column temperature was kept at 45 °C. Subsequent analysis with the QTrap6500 instrument was performed with the NanoESI III electron spray ionization source (ABSciex) equipped with PicoTip ESI emitters (New Objective) in the low mass hardware profile. The instrument was operated in positive mode, with a source voltage of 3,000 V, an ion source gas pressure of 15 psi and a curtain gas pressure of 30 psi. Collision gas (CAD) was set to high and the interface heater temperature was 150 °C. Precursor masses and theoretical fragment spectra used for the optimization of the measuring parameters for each peptide were generated online with the Protein prospector 5.18 web tool. LC-MRM3 acquisition methods were generated based on the manual MS3 optimization of compound-specific parameters. The LIT scan type was set to MS3 and the scan rate was set to 10,000 Da/s. The Q1 resolution was set to unit and the Q3 resolution to LIT. Moreover, the fill time was dynamic and the MS3 excitation time was 25 msec.
MS3 data review was performed manually in the Analyst® 1.6.2 (ABSciex) program. Chromatographic profiles as well as MS3 spectra obtained for the IP samples were compared to those obtained for the respective synthetic peptides.
Statistics
Statistical tests were used as indicated in the figure legends. All analyses were two-tailed and carried out using GraphPad Prism 6. n.s. (not significant): P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| APCs | = | Antigen-presenting cells |
| BMDC | = | Bone-marrow-derived dendritic cells |
| ConA | = | Concanavalin A |
| DCs | = | Dendritic cells |
| DNA | = | Deoxyribonucleic acid |
| ddPCR | = | Droplet digital PCR |
| gDNA | = | Genomic DNA |
| ICB | = | Immune checkpoint blockade |
| IP | = | Immunoprecipitation |
| IFN | = | Indian hedgehog |
| IL | = | Interleukin |
| i.t. | = | Intratumoral |
| LOD | = | Limit of detection |
| LC-MRM3 | = | Liquid chromatography–multiple reaction monitoring cubed |
| LC-MS2 | = | Liquid chromatography–tandem mass spectrometry |
| MHC | = | Major histocompatibility complex |
| MS | = | Mass spectrometry |
| MGBEQ | = | Minor Groove Binder-Eclipse Quencher |
| gp70 | = | Murine leukemia virus envelope glycoprotein 70 |
| Nav2 | = | Neuron navigator 2 |
| nsSNVs | = | Non-synonymous single nucleotide variations |
| PBS | = | Phosphate-buffered saline |
| PCR | = | Polymerase chain reaction |
| PD-1 | = | Programmed death-1 |
| PD-L1 | = | Programmed death-ligand 1 |
| RPKM | = | Reads which map per kilobase of transcript length per million mapped reads |
| RNA | = | Ribonucleic acid |
| Smc3 | = | Structural maintenance of chromosomes 3 |
| s.c. | = | Subcutaneous |
| TCRs | = | T cell receptors |
| TLR | = | Toll like receptor |
| TILs | = | Tumor infiltrating lymphocytes |
| UPLC | = | Ultra-Performance Liquid Chromatography |
| VAF | = | Variant allele frequency |
Supplemental Material
Download Zip (352 KB)Acknowledgments
The authors thank Imke Biermann, Bernadette Jesionek, Nele Brüne, Christian Stofft, Kathrin Thiemann, Stefania Gangi Maurici, Evelin Petscherskich and Ines Beulshausen for technical assistance, Tana Omokoko and Anke Oelbermann for help regarding identification and cloning of TCRs, Jutta Petschenka and Stefan Strobl for support regarding TLR7 agonist treatment as well as Lena M. Kranz for critical proofreading of this paper. This work was supported by the BMBF (ImmunoTransporter - 13N13338).
Supplementary Material
Supplemental data for this article can be accessed here.
Related Research Data
References
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–2017. doi:10.1056/NEJMoa1414428.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in advanced Melanoma. N Engl J Med. 2015;372(26):2521–3532. doi:10.1056/NEJMoa1503093.
- Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24(5):743–750. doi:10.1101/gr.165985.113.
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in Melanoma. N Engl J Med. 2014;371(23):2189–2199. doi:10.1056/NEJMoa1406498.
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi:10.1126/science.aad0095.
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi:10.1126/science.aaa1348.
- Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi:10.1016/S0140-6736(16)00561-4.
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581. doi:10.1038/nature13988.
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362:6411. doi:10.1126/science.aar3593.
- Vormehr M, Schrörs B, Boegel S, Löwer M, Türeci Ö, Sahin U. Mutanome engineered RNA Immunotherapy : towards patient-centered tumor vaccination. J Immunol Res. 2015;2015(Article ID 595363):6. doi:10.1155/2015/595363.
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi:10.1038/nature13954.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696. doi:10.1038/nature14426.
- Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi:10.1038/nature23003.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi:10.1038/nature22991.
- Vormehr M, Diken M, Boegel S, Kreiter S, Türeci Ö, Sahin U. Mutanome directed cancer immunotherapy. Curr Opin Immunol. 2016;39:14–22. doi:10.1016/j.coi.2015.12.001.
- Hotz C, Treinies M, Mottas I, Rötzer LC, Oberson A, Spagnuolo L, Perdicchio M, Spinetti T, Herbst T, Bourquin C. Reprogramming of TLR7 signaling enhances antitumor NK and cytotoxic T cell responses. Oncoimmunology. 2016;5(11):e1232219. doi:10.1080/2162402X.2016.1232219.
- Cook AM, Lesterhuis WJ, Nowak AK, Lake RA. Chemotherapy and immunotherapy: mapping the road ahead. Curr Opin Immunol. 2016;39:23–29. doi:10.1016/j.coi.2015.12.003.
- Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, Margulies DH, Pardoll DM. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13(4):529–538. doi:10.1016/S1074-7613(00)00052-2.
- Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15(1):190. doi:10.1186/1471-2164-15-190.
- Hamm S, Rath S, Michel S, Baumgartner R. Cancer immunotherapeutic potential of novel small molecule TLR7 and TLR8 agonists. J Immunotoxicol. 2009;6(4):257–265. doi:10.3109/15476910903286733.
- Wiedemann GM, Jacobi SJ, Chaloupka M, Krächan A, Hamm S, Strobl S, Baumgartner R, Rothenfusser S, Duewell P, Endres S, et al. A novel TLR7 agonist reverses NK cell anergy and cures RMA-S lymphoma-bearing mice. Oncoimmunology. 2016;5(7):e1189051. doi:10.1080/2162402X.2016.1189051.
- Ghiselli G, Iozzo RV. Overexpression of bamacan/SMC3 causes transformation. J Biol Chem. 2000;275(27):20235–20238. doi:10.1074/jbc.C000213200.
- Scherer A, Salathé MBS. High epitope expression levels increase competition between T cells. PLoS Comput Biol. 2006;8(18):2650–2657.
- Kreiter S, Selmi A, Diken M, Türeci Ö, Sahin U, Sebastian M, Osterloh P, Schild H, Huber C, Türeci O, et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. 2008;180(5):309–318. [pii]. doi:10.4049/jimmunol.180.1.309.
- Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, Kappler J. An inverse relationship between T cell receptor affinity and antigen dose during CD4 ؉ T cell responses in vivo and in vitro. Immunology. 1999;96(August):9781–9786. doi:10.1073/pnas.96.17.9781.
- Mcgranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–1469. doi:10.1126/science.aaf1490.
- Blatnik R, Mohan N, Bonsack M, Falkenby LG, Hoppe S, Josef K, Steinbach A, Becker S, Nadler WM, Rucevic M, et al. A targeted LC-MS strategy for low-abundant HLA class I-presented peptide detection identifies novel human papillomavirus T-cell epitopes. Proteomics. 2018 Mar;30:e1700390. doi:10.1002/pmic.201700390.
- Fortin T, Salvador A, Charrier JP, Lenz C, Bettsworth F, Lacoux X, Choquet-Kastylevsky G, Lemoine J. Multiple reaction monitoring cubed for protein quantification at the low nanogram/milliliter level in nondepleted human serum. Anal Chem. 2009;81(22):9343–9352. doi:10.1021/ac901447h.
- Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125(10):3981–3991. doi:10.1172/JCI82416.
- Linnemann C, Van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, Behjati S, Velds A, Hilkmann H, Atmioui D, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21(1):81–85. doi:10.1038/nm.3773.
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–342. doi:10.1038/nature10098.
- Harndahl M, Rasmussen M, Roder G, Dalgaard Pedersen I, Sørensen M, Nielsen M, Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol. 2012;42(6):1405–1416. doi:10.1002/eji.201141774.
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–576. doi:10.1038/nature14001.
- Khodadoust MS, Olsson N, Wagar LE, Haabeth OAW, Chen B, Swaminathan K, Rawson K, Liu CL, Steiner D, Lund P, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature. 2017; doi:10.1038/nature21433.
- Stadler CR, Bähr-Mahmud H, Celik L, Hebich B, Roth AS, Roth RP, Karikó K, Türeci Ö, Sahin U. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med. 2017;(May):3–8. doi:10.1038/nm.4356.
- Kurts C, Miller JF, Subramaniam RM, Carbone FR, Heath WR. Major histocompatibility complex class I-restricted cross-presentation is biased towards high dose antigens and those released during cellular destruction. J Exp Med. 1998;188(2):409–414. doi:10.1084/jem.188.2.409.
- Pihlgren M, Dubois PM, Tomkowiak M, Sjögren T, Marvel J. Resting memory CD8+ T cells are hyperreactive to antigenic challenge in vitro. J Exp Med. 1996;184(6):2141–2151. doi:10.1084/jem.184.6.2141.
- West MA, Wallin RPA, Matthews SP, Svensson HG, Zaru R, Ljunggren H-G, Prescott AR, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305(5687):1153–1157. doi:10.1126/science.1099153.
- Baker K, Qiao S-W, Kuo TT, Aveson VG, Platzer B, Andersen J-T, Sandlie I, Chen Z, de Haar C, Lencer WI, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc Natl Acad Sci. 2011;108(24):9927–9932. doi:10.1073/pnas.1019037108.
- Iborra S, Izquierdo HM, Martínez-López M, Blanco-Menéndez N, E Sousa CR, Sancho D. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. J Clin Invest. 2012;122(5):1628–1643. doi:10.1172/JCI60660.
- Zhang J-G, Czabotar PE, Policheni AN, Caminschi I, Wan SS, Kitsoulis S, Tullett KM, Robin AY, Brammananth R, van Delft MF, et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity. 2012;36(4):646–657. doi:10.1016/j.immuni.2012.03.009.
- Zeng B, Middelberg APJ, Gemiarto A, MacDonald K, Baxter AG, Talekar M, Moi D, Tullett KM, Caminschi I, Lahoud MH, et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J Clin Invest. 2018;128(5):1971–1984. doi:10.1172/JCI96791.
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396–401. doi:10.1038/nature18300.
- Paret C, Simon P, Vormbrock K, Bender C, Kölsch A, Breitkreuz A, Ö Y, Omokoko T, Hubich-Rau S, Hartmann C, et al. CXorf61 is a target for T cell based immunotherapy of triple-negative breast cancer. Oncotarget. 2015;6(28):25356–25367. doi:10.18632/oncotarget.4516.
- Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, Türeci O, Sahin U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–4017. doi:10.1182/blood-2006-04-015024.
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using stagetips. Nat Protoc. 2007;2(8):1896–1906. doi:10.1038/nprot.2007.261.