
Abstract
Objective: To determine the target population and optimize the study design of the phase 3 clinical trial evaluating reldesemtiv in participants with amyotrophic lateral sclerosis (ALS).
Methods: We evaluated the phase 2 study of reldesemtiv, FORTITUDE-ALS, to inform eligibility criteria and design features that would increase trial efficiency and reduce participant burden of the phase 3 trial.
Results: In FORTITUDE-ALS, the effect of reldesemtiv was particularly evident among participants in the intermediate- and fast-progressing tertiles for pre-study disease progression. These participants most often had symptom onset ≤24 months and an ALS Functional Rating Scale-Revised (ALSFRS-R) total score ≤44 at baseline. Compared with the overall FORTITUDE-ALS population, the subgroup meeting these criteria declined by fewer ALSFRS-R points at 12 weeks (difference of least-squares mean [SE] versus placebo 1.84 [0.49] and 0.87 [0.35] for the overall population). These inclusion criteria will be used for the phase 3 clinical trial, COURAGE-ALS, in which the primary outcome is the change in ALSFRS-R total score at week 24. We also measure durable medical equipment use and evaluate strength in muscles expected to change rapidly. To reduce participant burden, study visits are often remote, and strength evaluation is simplified to reduce time and effort.
Conclusions: In COURAGE-ALS, the phase 3 clinical trial to evaluate reldesemtiv, the sensitivity of detecting a potential treatment effect may be increased by defining eligibility criteria that limit the proportion of participants who have slower disease progression. Implementing remote visits and simplifying strength measurements will reduce both site and participant burden.
ClinicalTrials.gov identifiers: NCT03160898 (FORTITUDE-ALS) and NCT04944784 (COURAGE-ALS)
Introduction
In recent clinical trials for amyotrophic lateral sclerosis (ALS), much attention has been directed to designing inclusion and exclusion criteria that yield a study population expected to progress in a more homogeneous manner. The edaravone development program provides a good example of this approach. The penultimate study noted trends toward efficacy that were accentuated in subgroup analyses of more rapidly progressing participants; the pivotal studies focused on this group and showed a statistically significant and clinically meaningful effect (Citation1,Citation2). A recent trial of sodium phenylbutyrate and taurursodiol (AMX0035) took a similar approach, and recruited a population in which the placebo group progressed even more rapidly than in the edaravone studies. This study showed a statistically significant slowing of the loss in ALSFRS-R total score in the active-treatment arm compared with placebo in the double-blind phase and evidence in a post hoc analysis supporting the possibility of longer survival in the group randomized to start active drug before the group that received placebo prior to active drug in the open-label extension (Citation3,Citation4).
While these studies demonstrated that choice of inclusion criteria can modify the behavior of study populations, regulatory guidance emphasizes enrolling a study population that is as representative as possible of the overall disease state (Citation5). Thus, a balance must be struck between enrolling participants with the greatest chance of demonstrating a clinical response to therapy and enrolling a cohort that is generalizable to the disease at large. Furthermore, recruitment becomes difficult as inclusion criteria are made more stringent. For these reasons, despite the success of the AMX0035 study (Citation3), current studies have employed less stringent inclusion criteria (Citation6,Citation7).
Reldesemtiv is a selective, small-molecule fast skeletal muscle troponin activator that is being investigated in ALS. It acts by increasing the contractility of skeletal muscle, and is highly selective for fast skeletal muscle, with minimal effect on cardiac or slow skeletal muscle (Citation8). Reldesemtiv has been studied in a multicenter, placebo-controlled, double-blind, phase 2 study, FORTITUDE-ALS (Functional Outcomes in a Randomized Trial of Investigational Treatment with CK-2127107 to Understand Decline in Endpoints – in ALS). In this trial, 458 participants with ALS were randomized to receive either reldesemtiv at one of three doses or placebo for 12 weeks (Citation9). Inclusion criteria were quite broad, and subgroup analyses suggested a greater signal in more rapidly progressive participants, in keeping with the findings of the edaravone and AMX0035 development programs.
For the phase 3 study to demonstrate efficacy of reldesemtiv, we aimed to recruit a study population that focused on participants with at least intermediate rates of disease progression but did not entirely exclude those with more slowly progressive disease. Therefore, it was essential to establish eligibility criteria that would select these participants and to achieve this goal, we reviewed previously reported FORTITUDE-ALS data and performed additional post hoc analyses. We evaluated several novel endpoints of strength and function based on findings in FORTITUDE-ALS. We also recognized the burden that clinical trial participation places on participants and their caregivers, as well as study sites, and designed a trial that reduced such burdens as much as possible.
Methods
We performed subgroup analyses on the FORTITUDE-ALS primary dataset to determine how inclusion criteria might be modified to enrich a study population most likely to demonstrate clinical efficacy with reldesemtiv. To identify the subpopulation who were the best responders, prespecified subgroup analyses were used to examine the potential heterogenous treatment effect among baseline disease characteristics. All analyses were post hoc and p values were not adjusted for multiple comparisons.
For the design of the phase 3 trial, several exploratory and secondary outcome measures were modified to measure changes in muscle strength and specific aspects of disability in an efficient and nonburdensome manner. We also met with individuals with ALS as well as advocacy groups to help us ensure the design was as participant friendly as possible.
Results
FORTITUDE-ALS analyses
Analyses according to prespecified subgroups were performed to compare change from baseline in the ALSFRS-R total score in the FORTITUDE-ALS full analysis set, using a mixed-model for repeated measures as previously described (Citation9). These analyses suggested that a number of inclusion criteria might effectively minimize enrollment of slowly progressive participants in whom an effective treatment would be more difficult to ascertain. Short time from symptom onset, more rapid progression prior to study initiation, and lower vital capacity at study baseline all predicted faster progression rates. The prestudy rate of progression was estimated by assigning an ALSFRS-R value of 48 at the time of symptom onset reported by the participant and calculating a slope from that time to the ALSFRS-R value at baseline. Values were divided into tertiles, resulting in slow progressors (prestudy disease progression rate of ≤0.37 points per month), intermediate progressors (pre-study disease progression rate of >0.37–0.67 points per month), and fast progressors (prestudy disease progression rate of >0.67 points per month). We also reasoned that participants with high ALSFRS-R values at baseline might be expected to progress more slowly even with short times from symptom onset.
The analysis of change in ALSFRS-R total score for subgroups according to prestudy progression rate is shown in . For a combined subgroup of participants in the intermediate- and fast-progressing tertiles, those randomized to reldesemtiv declined by fewer points versus placebo at 12 weeks (LS mean difference: 1.15 points, p = 0.011). Although participants in the slowest progressing tertile accrued some benefit from reldesemtiv, the change from baseline at 12 weeks was not statistically significant versus placebo. This finding was consistent with results of other ALS trials, suggesting that focusing on patients with more rapid disease progression could potentially more readily demonstrate treatment benefit (Citation1–4).
Figure 1 Change in ALSFRS-R total score for subgroups of participants according to pre-study rate of progression in FORTITUDE-ALS. Analysis includes all participants with an ALSFRS-R total score measured at baseline and at least one timepoint after baseline. ALSFRS-R: Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; FPT: fastest-progressing tertile; LS: least squares; IPT: intermediate-progressing tertile; SPT: slowest-progressing tertile. Reprinted by permission of the publisher (Taylor & Francis. Ltd, http://www.tandfonline.com) from Shefner et al. [Citation9]
![Figure 1 Change in ALSFRS-R total score for subgroups of participants according to pre-study rate of progression in FORTITUDE-ALS. Analysis includes all participants with an ALSFRS-R total score measured at baseline and at least one timepoint after baseline. ALSFRS-R: Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; FPT: fastest-progressing tertile; LS: least squares; IPT: intermediate-progressing tertile; SPT: slowest-progressing tertile. Reprinted by permission of the publisher (Taylor & Francis. Ltd, http://www.tandfonline.com) from Shefner et al. [Citation9]](/cms/asset/39f3d07f-4c95-44f8-a069-5fb98382e524/iafd_a_2216223_f0001_c.jpg)
While in this post hoc analysis, excluding the slowest progressing tertile made the impact of reldesemtiv more evident, our goal remained to enroll a study population with a range of progression rates. Characteristics of participants in each tertile were examined to identify variables with potential to minimize but not exclude participants with slowly progressive disease. Of 43 participants with baseline ALSFRS-R total score ≥45, 41 were in the slowest progressing tertile. In addition, of among 143 participants with symptoms >24 months, 85 were slow progressors and only 9 were fast progressors. Those with symptom onset ≤24 months before baseline and baseline ALSFRS-R total score of ≤44 progressed more quickly than the overall study population, and the point estimate of the effect of reldesemtiv was also increased (). On the basis of these analyses, and the need to enroll participants with a reasonably broad spectrum of disease, we concluded that inclusion criteria of symptom onset ≤24 months before baseline and maximum baseline ALSFRS-R total score ≤44 would result in enrolling more participants with fast and intermediate progression of disease, without completely excluding those with slow progression.
Table 1 Change in ALSFRS-R total score in FORTITUDE-ALS, showing (A) the overall study population and (B) the subgroup with symptom onset ≤24 months and baseline ALSFRS-R total score ≤44.
Another approach that has been previously employed is to select participants according to El Escorial diagnostic criteria (i.e. definite vs probable vs possible ALS). However, although participants diagnosed according to these categories do differ modestly in their progression rates, the variability of rate is actually increased in the more diffuse categories compared with possible ALS (Citation10). This increased range of progression rates is in part what we wished to avoid. Secondarily, even in experienced hands, there is significant error in how people with ALS are categorized (Citation11).
Design of COURAGE-ALS: novel outcome measures and reduced burden for participants
Taken together, the efficacy and safety data from FORTITUDE-ALS supported evaluation of reldesemtiv in a clinical trial with longer treatment duration. Informed by the subgroup analyses discussed above, COURAGE-ALS (Clinical Outcomes Using Reldesemtiv on ALSFRS-R in a Global Evaluation in ALS) has been designed as a multicenter, randomized, double-blind, placebo-controlled trial with the primary objective of assessing the effect of reldesemtiv on functional outcomes. Secondary objectives include a combined assessment of function, respiratory insufficiency, and survival, as well as changes in ventilatory function, quality of life, and muscle strength.
The trial was designed by the COURAGE-ALS Executive Committee and the study sponsor, Cytokinetics, Incorporated, with input from caregivers and people living with ALS. The protocol minimized the number of in-clinic visits in favor of remote encounters. For remote visits, all participants receive a mobile device with connectivity to perform a tele-visit and pre-loaded with required applications, including spirometry and the disease-specific quality-of-life questionnaire, Amyotrophic Lateral Sclerosis Assessment Questionnaire (ALSAQ-40). Samples for safety laboratory tests are also collected at home. To maximize data acquisition, home vital capacity assessments are performed using a video link with a trained evaluator.
The trial is registered at ClinicalTrials.gov (NCT04944784) and EudraCT (2020-004040-29).
Study design
An overview of the design and schedule of visits is shown in . A 24-week randomized double-blind, placebo-controlled period is followed by a 24-week period when all participants receive reldesemtiv. During the second period, participants and site staff remain blinded to the initial treatment assignment. Those who complete dosing through week 48 may enter an open-label extension; alternatively, participants not entering the extension will have a follow-up visit after 4 weeks. Participants are enrolled from approximately 80 clinical trial sites in Australia, Canada, Europe, and the United States. Approximately 555 participants meeting eligibility requirements at screening will be randomized in a 2:1 ratio to reldesemtiv 300 mg twice daily (bid) or matching placebo tablets for the first 24-week double-blind period. Randomization is determined using a central interactive web-response system and stratified by use/nonuse of riluzole and use/nonuse of edaravone (either oral or intravenous in geographies where it is approved).
Figure 2 COURAGE-ALS study design schematic. Participants completing week 48 may enter a planned open-label extension instead of the 4-week follow-up. ALSAQ-40: Amyotrophic Lateral Sclerosis Assessment Questionnaire; ALSFRS-R: Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; EQ-5D-5L: EuroQol-5-dimension-5-level; EQ-VAS: EuroQol Visual Analogue Scale; FVC: forced vital capacity
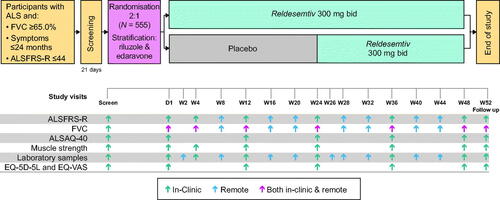
As shown in , there are up to eight clinic visits (Screening, day 1, weeks 4, 12, 24, 36, 48, and follow-up) and nine remote assessment visits (with an accompanying video call required except for visits only collecting safety laboratory test samples). For remote assessments, a trained evaluator performs the ALSFRS-R obtained via telemedicine and observes and coaches the participant as they perform the forced vital capacity (FVC) remotely, mirroring procedures for in-clinic assessment. After the randomization visit, in-clinic visits can be converted to remote visits at the discretion of the investigator, if deemed in the participant’s best interest.
In addition to study drug, participants are to receive standard of care for ALS according to the local region, as determined by the physician in discussion with the participant. For those receiving sodium phenylbutyrate/taurursodiol, edaravone or riluzole, participants must be on stable treatment before the trial and, following randomization, these therapies will ideally not be stopped or started. If additional agents are approved by local regulators for the treatment of ALS in specific regions, their use will be allowed in those regions during both the placebo-controlled and open-label periods.
Study population
The inclusion and exclusion criteria for COURAGE-ALS are summarized in and . As discussed above, these criteria have been modified from the FORTITUDE-ALS design, with the aim of minimizing the proportion of participants with slowly progressive disease without entirely excluding them. To help reduce the number of such participants, inclusion criteria restrict time from ALS symptom onset to ≤24 months and ALSFRS-R total score at screening to ≤44.
Table 2 COURAGE-ALS key inclusion criteria.
Table 3 COURAGE-ALS key exclusion criteria.
Study dose
The FORTITUDE-ALS trial discussed above was a dose-ranging study, in which participants with ALS were randomized 1:1:1:1 to 12 weeks of placebo or reldesemtiv at 150 mg bid, 300 mg bid, or 450 mg bid. All three reldesemtiv dose levels had similar effects versus placebo on changes in ALSFRS-R and slow vital capacity (SVC), although the more complete experience in the reldesemtiv clinical development program indicates that higher exposures are associated with greater increases relative to placebo in skeletal muscle force production. However, 450 mg bid was associated with higher rates of adverse events (AEs) related to nausea, decline in estimated glomerular filtration rate, elevations in alanine aminotransferase and aspartate aminotransferase and early termination of study drug than lower dose levels; consequently, 300 mg bid (for a total daily dose of 600 mg) of reldesemtiv was chosen for evaluation in COURAGE-ALS.
Study assessments
Key assessments are shown in . The ALSFRS-R and the FVC (safety permitting) will be completed at each study visit. Novel assessments include home FVC, which is measured both at scheduled remote visits and within 3 days of an in-clinic visit, to allow for direct comparison of clinic and home values. FVC was chosen rather than SVC given the paucity of home spirometers validated to perform SVC at the time we were selecting a device; studies have found that in those with ALS, the two measurements are strongly correlated (Citation14,Citation15). Quantitative muscle testing is assessed bilaterally for three intrinsic hand muscles. Evaluators who perform the ALSFRS-R, forced vital capacity, hand-held dynamometry, and grip strength receive trial-specific training and are required to pass associated tests that provide certification.
An additional novel measure assesses use of durable medical equipment, including manual or power wheelchairs, speech-generating devices, noninvasive ventilation, or a gastrostomy tube. When it is first prescribed and agreed to by the participant, and the reason why the equipment was obtained will be recorded and specific definitions will be used to capture timing and extent of use ().
Table 4 Dates for milestones of DME use in COURAGE-ALS.
Time to first hospitalization and number of hospitalizations will be recorded, and the investigator will determine if the hospitalization is related or unrelated to ALS. Hospitalizations unrelated to ALS are those that would have occurred even without the presence of ALS, such as acute appendicitis. Hospitalizations deemed related to ALS cover those that are related to disease progression, occur to address an ongoing ALS symptom or to be preventative (such as hospitalization for feeding tube), as well as those to address a complication of a treatment being administered for ALS.
Two standardized measures of health status, the EQ-5D-5L (EuroQol-5-dimension-5-level) and the EQ-VAS (EuroQol Visual Analogue Scale), as well as the ALSAQ-40 will be completed at all in-clinic visits except week 4. The BDI-FS (Beck Depression Inventory-Fast Screen) will be assessed at all in-clinic study visits. Time spent in each stage according to the Milano-Torino Staging (MiToS) system and number of participants whose transition stages over the course of the trial were selected as exploratory endpoints based on a post hoc analysis of the 12-week treatment period of FORTITUDE-ALS, which suggested that patients randomized to reldesemtiv may be at reduced risk for transitioning to a later stage (Citation16). Given the mechanism of action of reldesemtiv, it may be more likely to delay complete functional loss as measured by MiToS than delay progression of signs and/or symptoms of upper or lower motor neuron manifestations into a new region as measured by King’s staging (Citation16). Prespecified endpoints are summarized in .
Table 5 COURAGE-ALS study endpoints.
Safety will be assessed by monitoring AEs, physical examination, assessment of vital signs, 12-lead electrocardiograms, neurological exams, and clinical laboratory tests.
Statistical considerations
Efficacy analyses will be performed on the full analysis set, which includes all randomized participants who receive at least one dose of study drug and have a baseline efficacy assessment and at least one post-baseline efficacy assessment during the double-blind placebo-controlled period. Participants will be analyzed according to the treatment group to which they were assigned at randomization.
With 2:1 randomization to reldesemtiv and placebo, it was calculated that a sample size of approximately 555 participants will be required to achieve at least 90% power to detect a 1.8-point treatment difference between reldesemtiv 300 mg bid or placebo in the change from baseline to week 24 in ALSFRS-R total score. The treatment difference was estimated from the previous phase 2 study with 0.87-point mean treatment difference at Week 12, and assuming the separation trend continues to Week 24. This calculation is based on a two-sample t-test with two-sided alpha at 0.05 level, a common standard deviation of 5.5 points, and accounting for missing data due to early treatment terminations.
An independent, unblinded Data Monitoring Committee (DMC) will regularly review the data for safety and will also conduct two planned interim data reviews during this adaptive design study. First, 12 weeks after at least one-third of the planned study population has been randomized, they will assess the effect of reldesemtiv on ALSFRS-R total score change from baseline to week 12. If there appears to be a lack of effect in this first interim analysis, the DMC may recommend stopping the trial due to futility; if futility is not found, the trial will continue. Second, 24 weeks after at least one-third of the planned study population has been randomized, the DMC will assess whether the trial has adequate power to achieve a statistically significant effect on the primary endpoint in the final primary analysis, given the planned enrollment. The DMC may make a recommendation to (1) stop the trial if continuing is futile, (2) to increase the trial by a prespecified fixed number if continuing is promising, or (3) to continue as planned if the interim data do not suggest the first two options. This method is referred to as the CDL adaptive method and was initially proposed by Chen, DeMets, and Lan (Citation17), later extended by Gao, Ware and Mehta (Citation18), and Mehta and Pocock (Citation19).
Discussion
COURAGE-ALS is a phase 3 trial designed to confirm the efficacy of the fast skeletal muscle troponin activator reldesemtiv in ALS. When planning the clinical trial protocol, we assessed other recent ALS studies as well as subgroup analyses of the phase 2 study of reldesemtiv to identify factors that will enrich the study population with people whose ALS is progressing more rapidly and increase the probability of determining treatment benefit. Inclusion criteria have been carefully considered, with the aim of recruiting a participant cohort that provides good representation of all those living with ALS but progresses in a manner that allows for increased sensitivity to detect the impact of reldesemtiv on disease progression. The 24/44 criteria (symptoms of no more than 24 months and a maximum ALSFRS-R score of 44) are expected to enrich the population for intermediate and fast progressors and are practical from the standpoint of determining eligibility at screening. These inclusion criteria provide the opportunity for a broader population to be included in the trial compared with requiring a specific prestudy rate of disease progression or a run-in period requiring a set change in the ALSFRS-R total score. To avoid being too narrow—and wanting to balance the desire to identify a study population most likely to benefit with the practical need to complete recruitment in a reasonable time frame—COURAGE-ALS neither has inclusion criteria of a maximum FVC value of 80% nor does it exclude those with slow prestudy disease progression.
It should be noted that for vital capacity, it is possible that our calculation method could potentially impact trial eligibility related to the minimum FVC permitted (≥65.0%). In FORTITUDE-ALS, we instituted using the global lung initiative equation to determine the percent predicted value given it is based on a worldwide, large and recent compilation of normative data; this has continued in COURAGE-ALS. As well as trial eligibility, this has the potential to impact time to initiate noninvasive ventilation and to obtain a percutaneous endoscopic gastrostomy if these decisions are driven upon reaching a specific percent predicted FVC value (Citation20).
The study design also includes several nontraditional endpoints. Use of durable medical equipment reflects the level of disability reached by participants, and complements the functional assessment of the ALSFRS-R. We continue to assess strength, but in a manner that is less effortful than previous trials. Home testing of FVC offers the ability to obtain this measure without requiring monthly in-clinic visits and permits more rapid intervention when milestones are reached. Finally, the use of remote visits makes this trial as participant friendly as possible.
While the inclusion criteria for COURAGE-ALS are stricter than for FORTITUDE-ALS, they are still less stringent than those employed in either the edaravone or AMX0035 development programs (Citation2,Citation3), an approach that should increase the number of people with ALS who are eligible to participate in the trial. We have also endeavored to minimize the burden of clinical trial participation. Recognizing that frequent in-clinic visits become increasingly problematic with ALS progression, the trial was designed to reduce the number of in-clinic visits while continuing to assess disease progression monthly via tele-visits. We hope that the combination of the inclusion criteria and the participant-friendly design will allow for rapid recruitment and completion of this study.
It is important to recognize that the COURAGE-ALS inclusion criteria, as well as those of recent positive studies, are not intended to select for a disease phenotype that is uniquely responsive to any specific drug. Rather, such criteria are intended to identify a group in which a generally effective therapy may be efficiently evaluated. New insights into ALS pathophysiology may ultimately result in the identification of patient subgroups that respond to one form of therapy but not another. For example, drugs targeting the reduction of neuroinflammation might be assumed to be more effective in patients with high levels of inflammatory markers. Appropriate selection for such studies might rationally target some patients and exclude others. In contrast, we believe that, should COURAGE-ALS demonstrate efficacy, the effect could be reasonably expected to translate to the wide range of patients with ALS, including those who, for reasons of clinical trial efficiency, may not have been included in the trial itself.
Author contributions
All authors contributed to concept, interpretation of data and study design. LM and JW conducted the statistical analysis. JS drafted the manuscript, and all authors provided critical revision and review of the paper.
Acknowledgments
The authors thank the participants and their families for their contributions to the FORTITUDE-ALS and COURAGE-ALS clinical trials, the investigators, and members of the Data Monitoring Committees and Steering Committees. The authors acknowledge Geraldine Thompson, of Engage Scientific Solutions, Horsham, UK, for editorial assistance with the preparation of this manuscript, which was funded by Cytokinetics, Incorporated.
Disclosure statement
JMS reports compensation received as a consultant from Amylyx, Apic Biosciences, NeuroSense Therapeutics, Cytokinetics, Denali Therapeutics, GSK, Mitsubishi Tanabe Pharma America, Orphazyme, Orthogonal, Pinteon Therapeutics, RRD, SwanBio, Helixmith, Novartis, Sanofi, PTC, and EMD Serono; and research support from Amylyx, Biogen, Biotie Therapies (now Acorda Therapeutics), Cytokinetics, Mitsubishi Tanabe Pharma America, Alexion, MediciNova Inc, Ionis, Alector, and Orphazyme. AAC is an NIHR Senior Investigator (NIHR202421) and supported through the following funding organisations under the aegis of JPND - www.jpnd.eu (United Kingdom, Medical Research Council (MR/L501529/1; MR/R024804/1) and Economic and Social Research Council (ES/L008238/1)); and through the Motor Neurone Disease Association, My Name’5 Doddie Foundation, and Alan Davidson Foundation; and through the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London, and reports consultancies or participation in advisory boards for Amylyx, Apellis, Biogen, Brainstorm, Cytokinetics, GenieUs, GSK, Lilly, Mitsubishi Tanabe Pharma, Novartis, OrionPharma, Quralis, Sano, Sanofi, and Wave Pharmaceuticals. JAA reports research funding to their institution from Alexion, AZTherapies, Amylyx, Biogen, Cytokinetics, Orion, Novartis, MGH Foundation, Ra Pharma, Biohaven, Clene, and Prilenia and consulting fees from AL-S, Affinia, Amylyx, Apellis, Biogen, Cytokinetics, Denali, Orphazyme, Neurosense, Novartis, UCB, and Wave Life Sciences. AC reports participation on advisory boards for Biogen, Cytokinetics, Denali Pharma, Amylyx and Mitsubishi Tanabe. MdC reports compensation received as speaker/consultant from Cytokinetics, Kedrion, Biogen and GlaxoSmithKline; and research support from Cytokinetics, Biogen, and Pfizer. BMC is an employee of Sangamo Therapeutics, Inc., on the board of directors for Annexon Biosciences, Inc., and holds stock in Sangamo Therapeutics, Inc., Annexon Biosciences, Inc., and Cytokinetics, Incorporated. PCorica reports grant funding to his institution from Biogen and Cytokinetics, consulting fees from Kantar Health, and participation on a data safety monitoring board or advisory board for Cytokinetics, Amylyx, and VectorY. PCouratier reports grant funding to his institution from Cytokinetics and Agence nationale de la recherche (ANR), consulting fees from Amylyx and Biogen, support for travel for attending meetings from Amylyx and Biogen, and participation on a data safety monitoring board or advisory board for Amylyx and Biogen. MEC discloses potential conflicts of interest for Aclipse, Lilly, Immunity Pharm Ltd, Orion, Anelixis, Cytokinetics, Wave, Takeda, Avexis, Biogen, Denali, Helixsmith, Sunovian, Disarm, Als pharma, RRD, Transposon, quralis, Regeneron, Ab Sciences, Praxis Board of Director, Locust Walk, Neurosense, Faze, Arrowhead, Vector Y, Servier/Adiv, and Eledon. AG has served as a consultant for AB Science, AL-S Pharma, AveXis, Biogen, Cytokinetics, Incorporated, Mitsubishi Tanabe Pharma America, and Roche. OH reports consulting fees for participation in advisory boards for Biogen, Cytokinetics, Roche, and Pfizer; payment or honoraria for educational events from Biogen, participation on a data safety monitoring board or Advisory Board for Accelsior, and a leadership or fiduciary role as Editor in Chief, Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration Journal. TH-P reports grant funding to their institution from Cytokinetics for the study presented in the current manuscript, and other grant funding their institution from Cytokinetics, Samus, Orion, Bio 3, Mitsubishi Tanabe Pharma America, Healey Center, UCB Pharma, Alexion, AB Sciences; consulting fees from Samus, Alpha Insights, and Evidera; payment or honoraria for educational events from Platform Q Health, WebMD, MJH Holdings, IQVIA, P value, Vindico Medical Education, Projects in Knowledge; participation on a data safety monitoring board or advisory board for Mitsubishi Pharma America, Cytokinetics, AB Bio, Alexion, Biogen and Orphazyme; and a leadership or fiduciary role, unpaid, as the President of the ALS Hope Foundation. RDH reports compensation received from Biogen, Sanofi and has served on the advisory board for Cytokinetics. CI has received grant support from Pfizer, served on the data safety monitoring board for Appelis Therapeutics, and is a member of the ALS Publications Steering Committee for Cytokinetics. CEJ has served as a member on the data safety monitoring board for Anelixis Therapeutics Inc. and Mitsubishi Tanabe Pharma America. WJ reports grant funding to her institution from Cytokinetics, and personal payments from Cytokinetics for steering committee participation, both for the study reported in the present manuscript; grant funding to her institution from Brain Canada, Alexion Inc, ALS_Pharma Inc, Annexon Inc, Biogen, Calico, Medicinova, Mitsubishi-Tanabe Canada, Orion, Sanofi, and the University of Alberta Hospital Foundation; consulting fees from Amylyx, Biogen, and Mitsubishi-Tanabe Canada; and a leadership or fiduciary role for the ALS Society of Canada Board of Directors. NL is a consultant for Cytokinetics, Incorporated. AL reports grant funding to his institution from Cytokinetics, for the study reported in the present manuscript, and other grant funding to his institution from Amylyx, Ferrer International, Novartis Research and Development, Mitsubishi Tanabe, Apellis Pharmaceuticals, Alexion, Orion Pharme, Biogen, and Orphazyme; compensation for talks from Biologix, the German Society of Neurology, Biogen, Springer Medicine, Amylyx and Streamed Up! GmbH; support for attending meetings and/or travel from Biogen; participation on advisory boards of Roche Pharme, Biogen, Alector and Amylyx; and a leadership or fiduciary role as the president of the deutsche neurowissenschaftliche Gesellschaft NWG. NJM has received research support from Eledon, Apellis, Biogen Idec, Cytokinetics, Helixmith, Calico, Sanofi. He has served as a consultant or on advisory boards for Amylyx, Cytokinetics, Healey Center, Orion, Orphazyme, and Nura Bio. TMM is a consultant for Cytokinetics and Disarm Therapeutics, has licensing agreements with C2N Diagnostics and Ionis Pharmaceuticals; and serves on the advisory board and receives research support from Biogen. JSMP reports grant funding to his institution from Cytokinetics for the study reported in the present manuscript, serves in its advisory board, and reports other research grants from Ferrer International and Amylyx. SP has received honoraria as a speaker/consultant from Cytokinetics, Biogen, Roche, Desitin, Italfarmaco, Zambon, Amylyx, and grants from the German Neuromuscular Society, Federal Ministry of Education and Research, German Israeli Foundation for Scientific Research and Development, and EU Joint Programme for Neurodegenerative Disease Research. ZS reports grant funding to his institution from Cytokinetics, for the study reported in the present manuscript, and other research funding from MT Pharma; consulting fees from Amylyx or Cytokinetics, payment for participation on a data safety monitoring board or advisory board from Biogen and Corcept, and other personal payments from Wiley for his role as Editor of Muscle & Nerve. LHvdB reports participation on advisory boards for Cytokinetics, Ferrer, Amylyx, Sanofi, Biogen, Phoenix, Adore, and leadership or fiduciary role as Chair of ENCALS and Chair of TRICALS. LZ reports research support from the Focused Ultrasound Foundation, NIH, and ALS Canada; and consulting fees from Mitsubishi Tanabe Pharma, Amylyx, Biogen, and Cytokinetics. SK, FIM, LM, TJS, JW, AAW, and SAR are employees of Cytokinetics, Incorporated and hold stock in Cytokinetics, Incorporated.
Data availability statement
Data reported herein are part of a sponsor-led clinical development program that is ongoing, and thus study protocols and complete datasets for the trial will not be made available with this report.
Additional information
Funding
References
- Abe K, Itoyama Y, Sobue G, Tsuji S, Aoki M, Doyu M, et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:610–7.
- Edaravone (MCI-186) ALS 19 Study Group Writing Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16:505–12.
- Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383:919–30.
- Paganoni S, Hendrix S, Dickson SP, Knowlton N, Macklin EA, Berry JD, et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve. 2021;63:31–9.
- US Department of Health and Human Services, US Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Amyotrophic lateral sclerosis: developing drugs for treatment. Guidance for industry. https://www.fda.gov/media/130964/download. Accessed August 3, 2022.
- ClinicalTrials.gov. The PHOENIX trial: phase III trial of AMX0035 for amyotrophic lateral sclerosis treatment. NCT05021536. 2022. https://clinicaltrials.gov/ct2/show/NCT05021536
- ClinicalTrials.gov. The HEALEY ALS platform trial. NCT04297683. 2022. https://clinicaltrials.gov/ct2/show/NCT04297683
- Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, et al. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018;57:729–34.
- Shefner JM, Andrews JA, Genge A, Jackson C, Lechtzin N, Miller TM, et al. A phase 2, double-blind, randomized, dose-ranging trial of reldesemtiv in patients with ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22:287–99.
- de Jongh AD, Braun N, Weber M, van Es MA, Masrori P, Veldink JH, et al. Characterising ALS disease progression according to El Escorial and Gold Coast criteria. J Neurol Neurosurg Psychiatry. 2022;93:865–70.
- Johnsen B, Pugdahl K, Fuglsang-Frederiksen A, Kollewe K, Paracka L, Dengler R, et al. Diagnostic criteria for amyotrophic lateral sclerosis: a multicentre study of inter-rater variation and sensitivity. Clin Neurophysiol. 2019;130:307–14.
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
- Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40:1324–43.
- Pinto S, de Carvalho M. Correlation between forced vital capacity and slow vital capacity for the assessment of respiratory involvement in amyotrophic lateral sclerosis: a prospective study. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:86–91.
- Pinto S, de Carvalho M. SVC is a marker of respiratory decline function, similar to FVC, in patients with ALS. Front Neurol. 2019;10:109.
- Gebrehiwet P, Meng L, Rudnicki SA, Sarocco P, Wei J, Wolff AA, et al. MiToS and King’s staging as clinical outcome measures in ALS: a retrospective analysis of the FORTITUDE-ALS trial. Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(3-4):304–10.
- Chen YH, DeMets DL, Lan KK. Increasing the sample size when the unblinded interim result is promising. Stat Med. 2004;23:1023–38.
- Gao P, Ware JH, Mehta C. Sample size re-estimation for adaptive sequential design in clinical trials. J Biopharm Stat. 2008;18:1184–96.
- Mehta CR, Pocock SJ. Adaptive increase in sample size when interim results are promising: a practical guide with examples. Stat Med. 2011;30:3267–84.
- van Eijk RPA, Bakers JNE, van Es MA, Eijkemans MJC, van den Berg LH. Implications of spirometric reference values for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:473–80.