
ABSTRACT
Tumor cells activate the G2/M cell cycle checkpoint in response to ionizing radiation (IR) and effector immune cell-derived granzyme B to facilitate repair and survival. Wee1 kinase inhibition reverses the ability of tumor cells to pause at G2/M. Here, we hypothesized that AZD1775, a small molecule inhibitor of Wee1 kinase, could sensitize tumor cells to IR and T-lymphocyte killing and improve responses to combination IR and programmed death (PD)-axis immune checkpoint blockade (ICB). Multiple models of head and neck carcinoma, lung carcinoma and melanoma were used in vitro and in vivo to explore this hypothesis. AZD1775 reversed G2/M cell cycle checkpoint activation following IR, inducing cell death. Combination IR and AZD1775 induced accumulation of DNA damage in M-phase cells and was rescued with nucleoside supplementation, indicating mitotic catastrophe. Combination treatment enhanced control of syngeneic MOC1 tumors in vivo, and on-target effects of systemic AZD1775 could be localized with targeted IR. Combination treatment enhanced granzyme B-dependent T-lymphocyte killing through reversal of additive G2/M cell cycle block induced by IR and granzyme B. Combination IR and AZ1775-enhanced CD8+ cell-dependent MOC1 tumor growth control and rate of complete rejection of established tumors in the setting of PD-axis ICB. Functional assays demonstrated increased tumor antigen-specific immune responses in sorted T-lymphocytes. The combination of IR and AZD1775 not only lead to enhanced tumor-specific cytotoxicity, it also enhanced susceptibility to T-lymphocyte killing and responses to PD-axis ICB. These data provide the pre-clinical rationale for the combination of these therapies in the clinical trial setting.
Introduction
Immune checkpoint blockade (ICB) targeting the programmed death (PD)-axis is FDA approved for multiple recurrent/metastatic cancers, and multiple trials are underway to evaluate its role in the upfront treatment setting.Citation1 Response rates in patients with recurrent/metastatic malignancy for PD-1/PD-L1 blockade are 15–20% across multiple large trials.Citation2,Citation3 Enhanced understanding of why so few patients respond, and how to reverse these mechanisms of resistance in a translationally relevant way may enhance response rates to ICB resulting in improved tumor control and survival.
Development of antigen-specific anti-tumor immunity is a multi-step process that involves type I interferon-dependent innate immune priming of adaptive immunity.Citation4 Ionizing radiation (IR) enhances anti-tumor immunity through STING-dependent type I interferon production in response to cytosolic DNA and increased tumor cell immunogenicity.Citation5–Citation7 Accordingly, IR enhances responses to PD-axis ICB.Citation8 Following IR-induced DNA damage, tumor cells activate the G2/M cell cycle checkpoint to allow DNA repair before cell cycle progression, promoting resistance.Citation9 Inhibition of the master G2/M cell cycle checkpoint regulator Wee1 kinase abrogates the ability of tumor cells to pause their cell cycle and sensitizes them to IR.Citation10–Citation12 Our laboratory recently characterized similar tumor cell G2/M cell cycle pause in response to T-lymphocyte derived granzyme B and tumor necrosis factor (TNF).Citation13 Given this redundant, intrinsic tumor cell response to both IR and cytotoxic T-lymphocyte (CTL) products, we hypothesized that AZD1775, a small molecule inhibitor of Wee1 kinase, could lead to enhanced tumor cell cytotoxicity after IR and anti-tumor immunity when combined with IR in the setting of PD-axis ICB.
Here, using multiple syngeneic tumor models, we characterized timing and magnitude of G2/M cell cycle checkpoint activation after IR, and how to optimally combine AZD1775 with IR to induce mitotic catastrophe and maximize loss of tumor cell viability. We next demonstrated enhanced in vivo tumor control with combination AZD1775 and IR and that the on-target effects of systemic AZD1775 can be localized to tumor tissue with tumor-focused IR. We further demonstrated that AZD1775 and IR could cooperatively enhance antigen-specific T-lymphocyte killing of tumor cells. This enhanced susceptibility correlated with the ability of AZD1775 to reverse additive G2/M cell cycle block induced by IR and granzyme B. In vivo, combination AZD1775 and IR-enhanced CD8+ cell-dependent tumor control and the rate of complete rejection of established tumors following PD-axis ICB. Together, these data support the mechanism-based combination of AZD1775, IR, and PD-axis ICB and provide the pre-clinical rationale for investigation of this combination in the clinical trial setting.
Results
To first determine whether cell cycle alterations in LLC lung carcinoma, MOC1 oral carcinoma, and B16 melanoma cells are radiation dose- and time-dependent, we assessed cell cycle distribution by flow cytometry of DNA content following single doses of 2 Gy or 8 Gy over various time points (LLC , MOC1 and B16 supplemental Figure 1A&B). The higher single 8 Gy dose of IR produced greater G2/M accumulation compared to 2 Gy (quantified in ). Maximum G2/M accumulation occurred 12 h after IR for LLC and MOC1 and 24 h after IR for B16. G2/M accumulation was reversed at 24 h following 2 Gy IR but maintained at 24 h after 8 Gy IR. SubG1 accumulation, indicative of DNA fragmentation, was evident 24 h after IR and greater for 8 Gy compared to 2 Gy.
Figure 1. Ionizing radiation resulted in dose- and time-dependent G2/M cell cycle checkpoint activation.
(a), LLC cells were exposed to a single fraction of either low dose (2 Gy) or high dose (8 Gy) IR and assessed at 4, 8, 12, and 24 h after IR for cell cycle alterations by flow cytometry of DNA content. Representative data from one of at least two independent assays in technical triplicate shown. (b), quantification of cell cycle distribution of irradiated LLC, MOC1 and B16 cells following IR. Top panels quantify cells in the late S/G2/M gate, bottom panels quantify cells in the subG1 gate. (c), western blot analysis of p-cdc2 (Y15) and total cdc2 expression in LLC, MOC1 and B16 cells 8 h after exposure to a single fraction of 8 Gy IR with and without AZD1775 (250 nM) or DMSO (control). *, p < .05; **, p < .01; ***, p < .001 for all figures.
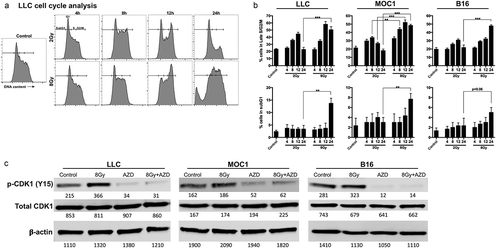
Protein analysis of cells irradiated with 8 Gy IR demonstrated significant phosphorylation of cyclin-dependent kinase I (CDK1; ), consistent with a DNA damage response and suggestive of a G2/M cell cycle block. The addition of AZD1775 (250 nM), a small molecule inhibitor of Wee1 kinase, significantly reduced baseline and IR-induced CDK1 phosphorylation. Thus, AZD1775 treatment would be predicted to reverse IR-induced G2/M cell cycle block. We next assessed the direct effects of AZD1775 at clinical peak and trough plasma concentrations.Citation14 Despite inhibition of CDK1 phosphorylation (), trough concentrations of AZD1775 (250 nM) produced little to no alteration in viability in the absence of IR as measured by real-time impedance analysis (Supplemental Figure 2). Peak doses (1500 nM) alone significantly altered the viability of LLC and MOC1 cells but not B16 cells.
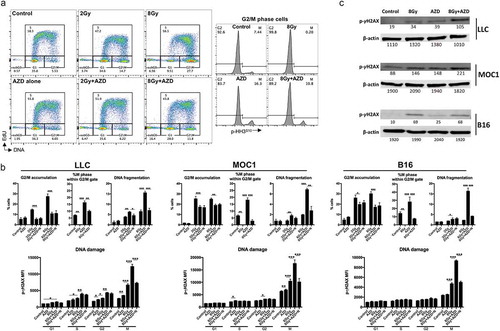
We next utilized cycle analysis designed to assess cells within each phase of the cell cycle following different treatment conditions. Treatment with IR-induced significant G2/M accumulation with very few cells in mitosis, indicative of a G2/M block (LLC , MOC1 and B16 supplemental Figure 3A&B). 8 Gy IR produced a greater G2/M block than 2 Gy IR. AZD1775 treatment produced a greater number of cells in mitosis, suggesting that Wee1 kinase inhibition pushed cells through the G2/M cell cycle checkpoint and reversed IR-induced G2/M block (quantified in ). This dual treatment with IR and AZD1775 produced significant subG1 accumulation compared to either treatment alone. As previously demonstrated by Lawrence and colleagues,Citation12,Citation15 the addition of exogenous nucleosides did not alter the ability of AZD1775 to reverse IR-induced G2/M block, but it did attenuate subG1 accumulation of cells. Assessment of phosphorylation of γH2AX, an early step in the DNA damage response, demonstrated greater accumulation of DNA damage within mitotic cells compared with G1/S/G2 in all three cell lines. The addition of exogenous nucleosides decreased phospho-γH2AX in mitotic cells. Greater accumulation of phospho-γH2AX within cells treated with combination IR and AZD1775 was verified by western blot (). Together, these data suggested that Wee1 kinase inhibition, in the setting of IR-induced DNA damage, pushed cells into mitosis without sufficient time to allow for DNA repair based upon available nucleoside pools, resulting in mitotic catastrophe.Citation16 To further validate this, we explored phospho-γH2AX localization within the nucleus by immunofluorescence in MOC1 cells (Supplemental Figure 4). Combination IR and AZD1775 significantly enhanced the development of cells with pan-nuclear phospho-γH2AX staining, consistent with induction of mitotic catastrophe. To verify that the DNA damage was directly a result of IR and not due general genomic instability from cell cycle pause caused by another mechanism, we assayed for phospho-γH2AX accumulation within cells following treatment with nocodazole, a microtubule inhibitor that produces a robust G2/M block (Supplemental Figure 5). IR, but not nocodazole, induced M-phase accumulation of phospho-γH2AX, indicating DNA damage is directly induced by IR.
Figure 2. Wee1 kinase inhibition reversed G2/M cell cycle checkpoint activation resulting in M phase DNA damage and cell death.
(a), LLC cells were exposed to 8 Gy IR with and without AZD1775 (250 nm) for 8 h and assessed for cell cycle distribution using flow cytometry. Left panels demonstrate gating strategy based upon DNA content and EdU uptake during S phase. Right panels demonstrate G2 and M phase segregation based upon phosphorylation (S10) of HH3. Representative data from one of at least two independent assays in technical triplicate shown. (b), top panels demonstrate quantification of cell cycle phase distribution of LLC, MOC1 and B16 cells after treatment as in a. Some cells were exposed to exogenous nucleoside supplementation (1×). Bottom panels demonstrate quantification of DNA damage as measured by phosphorylation (S139) of γH2AX within each cell cycle phase. (c), Western blot analysis of LLC, MOC1 and B16 cells 8 h after exposure to 8 Gy IR with and without AZD1775 (250 nM). AZD, AZD1775; N, nucleoside supplementation.
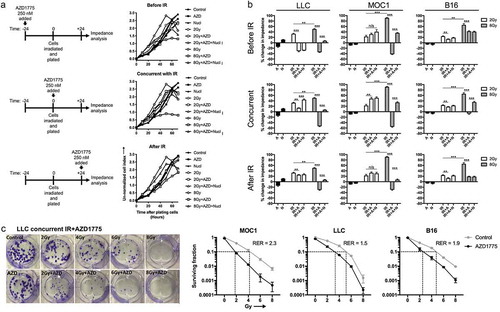
We next wished to determine if sequencing the exposure of irradiated cells to AZD1775 could alter loss of tumor cell proliferation and viability as measured by impedance analysis. Cells were exposed to AZD1775 24 h before, immediately after (concurrent), or 24 h after IR (, quantified in ). Exposure of cells to AZD1775 immediately after IR treatment resulted in the greatest decrease in impedance compared to exposure 24 h before or after IR in all three models. The addition of exogenous nucleosides reversed this decrease in impedance. To validate these concurrent treatment effects and to definitively demonstrate altered tumor cell viability, standard clonogenic assays were performed (). While AZD alone had little effect, AZD enhanced loss of viability across IR doses with radiation enhancement ratios ranging from 1.5 to 2.3.
Figure 3. Concurrent Wee1 kinase inhibition and ionizing radiation produced the greatest loss of tumor cell viability.
(a), LLC cells were exposed to AZD1775 (250 nM) 24 h before (before IR), immediately after (concurrent) or 24 h after (after IR) a single fraction of 2 or 8 Gy IR and assessed for viability over time via impedance analysis. Some cells were exposed to nucleoside supplementation (1×). Schema of sequenced treatments on left, representative cell index impedance plots on right. (b), quantification at 48 h of change in impedance (cell index) for LLC, MOC1 and B16 cells with variable sequencing of AZD1775. C, LLC, MOC1 or B16 cells were treated with single fractions of IR, treated with concurrent AZD1775 (250 nM), and assessed for colony formation potential via clonogenic assays. Representative colony photographs on left, with quantification on right. Dashed lines correlate to radiation doses, with and without AZD1775, used to calculate the radiation enhancement ratio (RER). Representative data from one of at least two independent assays in technical triplicate shown.
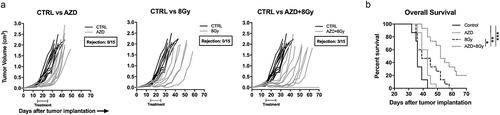
To explore whether these in vitro findings could be translated in vivo, we established MOC1 tumors in WT B6 mice and treated established (5–6 mm diameter) tumors with a single dose of 8 Gy IR alone, AZD1775 alone, or the combination. Treatment with AZD1775 alone produced an initial growth delay but tumors rebounded and survival was minimally extended compared to control (). A single IR dose of 8 Gy to the tumor produced a more robust growth delay in a subset of tumors, moderately extending survival. The combination of 8 Gy IR and AZD1775 produced growth delay in the majority of tumors, and complete rejection of 3/15 (20%) established tumors, significantly prolonging survival over control or either treatment alone.
Figure 4. Combination ionizing radiation and Wee1 kinase inhibition enhances tumor growth control and survival over either treatment alone.
WT B6 mice bearing established (0.1 cm3) MOC1 tumors were treated with a single fraction of 8 Gy IR alone (d 14) or in combination with systemic AZD1775 (120 mg/kg daily for 10 d, starting at d 14). Cumulative results from two independent experiments shown. (a), individual tumor growth curves. (b), survival analysis.
Previous work in our laboratory has demonstrated that Wee1 kinase inhibition sensitizes tumors to TNFα-mediated cell death.Citation13 We explored whether the enhanced tumor control observed with combination IR and AZD1775 is dependent upon TNFα production. Cells were first assessed for TNFα production following irradiation in vitro. IR had little effect on cell surface bound (Supplemental Figure 6A) or secreted (Supplemental Figure 6B) TNFα with the exception of a modest increase in secreted TNFα in B16 cells. Established MOC1 tumors were treated with 8 Gy IR and qPCR was used to assess whole tumor TNFα production. IR produced a significant increase in TNFα 24 h after IR, but this increase normalized 7 d later (Supplemental Figure 6C). To definitely assess the role of TNFα in MOC1 growth control following combination IR and AZD1775, mice were treated with this combination with and without systemic administration of a TNFα depleting antibody (Supplemental Figure 6D). TNFα depletion did not alter the enhanced tumor growth control observed with combination IR and AZD1775, suggesting these effects are independent of TNFα.
To investigate on-tumor and on-target effects of combination IR and AZD1775, tumor (exposed to both IR and systemic AZD1775) or oral mucosa from the same mice (exposed to systemic AZD1775 only) were assessed for phospho-γH2AX and phospho-CDK1 by intracellular flow cytometry to allow lineage-specific analysis. AZD1775 alone modestly induced phospho-γH2AX in tumor-associated fibroblasts (TAFs) but not other cell types (). IR alone induced phospho-γH2AX in tumor cells and tumor-infiltrating leukocytes (TILeu). Combination treatment induced significant increases in phospho-γH2AX in all cell types within the tumor. Notably, oral mucosa obtained from the same mice lacked any significant accumulation of phospho-γH2AX despite exposure to systemic AZD1775. IR alone induced significant CDK1 phosphorylation in tumor cells, TAFs and tumor endothelial cells, with little alteration of TILeu (). The addition of AZD1775 to IR reversed this phospho-CDK1 increase. No significant alterations in phospho-CDK1 were observed in the non-irradiated oral mucosa. These results demonstrate that despite systemic administration of AZD1775, on-target effects of cell cycle alteration and subsequent induction of DNA damage to tumor cells, TAFs and tumor endothelial cells can be localized to tumor tissue via delivery of IR.
Figure 5. On-target effects of Wee1 kinase inhibition were localized to tumors following ionizing radiation.
WT B6 mice bearing MOC1 tumors were treated at d 14 with a single fraction of 8 Gy IR and AZD1775 (120 mg/kg) at 1 and 24 h after IR. Four hours later (28 h after IR), tumors or oral mucosa from the same mice (n= 3/group) were assessed for cell lineage-specific intracellular phospho-γH2AX (S139) (a) or phospho-CDK1 (Y15) (b) in CD45.2−CD31−PDGFR− tumor cells, CD45.2−CD31−PDGFR+ fibroblasts, CD45.2−CD31+ endothelial cells or CD45.2+CD31− immune cells by flow cytometry. For A&B, representative dot plots or histograms shown on left, with quantification of phospho-γH2AX (a) or phospho-CDK1 (b) on the right. (c), MOC1 tumors (n= 3/time point) were exposed to a single dose of 8 Gy IR and a time course of TIL infiltration was assessed by flow cytometry. Representative dot plots on top, quantification of CD8+ and CD4+ TIL on bottom. (d), PD-1 expression on CD8+ TIL after IR was assessed by flow cytometry.
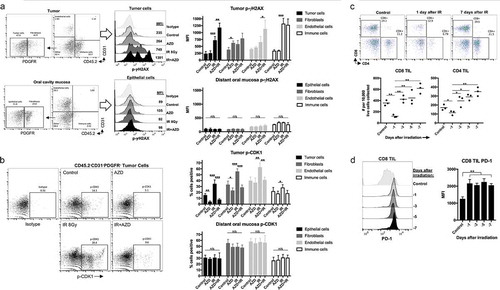
Tumor IR-induced significant DNA damage in TILeu. Thus, we performed a time course analysis of CD8+ and CD4+ tumor-infiltrating lymphocytes (TIL) accumulation after IR. TIL were significantly reduced 1 d after IR but rebounded to baseline levels 3 d after IR and trended toward increased accumulation from d 3 to 7 (). CD8+ TIL PD-1 expression was increased after IR at all time points (). Thus, while TIL appeared to be sensitive to IR-induced DNA damage, accumulation of PD-1 high TIL occurred between 1 and 3 d after IR, supporting previous work demonstrating the ability of 8 Gy IR to stimulate anti-tumor immunity.Citation7
Wee1 kinase inhibition and IR enhance tumor cell susceptibility to CTL killing through distinct mechanisms.Citation7,Citation13 Using tumor cells engineered to express the MHC class I-restricted epitope of ovalbumin as a model antigen, and OT-I CTL as effectors, we explored the effects of combination IR and AZD1775 on CTL killing using impedance analysis. While IR or AZD1775 alone enhanced CTL killing of tumor cells to variable degrees between models, combination IR and AZD1775 together consistently and significantly enhanced early (4 h) killing of tumor cells at low E:T ratios (). This early CTL killing of tumor cells was reversed in the presence of concanamycin A, demonstrating the requirement for perforin/granzyme B (). LLC, MOC1, and B16 are wild-type Trp53 or harbor non-synonymous Trp53 mutations that encode functional protein.Citation17–Citation19 To determine if AZD1775 and IR enhance CTL killing in cell lacking functional tp53, similar experiments were performed in MOC2 cells that harbor a non-sense Trp53 mutationCitation18 (Supplemental Figure 7). AZD1775 and IR significantly enhanced CTL killing over either treatment alone, demonstrating that the mechanism of enhancement may be independent of functional tp53 protein.
Figure 6. Combination ionizing radiation and Wee1 kinase inhibition enhance granzyme B-dependent CTL killing.
(a), SIINFEKL-expressing LLC, MOC1 or B16 cells were exposed to a single fraction of 8 Gy IR and AZD1775 (250 nM) alone or in combination and assessed for sensitivity to OT-I CTL killing (1:1 E:T ratio) via impedance analysis. Representative cell index impedance plots on left, with quantification of loss of cell index at 4 h on the right. (b), CTL killing following combination 8 Gy IR and AZD1775 was repeated in the presence and absence of concanamycin A (100 nM, OT-I CTL pre-treated for 4 h prior to addition to impedance assay). (c), G2/M block in tumor cells following exposure to granzyme B (3μg/mL)/streptolysin-O (40 ng/mL), AZD1775 (250 nM) and/or a single fraction of 8 Gy IR as detailed was assessed via flow cytometry 8 h after treatment. Representative dot plots from MOC1 cells shown on top and quantification of subG1 accumulation in MOC1, LLC and B16 cells on bottom. ConA, concanamycin A; GzmB, granzyme B; SLO, streptolysin-O.
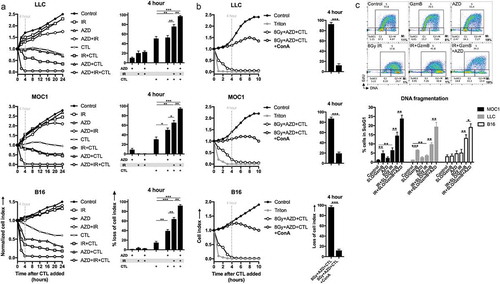
To explore possible mechanisms of this enhanced granzyme B-dependent CTL killing of tumor cells with combination IR and AZD1775, we measured components of immunogenicity on treated tumor cells (Supplemental Figure 8A&B). IR alone consistently enhanced cell surface expression of proteins required for CTL recognition and engagement of tumor cells, such as MHC class I, ICAM and CD86, but also enhanced PD-L1 expression. AZD1775 treatment alone produced fewer, inconsistent changes in these markers. Similarly, IR alone induced cell surface calreticulin translocation and HMGB1 release from cells, both markers of immunogenic cell death. This was not enhanced with the addition of AZD1775. These data suggested that changes in tumor cell immunogenicity or enhancement of immunogenic cell death could be one explanation for IR-enhanced CTL killing of tumor cells but did not explain why combination IR and AZD1775 treatment-enhanced CTL killing over IR alone. Both IR and streptolysin O/granzyme B (SLO/GzmB) induce G2/M cell cycle pause in tumor cells.Citation13,Citation20 While IR alone produced a dramatic G2/M cell cycle block, the addition of SLO/GzmB induced G2/M block in a greater percentage of cells (), suggesting at least a combinatorial effect. The addition of AZD1775 appeared to reverse this G2/M block and increase subG1 accumulation of tumor cells. These data suggested that both IR and CTL-derived SLO/GzmB dually induced G2/M pause within rapidly dividing tumor cells, and that AZD1775 reverses this G2/M block to induce additional loss of cell viability.
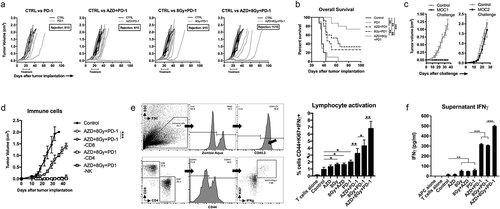
Given the above data, we hypothesized that combination IR and AZD1775 could enhance anti-tumor immunity following PD-1 ICB. Treatment of established MOC1 tumors with PD-1 mAb alone had little effect (). Treatment with a single dose of 8 Gy IR or AZD1775 in combination with PD-1 mAb produced variable tumor growth delay and rejection of established tumors in 27–33% of treated mice. Treatment with combination IR and AZD1775 plus PD-1 mAb induced rejection of established tumors in 73% of treated mice, and significant growth delay in the remaining treated mice, leading to significantly prolonged survival in the triple treatment group (). Treated mice demonstrated no weight loss or evidence of blood or organ toxicity (Supplemental Figure 9). Mice cured following triple agent treatment did not engraft tumors following rechallenge with MOC1 tumor cells in the contralateral flank 60 d after tumor rejection () but readily engrafted tumors when mice that rejected MOC1 rechallenge were subsequently rechallenged with MOC2 cells, demonstrating the presence of immunologic memory. Tumors were assessed for alterations in accumulation of neutrophilic-myeloid-derived suppressor cells (PMN-MDSC) or regulatory T cells (Tregs) following AZD1775 and IR treatment (Supplemental Figure 10). IR alone reduced PMN-MDSC and increased Treg tumor accumulation, but these cell subsets were not further altered with the addition of AZD1775. Next, tumor-bearing mice were treated with the triple combination in the presence or absence of systemic CD8, CD4 or NK1.1 depleting antibodies (). CD8 but not CD4 or NK1.1 depletion sub-totally reversed the triple agent treatment effect, demonstrating that rejection of established tumors is dependent on CD8+ cells under these experimental conditions. As a more direct measure of in vivo T-cell activation, T-cells from tumor-draining lymph nodes (DLNs) were sorted and, after combining with IFNγ-pretreated and irradiated tumor MOC1 tumors cells to induce tumor antigen-specific responses, assessed for activation. T-cells from mice treated with the triple therapy demonstrated the greatest activation, as measured by CD44, Ki67 and IFNγ positivity (), and produced the greatest amount of soluble IFNγ () upon exposure to MOC1 tumor antigen. These data support that a single dose of 8 Gy IR and systemic AZD1775 not only induce direct anti-tumor effects but also sensitize MOC1 tumors to immune activation and CD8+ cell-dependent tumor control following PD-1 ICB.
Figure 7. Ionizing radiation and Wee1 kinase blockade enhanced CD8+ cell-dependent responses to PD-1 ICB.
Established MOC1 tumors (0.1 cm3) were treated with combinations of 8 Gy IR alone (d 14), systemic AZD1775 (120 mg/kg daily for 10 d, starting at d 14) and PD-1 mAb (200 μg twice weekly for 2 weeks) as detailed. Cumulative results from two independent experiments shown. (a), individual tumor growth curves. (b), survival analysis. (c), control (WT B6) mice (n= 5) or mice cured following triple therapy (n= 11) were rechallenged in the contralateral flank at tumor d 100 with MOC1 cells (3e6 cells) and tracked for tumor growth. Forty days following challenge with MOC1, the same mice were rechallenged with MOC2 cells (1e5 cells) in the contralateral flank. (d), established MOC1 tumors were treated with triple therapy as in a, in the presence or absence of CD8, CD4 or NK depleting mAbs, and followed for tumor growth (n= 5/group). Established MOC1 tumors were treated with IR, AZD1775, and PD-1 mAb, alone or in combination, and CD3+ TIL sorted from tumor-draining lymph nodes were assessed for individual T-cell activation (CD44+Ki67+IFNγ+ by flow cytometry) (e) and cumulative IFNγ production (by ELISA) (f) following 48 h of exposure to MOC1 tumor antigen. APC alone refers to IFNγ-pretreated and irradiated MOC1 cells alone.
Discussion
Immune checkpoint blockade targeting the PD-axis is a promising treatment modality that can induce durable tumor control.Citation2,Citation3 Yet, the majority of patients with recurrent/metastatic cancer do not respond to PD-1/PD-L1 blockade. Combination PD-1-axis ICB with cytotoxic treatments designed to enhance anti-tumor immunity through induction of immunogenic cell death may enhance response rates.Citation21 Here we explore an alternative combination treatment strategy by addressing a tumor cell-intrinsic mechanism of resistance to IR and CTL killing. Tumor cells activate the G2/M cell cycle checkpoint, independent of underlying TP53 genomic status,Citation10,Citation13 in response to the genotoxic effects of IR and the cytotoxic effects of Granzyme B. This gives tumor cells time to repair cellular and DNA damage before cell cycle progression, preventing mitotic catastrophe and cell death.Citation16,Citation22,Citation23 Wee1 kinase inhibition abrogates the ability of tumor cells to activate the G2/M cell cycle checkpoint, reversing this mechanism of resistance. Administration of AZD1775, a Wee1 kinase inhibitor in clinical development, directly sensitized tumor cells to DNA damage and cell death after IR both in vitro and in vivo. Further, combination IR and AZD1775 sensitized tumor cells to CTL killing in vitro, as well as significantly enhanced the rate of immune-dependent rejection of established tumors following PD-1 ICB in vivo. IR is widely used for many cancer types, PD-1-axis ICB is FDA approved for many cancer types, and AZD1775 is progressing in clinical development.Citation14,Citation24 The use of three clinically relevant treatments targeting complementary mechanisms is a major strength of this work. IR and activated antigen-specific CTLs certainly kill tumor cells through mechanisms independent of mitotic catastrophe, but this work defines a newly described mechanism of tumor cell killing through AZD1775 reversal of G2/M block induced by both IR and CTL-derived granzyme B.
At concentrations similar to or below trough plasma concentrations achieved in phase I studies (250 nM),Citation14 AZD1775 induced little direct cytotoxicity in these tumor cells lines. This is consistent finding across multiple studies.Citation11,Citation13,Citation25 Inhibiting multiple component of the DNA damage response such as Wee1 kinase plus ATR or PARP enhances direct tumor cell cytotoxicityCitation11,Citation25 and would likely do so in the cell lines studied here. In this study, AZD1775 monotherapy was used to abrogate cell cycle checkpoint activation in response to cytotoxic insults mediated by IR and granzyme B. Other mechanisms underlying the ability of AZD1775 to enhance tumor cell cytotoxicity in response to IR and granzyme B may be playing a role. AZD1775 also inhibits the function of polo-like kinase-1 (PLK1),Citation26 a component of the cell cycle machinery that also regulates cell cycle progression in the setting of a DNA damage response.Citation27 Whether specific inhibition of PLK1 can reverse granzyme B-induced G2/M cell cycle pause to enhance cytotoxicity mediated by CTLs requires further study. We demonstrated that inhibition of Wee1 kinase with AZD1775 abrogated the G2/M cell cycle checkpoint and forced cells prematurely into mitosis. Whether these tumor cells prematurely entered mitosis after accumulating DNA damage in G2 or were directly pushed into mitosis from S phase as described previously with Wee1 kinase abrogation also requires further study.Citation28 Checkpoint kinase 1 (CHK1) is involved in G2/M cell cycle progression through inhibitory phosphorylation of cdc25.Citation29 Recent work has demonstrated that CHK1 inhibition can induce STING-dependent type I interferon, tumor cell PD-L1 expression and activation of innate immunity that can enhance the anti-tumor effects of PD-L1 blockade.Citation30 Our work demonstrated that AZD1775 did not induce immunogenic cell death, evidence of interferon signaling or directly enhance PD-L1 expression on tumor cells. It is likely that multiple mechanisms, in addition to direct abrogation of the G2/M cell cycle checkpoint, contribute to enhancement of anti-tumor immunity when small molecules that perturb cell cycle regulatory machinery are combined with immunotherapies such as immune checkpoint blockade.
Enhanced cytotoxic activity of IR after Wee1 kinase inhibition has been demonstrated in models of lung,Citation15 pancreatic,Citation11 hepatocellular,Citation12 central nervous systemCitation31,Citation32 and bone cancer.Citation33 This work demonstrated similar findings in head and neck and lung carcinoma and melanoma, suggesting that enhanced cell death following IR with G2/M cell cycle checkpoint inhibition may be a universal finding among cancer types. Previous work revealed sensitization of xenografts to IR with AZD1775.Citation11,Citation31 While xenograft models established that IR sensitization with AZD1775 translates in vivo, they did not allow the study of how these agents alter anti-tumor immunity. Through the use of syngeneic models, this work is the first to demonstrate that IR and AZD1775 cooperate to sensitize tumor cells to CTL killing, and that combination IR and AZD1775 sensitizes tumors in wild-type mice to PD-axis ICB. Previous work from our laboratory had demonstrated that two fractions of 8 Gy IR alone could sensitize tumors to PD-axis ICB resulting in systemic anti-tumor immunity and abscopal effects,Citation8 and that 3 weeks of daily Wee1 kinase inhibition alone could reverse cell cycle checkpoint activation induced by granzyme B leading to enhanced PD-axis ICB.Citation13 Our strategy here was to de-intensify both IR (one fraction of 8 Gy) and AZD1775 (2 weeks) and show that the combination enhanced CTL killing in vitro and PD-axis ICB in vivo to a greater degree than either treatment alone. Combination treatment with AZD1775, IR and PD-1 blockade would need to be assessed clinically in phase I setting initially, and most patients will have received and developed recurrent disease after definitive fractionated IR. A single, higher dose fraction of IR may more appropriately mirror IR that would be delivered in this setting.Citation34
Wee1 kinase blockade with AZD1775 is under evaluation in numerous clinical trials, mainly in combination with genotoxic chemotherapies.Citation35 This approach is supported with valid and extensive pre-clinical data, but most of these studies have been performed primarily in vitro or with the use of immunodeficient mouse models. Systemic genotoxic chemotherapy can cause long-standing systemic immunosuppression, and the balance of possible induction of immunogenic tumor cell death with negative effects on immune cell subsets needs clarification.Citation36,Citation37 This data and the work of Lawrence and colleaguesCitation11,Citation12,Citation15 support the use of localized IR as the source of DNA damage instead of systemic chemotherapy. Daily, low-dose fractionated IR can also cause profound immunosuppression, but recent and ongoing research has demonstrated that single dose or hypofractionated IR are immunostimulatory.Citation7,Citation8 IR enhances tumor cell immunogenicity, may increase pools of available neoantigens for presentation and detection by CTL, and induces STING-dependent type I interferon production required for innate immune priming.Citation5,Citation6,Citation38 Given the lack of immunosuppression, ability to selectively target tumor tissues and direct enhancement of anti-tumor immunity, single dose or hypofractionated IR may be a preferred alternative to the use of genotoxic chemotherapy in combination with Wee1 kinase inhibition. Additionally, combination treatment with AZD1775, IR and PD-1 blockade would need to be assessed clinically in phase I setting initially, and most patients will have received and developed recurrent disease after definitive fractionated IR. A single, higher dose fraction of IR may more appropriately mirror IR that would be delivered in this setting.Citation34
AZD1775 is administered systemically, raising the concern for on-target but off-tumor effects that could result in adverse side effects in patients. Inhibition of CDK1 phosphorylation in hair follicles is used as a biomarker of on-target effects in clinical trials.Citation39 Here we demonstrated greater accumulation of DNA damage (γH2AX phosphorylation) and enhanced tumor cell death after IR with the addition of AZD1775, likely due to induction of mitotic catastrophe. In tumors, we demonstrated reduced CDK1 phosphorylation and enhanced γH2AX phosphorylation in irradiated tumor but not distant, non-irradiated tissue. This suggests that the on-target effects of Wee1 kinase blockade with AZD1775 can be preferentially localized to tumor tissues with targeted IR. Systemic administration of PD-1 mAb also carries concern for on-target but off-tumor effects, but susceptibility to CTL killing was greatest with Wee1 kinase inhibition plus IR. This suggests that enhanced anti-tumor immunity following IR, Wee1 kinase inhibition and PD-1 blockade could similarly be focused on tumor tissue by using targeted IR. Carefully controlled clinical trials will be needed to ultimately assign causality and to determine if targeted IR and systemic AZD1775 can selectively sensitize tumor cells to immune destruction following PD-axis ICB.
In conclusion, by reversing G2/M cell cycle checkpoint activation, Wee1 kinase inhibition sensitized tumor cells to IR leading to enhance tumor control in vivo. This combination also sensitized tumor cells to CTL killing, at least through AZD1775’s ability to reverse redundant G2/M cell cycle block in cells exposed to IR and CTL-derived granzyme B. This appeared to be independent of induction of immunogenic cell death or interferon signaling within treated tumor cells. AZD1775 treatment translated in vivo to enhanced anti-tumor immunity and CD8+-dependent growth control or rejection of established tumors with combination IR, AZD1775, and PD-axis ICB. The ability of AZD1775 to sensitize tumor cells to IR is generalizable across many tumor types and supports the use of IR as an alternative to genotoxic chemotherapy in combination with Wee1 kinase inhibition. These data provide important mechanistic insights into how Wee1 kinase inhibition can be rationally combined with other anti-cancer strategies and provide the pre-clinical rationale for the combination of these treatments in clinical trials designed to enhance response rates to PD-axis ICB.
Methods
Cells and treatments
Lewis lung carcinoma (LLC, acquired from J. Hodge, National Cancer Institute), murine oral cancer-1 (MOC1, acquired from R. Uppaluri, Dana-Farber), and B16F10 (B16) melanoma cells (acquired from C. Hinrichs, National Cancer Institute) were cultured and validated as described.Citation40 AZD1775 was purchased from SelleckChem. In vitro, AZD1775 and Nocodazole (Sigma) were suspended in DMSO, EmbryoMAX nucleosides (Sigma), granzyme B (Biolegend) and streptolysin-O (Sigma) were suspended in culture media. For some experiments, tumor cells were engineered to express an MHC class I-restricted epitope of ovalbumin (SIINFEKL) via stable retroviral transduction as described.Citation13 A CsCitation1,Citation37, source irradiator (Gammacell-1000l 0.74 Gy/min) was used to irradiate cells in vitro. All in vivo experiments were approved by the NIDCD animal care and use committee. Tumors were established by subcutaneous injection of MOC1 cells (5x106 cells/mouse) in matrigel (Trevigen) into the lateral aspect of the right leg. Tumors were allowed to reach a volume of 0.1 cm3 (5–6 mm diameter) before the start of treatment. Volume calculated as (length x width2)/2. Mice were irradiated as previously described.Citation8 Mice were administered AZD1775 suspended in 0.5% methylcellulose via oral gavage. In some experiments, mice were treated with systemic PD-1 (clone RMP1-14), TNFα (clone XT3.11), CD8 (clone YTS169.4), CD4 (clone GK1.5) or NK1.1 mAbs (PK136) from BioXCell administered via intraperitoneal injection. For challenge experiments, MOC1 cells were injected subcutaneously in matrigel into the contralateral flank.
Flow cytometry and cell cycle analysis
For cell cycle analysis, EdU uptake was assessed using a Click-iT EdU assay kit (Thermo) per manufacturer recommendations, with the addition of FxCycle DNA stain (Thermo) and primary-conjugated antibodies for p-HH3S10 (clone 11D8) and p-γH2AXS139 (clone 2F3) from Biolegend after fixation and permeabilization. Tumor or oral mucosa tissues were digested into a single cell suspension as previous described.Citation41 Cell viability markers (sytox, zombie dye or 7AAD) were used in all assays to exclude dead cells. Isotype controls and “fluorescence minus one” methods were used to determine staining specificity.Citation42 Cells were analyzed via flow cytometry using a BD Canto I or BD Fortessa.
Western blot analysis
Standard lysate acquisition and blotting techniques were used. Anti-mouse total CDK1, p-CDK1Y15, p-γH2AXS139, and β-actin antibodies were purchased from Cell Signaling Technologies. Blots were imaged using Image Studio software (LI-COR Biosciences).
Immunofluorescence
Staining was performed as describedCitation41 using an unconjugated p-γH2AXS139 primary antibody (Biolegend) and secondary antibody conjugated to AF594 (Life Technologies). Images were acquired on an LSM 780 confocal microscope (Zeiss).
Real-time impedance assay
Parental MOC1, LLC, and B16 cells were plated at 1–2 × 104 cells/well in 96-well E-Plates (ACEA Biosciences), and real-time assessment of impedance was acquired using the xCELLigence Real-Time Cell Analysis (RTCA) platform per manufacturer recommendations and as described.Citation13 For other experiments, tumor cell variants expressing SIINFEKL were plated and allowed to adhere and gain impedance overnight before the addition of effector immune cells. Cytotoxic T-lymphocytes were derived ex vivo from OT-I mouse splenocytes as previously described.Citation13 Triton X-100 (0.2%) was added to some wells to induce complete loss of cell index with total cell lysis. Percent loss of cell index for a given time point was calculated as 1 − (experimental cell index/control cell index).
Clonogenic assay
Colony formation assays were performed as described.Citation43 Radiation enhancement ratio (RER) defined as the ratio of the radiation dose that resulted in 10% survival in the absence or presence of AZD1775.
Elisa
TNFα and IFNγ ELISA kits from R&D Systems were used per manufacturer recommendations.
qRT-PCR
Performed as previously described.Citation8 TNFα and GAPDH primers were purchased from Thermo.
Tumor antigen-specific T-lymphocyte stimulation assay
Tumor-draining lymph nodes were harvested into a single cell suspension and T-lymphocytes were sorted via negative magnetic selection (Pan T Cell Isolation Kit II) on an AutoMACS (Mitenyi). Sorted T-lymphocytes were added to IFNγ-pretreated (20 ng/mL × 24 h) and irradiated (50 Gy) MOC1 cells serving as antigen presenting cells at a 10:1 E:T ratio for 48 h, with the addition of brefeldin A for the final 4 h of stimulation. T-lymphocytes were assessed for activation via flow cytometry and supernatants were assessed for IFNγ via ELISA.
Statistical analysis
Tests of significance between pairs of data were reported as p-values, derived using a student’s t-test with a two-tailed distribution and calculated at 95% confidence. Comparison of multiple sets of data was achieved with analysis of variance (ANOVA) with Tukey’s multiple comparisons. All error bars indicate standard deviation. Statistical significance was set to p < .05. All analysis was performed using GraphPad Prism v7.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download MS Word (4.8 MB)Acknowledgments
The authors thank Drs. Nicole Schmitt and Larissa Sweeney for their critical review of this manuscript.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi:10.1126/science.aar4060.
- Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–1867. doi:10.1056/NEJMoa1602252.
- Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–965. doi:10.1016/S1470-2045(16)30066-3.
- Allen CT, Judd NP, Bui JD, Uppaluri R. The clinical implications of antitumor immunity in head and neck cancer. Laryngoscope. 2012;122:144–157. doi:10.1002/lary.21913.
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–852. doi:10.1016/j.immuni.2014.10.019.
- Gameiro SR, Malamas AS, Bernstein MB, Tsang KY, Vassantachart A, Sahoo N, Tailor R, Pidikiti R, Guha CP, Hahn SM, et al. Tumor cells surviving exposure to proton or photon radiation share a common immunogenic modulation signature, rendering them more sensitive to T cell-mediated killing. Int J Radiat Oncol Biol Phys. 2016;95:120–130. doi:10.1016/j.ijrobp.2016.02.022.
- Morisada M, Chamberlin M, Allen C. Exploring the rationale for combining ionizing radiation and immune checkpoint blockade in head and neck cancer. Head Neck. 2018;40:1321–1334. doi:10.1002/hed.25101.
- Morisada M, Clavijo PE, Moore E, Sun L, Chamberlin M, Van Waes C, Hodge JW, Mitchell JB, Friedman J, Allen CT. PD-1 blockade reverses adaptive immune resistance induced by high-dose hypofractionated but not low-dose daily fractionated radiation. Oncoimmunology. 2018;7:e1395996. doi:10.1080/2162402X.2018.1490854.
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi:10.1038/nature08467.
- Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi:10.1158/1535-7163.MCT-09-0463.
- Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR, Marsh KB, Davis MA, Zhao L, Maybaum J, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20:5085–5096. doi:10.1158/1078-0432.CCR-14-1038.
- Cuneo KC, Morgan MA, Davis MA, Parcels LA, Parcels J, Karnak D, Ryan C, Liu N, Maybaum J, Lawrence TS. Wee1 kinase inhibitor AZD1775 radiosensitizes hepatocellular carcinoma regardless of TP53 mutational status through induction of replication stress. Int J Radiat Oncol Biol Phys. 2016;95:782–790. doi:10.1016/j.ijrobp.2016.01.028.
- Sun L, Moore E, Berman R, Clavijo PE, Saleh A, Chen Z, Van Waes C, Davies J, Friedman J, Allen CT. WEE1 kinase inhibition reverses G2/M cell cycle checkpoint activation to sensitize cancer cells to immunotherapy. Oncoimmunology. 2018;7:e1488359. doi:10.1080/2162402X.2018.1490854.
- Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol. 2015;33:3409–3415. doi:10.1200/JCO.2014.60.4009.
- Parsels LA, Karnak D, Parsels JD, Zhang Q, Vélez-Padilla J, Reichert ZR, Wahl DR, Maybaum J, O’Connor MJ, Lawrence TS, et al. PARP1 trapping and dna replication stress enhance radiosensitization with combined WEE1 and PARP inhibitors. Mol Cancer Res. 2018;16:222–232. doi:10.1158/1541-7786.MCR-17-0455.
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi:10.1038/sj.onc.1207528.
- Stankevicius V, Kuodyte K, Schveigert D, Bulotiene D, Paulauskas T, Daniunaite K, Suziedelis K. Gene and miRNA expression profiles of mouse Lewis lung carcinoma LLC1 cells following single or fractionated dose irradiation. Oncol Lett. 2017;13:4190–4200. doi:10.3892/ol.2017.5877.
- Onken MD, Winkler AE, Kanchi K-L, Chalivendra V, Law JH, Rickert CG, Kallogjeri D, Judd NP, Dunn GP, Piccirillo JF, et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res. 2014;20:2873–2884. doi:10.1158/1078-0432.CCR-14-0205.
- Melnikova VO, Bolshakov SV, Walker C, Ananthaswamy HN. Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene. 2004;23:2347–2356. doi:10.1038/sj.onc.1207405.
- Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer. 2007;7:861–869. doi:10.1038/nrc2248.
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44:343–354. doi:10.1016/j.immuni.2015.11.024.
- De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin Cancer Res. 2011;17:4200–4207. doi:10.1158/1078-0432.CCR-10-2537.
- Chen G, Shi L, Litchfield DW, Greenberg AH. Rescue from granzyme B-induced apoptosis by Wee1 kinase. J Exp Med. 1995;181:2295–2300. doi:10.1084/jem.181.6.2295.
- Méndez E, Rodriguez CP, Kao MC, Raju S, Diab A, Harbison RA, Konnick EQ, Mugundu GM, Santana-Davila R, Martins R, et al. A phase i clinical trial of AZD1775 in combination with neoadjuvant weekly docetaxel and cisplatin before definitive therapy in head and neck squamous cell carcinoma. Clin Cancer Res. 2018;24:2740–2748. doi:10.1158/1078-0432.CCR-17-3796.
- Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, Gamper AM. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J Clin Invest. 2019;129:1329–1344. doi:10.1172/JCI122622.
- Wright G, Golubeva V, Remsing Rix LL, Berndt N, Luo Y, Ward GA, Gray JE, Schonbrunn E, Lawrence HR, Monteiro ANA, et al. Dual targeting of WEE1 and PLK1 by AZD1775 elicits single agent cellular anticancer activity. ACS Chem Biol. 2017;12:1883–1892. doi:10.1021/acschembio.7b00147.
- van Vugt MA, Bras A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. 2004;15:799–811. doi:10.1016/j.molcel.2004.07.015.
- Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A, Turner NC. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–539. doi:10.1158/2159-8290.CD-11-0320.
- Zeng Y, Forbes KC, Wu Z, Moreno S, Piwnica-Worms H, Enoch T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature. 1998;395:507–510. doi:10.1038/26766.
- Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–661. doi:10.1158/2159-8290.CD-18-1020.
- Caretti V, Hiddingh L, Lagerweij T, Schellen P, Koken PW, Hulleman E, van Vuurden DG, Vandertop WP, Kaspers GJL, Noske DP, et al. WEE1 kinase inhibition enhances the radiation response of diffuse intrinsic pontine gliomas. Mol Cancer Ther. 2013;12:141–150. doi:10.1158/1535-7163.MCT-12-0735.
- Sarcar B, Kahali S, Prabhu AH, Shumway SD, Xu Y, Demuth T, Chinnaiyan P. Targeting radiation-induced G(2) checkpoint activation with the Wee-1 inhibitor MK-1775 in glioblastoma cell lines. Mol Cancer Ther. 2011;10:2405–2414. doi:10.1158/1535-7163.MCT-11-0469.
- PosthumaDeBoer J, Würdinger T, Graat HC, van Beusechem VW, Helder MN, van Royen BJ, Kaspers GJ. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer. 2011;11:156. doi:10.1186/1471-2407-11-156.
- Baliga S, Kabarriti R, Ohri N, Haynes-Lewis H, Yaparpalvi R, Kalnicki S, Garg MK. Stereotactic body radiotherapy for recurrent head and neck cancer: A critical review. Head Neck. 2017;39:595–601. doi:10.1002/hed.24633.
- Fu S, Wang Y, Keyomarsi K, Meric-Bernstein F. Strategic development of AZD1775, a Wee1 kinase inhibitor, for cancer therapy. Expert Opin Investig Drugs. 2018;27:741–751. doi:10.1080/13543784.2018.1511700.
- Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, Magrath IT, Wexler LH, Dimitrov DS, Gress RE. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. 1997;89:3700–3707.
- Wu J, Waxman DJ. Immunogenic chemotherapy: dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018;419:210–221. doi:10.1016/j.canlet.2018.01.050.
- Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–1271. doi:10.1084/jem.20052494.
- Leijen S, van Geel RMJM, Pavlick AC, Tibes R, Rosen L, Razak ARA, Lam R, Demuth T, Rose S, Lee MA, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34:4371–4380. doi:10.1200/JCO.2016.67.5991.
- Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS Jr., Dunn GP, Bui JD, Sunwoo JB, Uppaluri R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012;72:365–374. doi:10.1158/0008-5472.CAN-11-1831.
- Clavijo PE, Moore EC, Chen J, Davis RJ, Friedman J, Kim Y, Van Waes C, Chen Z, Allen CT. Resistance to CTLA-4 checkpoint inhibition reversed through selective elimination of granulocytic myeloid cells. Oncotarget. 2017;8:55804–55820. doi:10.18632/oncotarget.18437.
- Herzenberg LA, Tung J, Moore WA, Herzenberg LA, Parks DR. Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol. 2006;7:681–685. doi:10.1038/ni0706-681.
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi:10.1038/nprot.2006.339.