
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of this study was to optimize the cationic PEGylated niosome-containing anti-cancer drugs and siRNA to enhance the therapeutic response. Therefore, various surfactant-based (tween-60) vesicles of doxorubicin (DOX; a chemotherapeutic drug) and quercetin (QC; a chemosensitizer) were prepared. To load siRNA on niosomes, 1, 2-dioleoyl-3-trimethylammonium-propane (DOTAP) was used as a cationic lipid. The optimum formulation containing tween-60:cholesterol:DPPC:DOTAP:DSPE-PEG2000 at 49.5:5.5:15:25:5 demonstrated that the vesicle size and zeta potential were 52.8 ± 2.7 nm and +27.4 ± 2.3 mV, respectively. Entrapment efficiency (EE%) for DOX and QC was 86.4 ± 2.1% and 94.9 ± 3.9%, respectively. Moreover, the drug release during 6 h was 32.1 ± 1.6% and 30.5 ± 1.3% for DOX and QC, respectively denoted on the controlled release. The gel retardation assay demonstrated that siRNA could be successfully loaded into a cationic niosome:siRNA in a weight ratio 40:1. Additionally, noisome-encapsulated drugs had a higher toxicity against cancer cells when compared with un-encapsulated forms and the synergistic effects of co-delivery of siRNA and DOX with QC on gastric, prostate, breast cancer cells as well as human foreskin fibroblast as a normal cell line was shown. The results showed that the co-delivery of drugs and siRNA using cationic PEGylated niosomes exhibited an increased anti-cancer activity against the tumor cell death. It seems that cationic PEGylated niosomes have opened up a new avenue to enrich the armamentarium of therapeutic agents to fight cancer.
Graphical Abstract
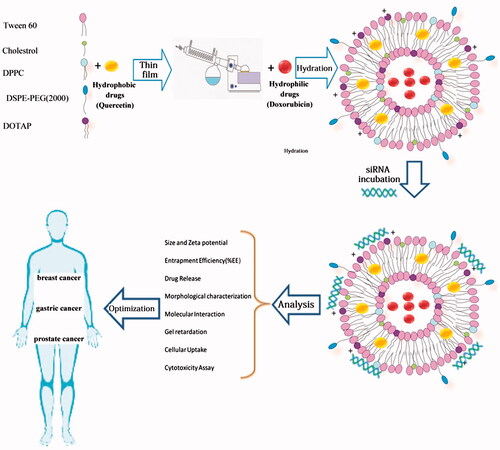
Introduction
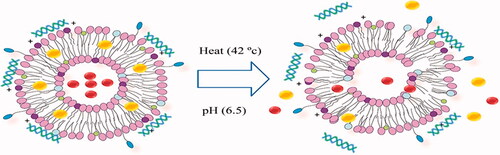
Cancer is the major cause of death both in developed and developing countries [Citation1]. The therapeutic approaches taken into account for the treatment of cancer are the surgery, chemotherapy and radiation. Most of the time, combination therapy is applied to reach a desired outcome and avoid the resistance during cancer therapy. The main obstacle of chemotherapeutic agents use is systemic toxicity and side effects on the normal cells. Drug resistance and extensive heterogeneity of cancer cells are the greatest impediment in cancer treatment. The efflux of anticancer drugs from tumor cells and the overexpressing the anti-apoptotic proteins result in chemo-resistance. The epigenetic differences and the DNA instability during clonal evolution of tumor cells generate tumor heterogeneity [Citation2]. Heterogeneity of cancer cells can lead to different responses to therapy and some tumor cells remain alive after the treatment. Therefore, combination treatment would more effectively inhibit cancer growth than the single-agent therapy and target multiple pathways of cancer cells. Combination therapy by either herbal medicine or chemotherapeutic agents with siRNA/miRNA exhibit synergistic enhances the tumor suppression [Citation3,Citation4]. Given the numerous advantages of nano-carriers including the ability to carry multiple drugs simultaneously and the reduction in systemic toxicity of the drugs in treating the cancer cells, these structures have attracted the attention of many researchers [Citation5]. Doxorubicin (DOX) is a chemotherapeutic agent that blocks the topoisomerase function and inhibits the tumor growth. Quercetin (QC), as an herbal medicine and chemo-sensitizer, is a phenolic compound possessing the antioxidant properties and anti-proliferative activity against many cancer cells [Citation6]. QC is capable of averting the tumor proliferation through activating the intrinsic pathway of apoptosis and arresting S phase during the cell cycle progression [Citation7]. It has been also shown that QC attenuates DOX-induced cardiotoxicity in mice [Citation8]. Small interfering RNA (siRNA) provides a promising approach for cancer therapy which inhibits the gene expression or translation. Oncogenes involved in different crucial steps of cell growth and development in cancer cells could be considered as the targets of these types of small RNAs [Citation9]. The siRNA can be designed against the oncogenes to inhibit the tumor growth. Cell division cycle 20 (CDC20) is a regulatory protein in the cell-cycle checkpoint (G2/M) interacting with anaphase promoting complex/cyclosome (APC/C) resulting in anaphase initiation [Citation10]. The up-regulation of CDC20 in cancer cells has been reported by many studies. Such overexpression is expected to increase the cell proliferation, tumor initiation and the development of cancer [Citation11,Citation12]. CDC20 siRNA can arrest the G2/M-phase and suppress the cell growth. There are some limitations for siRNA delivery into cells including a rapid degradation by serum nucleases, cellular levels of transport barriers, immunogenicity, systemic safety and stability [Citation13,Citation14]. Hence, in last decades, various types of nano-carriers have been extended. Niosome, a bilayer non-ionic surfactant-based vesicle, is a novel drug delivery system. Having the nano-metric scale size, biodegradability, relatively nontoxic property, remarkable stability, long storage time and low cost has enabled niosomes to be forerunners of other nano-carriers [Citation15]. According to , when the present delivery system is accompanied with the external stimuli such as pH and the temperature, it triggers the release of the drugs named thermo- and pH-sensitive niosome. In this study, to overcome the multidrug resistance and provide synergistic antitumor effects, we sought to develop cationic PEGylated niosomes that were able to encapsulate siRNA against CDC20 as well as DOX and QC. We also optimized the entrapment efficiency (EE%), the size of particles, the controlled release behavior and the yield of siRNA delivery. The co-encapsulation effect of DOX and siRNA on gastric, prostate and breast cancer cells was investigated when accompanied by co-administration of QC.
Figure 1. Schematic diagram of temperature and pH effects on cationic PEGylated niosome.
Materials and methods
Materials
Doxorubicin HCl (DOX) and QC (purity >95%) were obtained from Ebewe Pharma (Austria) and Sigma-Aldrich (St Louis, MO), respectively. Tween-60 was purchased from DaeJung Chemicals & Metals (South Korea). DSPE-PEG2000 (distearoyl phosphoethanolamine-polyethylene glycol), DPPC (1, 2-dipalmitoyl-sn-glycero-3-phosphocholine phospholipid) were obtained from Lipoid GmbH (Ludwigshafen, Germany). Cholesterol and DOTAP (1, 2-dioleoyl-3-trimethylammonium-propane) were supplied by Sigma-Aldrich (St. Louis, MO) and Avanti Polar Lipids (Alabaster, AL), respectively. PBS tablets, dialysis bag (MW = 12 kDa), DMSO (dimethyl sulfoxide), MTT (3–(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide) and paraformaldehyde solution were procured from Sigma-Aldrich (St. Louis, MO). DAPI (4′, 6-diamidino2-phenylindole) and DIL Stain (1, 1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine Perchlorate) were supplied from Thermo Fisher Scientific (Waltham, MA). All other chemicals, solvents and salts were of the analytical grade and used without further purification unless specified. The sequences of the siRNA targeted the CDC20 mRNA were sense 5′-GGGAAUAUAUAUCCUCUGUTT-3′, antisense 5′-ACAGAGGAUAUAUAUUCCCTT-3′. The sequences of the siRNAs were synthesized by Eurofins Genomics Ebersberg. The 5′ end was labeled with FAM to allow tracking using a florescence microscopy.
Methods
Preparation of drug-loaded nano-niosome
DOX-and QC-loaded nano-niosomes were synthesized and analyzed for particle size, zeta potential, EE% and controlled release parameters. To optimize these parameters and ascertain the optimal results following outlines were accomplished:
Evaluation of the influence of different molar ratios of tween-60:cholesterol and lipid to drug (lipid/drug) were performed on drug-loaded nano-carrier.
In order to be nano-niosome sensitive to temperature, we added 15% DPPC (transition temperature =41 °C).
To achieve long blood circulation and improve the drug bioavailability, the preparation stealth nano-niosomes was investigated by addition of 5% DSPE-PEG.
After achieving the best conditions of particle size, zeta potential, EE% and release, in order to load siRNA, 25% DOTAP was added to cationic nano-niosomes.
DOX and QC-loaded nanoparticles were prepared by thin film technique. In brief, tween-60, cholesterol, DPPC and DOTAP in the various mole ratios were dissolved in chloroform that was later evaporated. The present formulation contained 0 or 5% DSPE-PEG2000. QC stock solution was dissolved in methanol (0.5 mg ml−1) and added to the mixture of surfactant and lipids. To evaluate the cellular uptake, fluorescent label DIL was added to lipid phase at 0.1% mol. The thin films were dried to remove chloroform. To prepare niosomal DOX, the lipid-formed film was hydrated with DOX (0.5 mg ml−1) in phosphate-buffered saline (PBS) for 60 min at 60 °C using rotary instrument (Heidolph, Germany). Then, nanoparticles were sonicated over an ice bath for 30 min to produce small vesicles using microtip probe sonicator (E–Chrom Tech Co, Taiwan). Afterwards, free DOX and QC (unloaded) were separated from niosomal DOX and QC (Nio-DOX and Nio-QC) by dialysis bags that had a cut-off of 12 kDa. Then, they were analyzed in terms of the particle size, high EE% and controlled release parameters. The cationic niosomes were complexed with siRNA at various ratios and the formation of these complexes was confirmed by a gel retardation assay.
Cell lines and preparation of biological samples
The cell lines of human gastric cancer (AGS), prostate cancer (PC3) and breast cancer (MCF7) were supplied from Pasteur Institute (Tehran, Iran) and human foreskin fibroblast (HFF) cell line as a normal cell line were obtained from Stem Cell Biology Research Center (Yazd, Iran). They were cultured in DMEM medium (Gibco, Grand Island, NY). All cells were supplemented with 10% FBS (fetal bovine serum) (Gibco, Grand Island, NY) and penicillin–streptomycin (Gibco, Grand Island, NY) under standard condition (37 °C and 5% CO2 in a humidified incubator).
Particle size, zeta potential and morphological characterization
The particle size of niosomes, polydispersity index (PDI) and zeta potential was determined using dynamic laser scattering technique (DLS) by Brookhaven Corp Instruments (Holtsville, NY). The structure and surface morphology of nano-niosome were analyzed using scanning electron microscope (SEM). For SEM analysis, 5 μl of suspension was poured on the glass plate to form a thin layer film and air dried. The sample was sputter coated with a gold layer, and images were obtained using scanning electron microscopy (model EM3200, KYKY, China).
Entrapment efficiency
To determine DOX and QC content in the niosomal formulations, unentrapped drug was separated from the niosome-retained drug using membrane dialysis method against PBS at 4 °C. Afterwards, Nio-DOX and Nio-QC particles were lysed with isopropanol to analyze DOX and QC concentrations. The concentration of DOX and QC was determined by measuring absorption at 481 nm and 380 nm, respectively with a UV–Vis spectrophotometer (Perkin Elmer Germany). The EE% was calculated by the following equation:
EE% = Loaded drug in niosome (mg ml−1)/Total drug (mg ml−1) ×100
In vitro thermo- and pH-sensitive DOX and QC release assay
The release of DOX and QC from niosomes was carried out by a 12 kDa cut-off dialysis tube against PBS for 72 h at 37 °C and 42 °C temperature and pH 7.4, 6.5. To calculate the released DOX, the vessel was placed over magnetic stirrer (70 rpm). The dialysis media were collected at different times and immediately replaced with fresh PBS to maintain the sink condition throughout the experiment. Samples were analyzed using the UV–Vis spectrophotometer. According to the total drug concentration of the niosome formulation, percentage of release was calculated at each time interval.
Mathematical modeling of drug delivery
Drug released (cumulative percentage) from the nano-carrier was evaluated using the Equation as below:
(1)
(1)
Mt and Mf are the cumulative percentage of drug at t and final amount of drug released, respectively.
Zero-order rate correlation is as below:
(2)
(2)
where Qt and Q0 are the remaining drug at t and the initial value of drug, respectively.
For first order rate relation:
(3)
(3)
K is the first order release constant.
Higuchi’s model is as below:
Q is the value of drug released in t per surface, and KH is constant.
Functional group characterization
Fourier transform infrared (FTIR) spectroscopy (Model 8300, Shimadzu Corporation, Tokyo, Japan) was applied to analyze the molecular interaction between drugs and carrier for pure DOX, pure QC, and blank niosome, Nio-DOX, Nio-QC and Nio-siRNA, Nio-DOX-QC-siRNA. Samples were lyophilized and prepared as dry powder and mixed separately with potassium bromide (KBr), and the pellets were formed by placing samples in a hydraulic press.
Complex formation of cationic niosome with siRNA (gel retardation)
In order to optimize of the niosome:siRNA delivery system, cationic PEGylated niosomes were mixed with siRNA at various weight ratios (niosome/siRNA:10:1, 20:1, 30:1, 40:1, 50:1) and incubated at 25 °C for 30 min. Then complexes were loaded on ethidium bromide-containing 2% agarose gel and detected by electrophoresis at 90 V for 50 min and the images were obtained after UV exposure with a gel visualization system (Syngene GBOX, 680X) and analyzed to choose the maximum loading capability of siRNA in cationic PEGylate niosome.
In vitro cellular uptake
To determine the distribution of DOX, QC and FAM-labeled siRNA in the cells, the fluorescence intensity was detected. Briefly, AGS, PC3, MCF7 and HFF cells (1.5*105 per well) were seeded in 6-well plates, then were treated with DIL Nio, free DOX (0.3 µg ml−1), DIL-Nio-DOX (0.3 µg ml−1), free QC (40 µg ml−1), DIL-Nio-QC (40 µg ml−1), free FAM-labeled siRNA (100 nM), Nio-FAM-labeled siRNA (100 nM), free DOX-QC, Nio-DOX-QC. After 30 and 180 min of incubation, cells were washed twice with PBS (pH 7.4) and fixed with a 95% ethanol solution. The nuclei of cells were counterstained with DAPI (1 mg ml−1) for 15 min. Images were obtained by fluorescence microscopy (Olympus, Japan).
Cytotoxicity study
MTT assay was performed to calculate the IC50 doses of free DOX and niosomal DOX (0.039, 0.078, 0.156, 0.312 and 0.625 µg ml−1) as well as free QC and niosomal QC (7.8, 15.6, 31.2, 62.5 and 125 µg ml−1), which were incubated for 24, 48 and 72 h. MTT assay was also carried out to study the anti-proliferative activity of blank niosome, free siRNA (100 nM) and Nio-siRNA (100 nM and 200 nM). In order to find out whether there was a synergism/antagonism in co-administration of niosomal forms of DOX and QC, we used the different concentrations of QC (0, 10, 20, 30, 40 and 60 µg ml−1) with a fixed concentration of DOX (0.156 µg ml−1) and also performed combination experiments with a series of different DOX concentrations (0.039, 0.078, 0.156 and 0.312 µg ml−1) plus constant concentrations of QC (20 µg ml−1). The inhibitory effects of triple combination therapy consisted of Nio-siRNA (100 nM)-DOX (0.156 µg ml−1) with Nio-QC (40 µg ml−1) which were incubated for 72 h with 104 cells in a 96-well plate was also executed. After 72 h, the contents of wells were removed and incubated with 10 µl of 5 mg ml−1 MTT and 90 µl medium for 3 h. The resultant formazan crystals were dissolved in DMSO. The absorbance of obtained samples was measured using EPOCH microplate spectrophotometer (synergy HTX, Bio-Tek, Winooski, VT) at 570 nm.
Statistical analysis
The statistical analysis was calculated using GraphPad Prism version 6 (GraphPad, San Diego, CA). The experimental values were expressed as the mean ± standard deviation (SD). A Student's t test was used when comparing two independent groups, and an ANOVA test was used when comparing multiple samples. The significance of difference between groups was shown as p < .05 (*), p < .01 (**), p < .001 (***) and p < .0001 (****).
Results and discussion
Characterization of optimized formulation
Particle size, zeta potential and morphological characterization
As shown in , the average diameters were less than 100 nm, which are enabling them to pass through the blood barrier and be accumulated in purpose tissue. The addition of negatively charged DSPE-PEG by making repulsive forces between niosomes, reduced the vesicle size to 52.8 ± 2.7 nm versus F10 with 59.7 ± 2.4 nm. These results are in agreement with previous studies that addition of DSPE-PEG into niosomes results in the reduction of niosomes aggregation and vesicle size (average 11.6%) [Citation16]. The size of cationic noisome is slightly increased when loaded with siRNA (average 20.1%). The zeta-potential of niosomes containing tween-60:cholesterol was −6.5 ± 1.8 mV and for the positively charged DOTAP niosomes (F12) was +35.2 ± 1.6 mV. Incubating the siRNA with niosomal formulation (F12) reduced the zeta potential to +26.8 ± 2.5 mV (average 23.9%) (). These results confirmed that the siRNA was successfully incubated with niosomes. As shown in , the SEM photograph of the Nio-siRNA-DOX showed that cationic PEGylated niosomes were rounded without having a formation of aggregation. The photographs also depicted that niosomes had a smooth surface. As expected, the average diameter of nanoparticle was consistent with the sizes measured by the DLS.
Figure 2. The scanning electron microscopy (SEM) of Nio-siRNA-DOX.

Effect of tween-60:cholesterol and lipid/drug ratios on DOX-QC niosome formulations
In order to optimize nano-niosome, we surveyed the effects of different ratios of tween-60:cholesterol and lipid/drug (L/D) on EE% and the release of niosomes. In , it has been shown that the EE% of DOX and QC was significantly increased together with the increment of tween-60:cholesterol ratio. The maximum EE% was found to be 82.5 ± 3.7% for DOX and 91.7% ± 3.2 for QC at a molar ratio of 90:10 (tween-60 to cholesterol). The results indicated that tween-60, has a larger hydrophilic head group than the cholesterol thus entrapping higher levels of DOX. The long chain of tween-60 decreases the drug leakage leading to the lower rate of the drug release. The EE% of QC reduced when the cholesterol content was increased since the interference of cholesterol would decrease the drug entrapment by competing with the drug for entry into the bilayer, thus preventing lipophilic drug to be imbedded into the vesicles [Citation17]. On the other hand, the hydrophilic head group of tween-60 can dissolve a higher level of QC due to the existence of phenolic groups suggesting a higher chance for the substance to form hydrogen bonds [Citation18]. The fatty acid chain length of tween-60 influences on the drug release and as the higher ratio of tween-60 to cholesterol diminishes the release rate [Citation19]. As summarized in , the L/D molar ratio has an impact on drug loading. Changing the L/D molar ratio from 5 to 10, while keeping other factors constant, elevates the EE% of DOX from 77.6 ± 4% to 82.5 ± 3.7%. In contrary to QC, changing the L/D molar ratio from 5 to 10, while keeping other factors unchanged, lowered the EE% of QC from 95.1 ± 3.9% to 91.7 ± 3.2%. Increasing the L/D molar ratio results in increasing the concentration of bilayer-forming materials, which in turn increases the number of vesicles in a given volume that can lead to increase in the amount of DOX entrapped in the vesicles [Citation20]. In contrary to QC, increased in the L/D molar ratio leads to the lower amount of EE% because the high lipid concentrations compete with QC to entry into the lipid bilayer. In all formulations, the release of drug from niosomes was followed by a triphasic pattern consisting of an initial burst release, a constant and continuous release followed by an apparent zero-order release phase. The initial burst release is due to diffusion from higher to lower concentration. shows that niosomal formulations of tween-60 containing 10% cholesterol for DOX (22.4 ± 1.6%) and QC (40.5 ± 1.8%) displayed the lowest extent of drug release after 72 h as compared with 50% cholesterol that exhibited the drugs release 29.1 ± 1.4% and 47.6 ± 1.7% for DOX and QC, respectively (p < .05). This phenomenon suggests that increasing the cholesterol content can interrupt the regular linear structure of the vesicular membrane and increase the rate of drug release [Citation21,Citation22]. The increase in lipid to drug ratio would decrease the drug release owing to increasing the interaction between hydrophilic end and hydrophobic acyl chain of tween-60 and drugs. displays that the burst release rate of DOX and QC within 6 h reached 32.1 ± 1.6% and 30.5 ± 1.3%, respectively suggesting that niosomes provide an excellent release from the carriers. The best formulation for further studies was found to be F6 that provided a high EE% and continuous drug release.
Table 1. The effect of tween-60:cholesterol and L/D ratio on EE%, the long-term and short-term release.
Table 2. The effect of various types of phospholipids on EE%, size, zeta potential and the release.
Effect of phospholipid on DOX-QC niosome formulations
For the development of a biocompatible and optimum niosomal delivery system, various phospholipids used. As shown in , the formula containing phospholipids showed the higher drug EE%. Adding DPPC:DOTAP in the ratio of 15:25 increases the EE% of DOX and QC 91.6 ± 3.5% and 99.3 ± 3.3%, respectively since DOX and QC can make an interlink to phospholipids by hydrogen bond established between their own hydroxyl groups existing in DOX and QC and lipid polar head groups in phospholipids [Citation23]. Upon the addition of DOTAP, the flexibility of binding was enhanced because of the unsaturated alkylchain of DOTAP has made it more mobile and flexible that increases the drug release [Citation24]. According to the results represented in , the formula of cationic PEGylated niosomes containing DPPC, DOTAP, and DSPE-PEG2000 at a molar ratio of 15:25:5 (F11) indicated that the EE% of DOX and QC could achieve 86.4 ± 2.1% and 94.9 ± 3.9%, respectively. Similarly, Li Y-P was found that the EE% was slightly reduced by the addition of DSPE-PEG [Citation25]. The release of DOX and QC was about 32.1 ± 1.6% and 30.5 ± 1.3%, respectively at the first hours and 27.4 ± 1.4% and 45.3 ± 1.1% for DOX and QC, respectively after 72 h. Comparison of F10 release with F11 has shown that addition of DSPE-PEG lowers the drug release that was aligned with the results of similar studies [Citation25,Citation26]. Also addition of DSPE-PEG2000 enhances the antitumor activity of drugs through prolonging the circulation time of drugs in blood circulation, solubilizing the hydrophobic drugs in water and decreasing the size of particles which in turn results in increasing the cellular uptake [Citation27,Citation28] decreasing the reticuloendothelial system (RES) and renal clearance [Citation29,Citation30]. For siRNA delivery, we synthesized cationic niosomes bearing a high positively charged (zeta potential +35.2 ± 1.6 mV by addition of 25% DOTAP. Notably, the measurement of PDI (0.16 ± 0.02) indicated that there was no agglomeration. It can be inferred that the optimal formula contains 25% DOTAP, 15% DPPC and 5% DSPE-PEG 2000, which led to a positive zeta potential, small size, highest EE% and optimal controlled release.
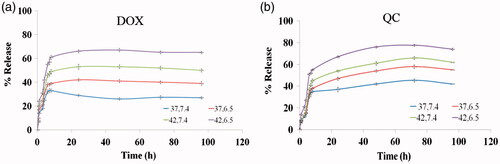
In vitro thermo- and pH-sensitive release assay
The alteration occurs in cancer cells leads to a lower pH and a higher temperature when compared to normal cells [Citation31]. The structure of the nano-niosome membrane, pH of surrounding buffer and temperature influence on the rate of drugs release, therefore, we designed a thermo- and pH-sensitive formulation to improve passive targeting of anti-cancer drugs into tumor cells and subsequently reduce the side effects on normal cells. shows the in vitro release profiles of DOX and QC at physiological condition i.e. pH 7.4 and temperature 37 °C and for the tumor tissue i.e. pH 6.5 and temperature 42 °C. The results demonstrated that DOX and QC at pH 6.5 and temperature 42 °C had the fastest release rate in which the release rates of 65.1 ± 1.3% for DOX and 77.6 ± 1.5% for QC after 72 h. However, at physiological pH and temperature, the least release rates of these drugs, 27.4 ± 1.4% for DOX and 45.3 ± 1.1% for QC, were observed during the same period (p < .05). In fact, in a higher temperature above ∼40 °C, the permeability of bilayer membrane of noisomes is enhanced; leading to the higher levels of the drug release. Also at acidic pH, a proton gradient is created across the membrane and protonation of the amino group on DOX enhances the drug release [Citation32]. Interestingly, the cytotoxicity assay showed that drug-loaded niosomes have a higher cytotoxicity in comparison to un-encapsulated drugs. The obtained result is consistent with the observations of a study performed by Naderinezhad et al. [Citation33]. In line with this, the results of the drug release revealed that it was just 65.1 ± 1.3% for DOX and 77.6 ± 1.5% for QC during 72 h at the concentrations of 0.156 µg ml−1 Nio-DOX and 40 µg ml−1 Nio-QC lead to killing approximately 67% of AGS, 74% of PC3 and 72% of MCF7 cells. Furthermore, the total release of free DOX and QC lead to killing 59% of AGS, 66% of PC3 and 63% of MCF7 cells. It can be concluded that Nio-DOX and Nio-QC at lower dose show a higher toxicity against cancer cells.
Figure 3. The drug release profile in various temperatures and pH.
Modeling of drug release
shows the correlation coefficient (R-square) evaluated for nano-carrier formulation. The results revealed that the DOX and QC from nano-carrier film are most fitted to Higuchi’s correlation due to higher R2. illustrates the comparison of experimental data and theoretical results for release of DOX and QC according to Higuchi’s model. Also, shows the Higuchi release kinetics for DOX and QC. confirms the Zero order release kinetics for DOX and QC as well. The first order model of DOX and QC release was shown in . These results suggested that DOX and QC molecules were disseminated in the niosome matrix and that there were limited interactions between drugs and the components of niosome [Citation34]
Figure 4. (a) Plot of theoretical amount of DOX release versus experimental data based on the Higuchi’s model; (b) plot of theoretical value of QC release versus experimental data versus based on the Higuchi’s model; (c) Higuchi release kinetics for DOX and QC; (d) zero order release kinetics for DOX and QC and (e) first order release kinetics for DOX and QC.
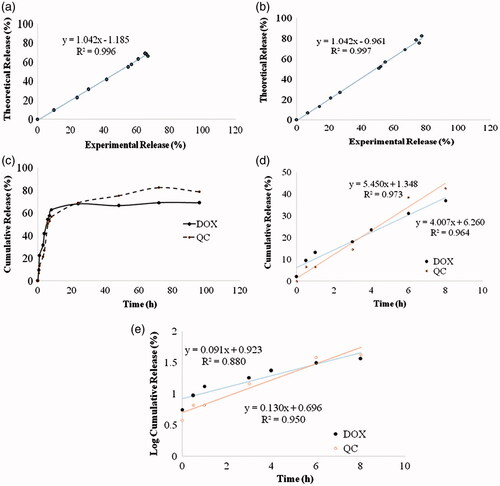
Table 3. Release kinetics data of DOX and QC from the niosomal optimum formulae.
Chemical interaction
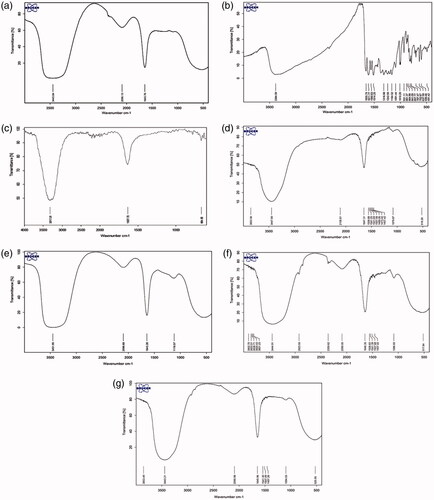
As depicted in the FTIR results of the pure DOX, pure QC, blank niosome, a single drug carrier and Nio-siRNA-DOX-QC were represented. illustrates the FTIR of pure DOX and pure QC. As displayed in (blank niosome), there were special peaks for tween-60, DSPE-PEG and cholesterol at 3317.01 cm−1 that represent O–H bands vibrating and N–H stretching in 2° amines. The peaks at 1637.72 cm−1 verified the existence of C=O stretching of the ester group in tween-60, DOTAP and cholesterol. illustrates that there was no chemical interaction between siRNA and cationic PEGylated niosomes. Additionally, the siRNA loading on cationic niosomes had no effect on its structural characterization. According to , the out-of-plane bending peaks occurs within the range of 900–500 cm−1 that can be referred to mono-substitution occurred on the DOX ring that confirms the encapsulation of DOX with the cationic niosome. Also shows the out-of-plane bending peaks occurred in the range of 800–500 cm−1 that allude to an appropriate ortho substitution on the QC ring verifying that the QC loading on the cationic niosome formulation. Hence, it could be conceivable that all of peaks repeatedly recorded in above experiments would imply that there was no chemical interaction between the drugs and other components of the formula indicating a successful loading on cationic PEGylated niosomes ().
Figure 5. FTIR spectra of (a) DOX; (b) QC; (c) Blank niosome; (d) Nio-siRNA; (e) Nio-DOX; (f) Nio-QC; and (g) Nio-siRNA-DOX-QC.
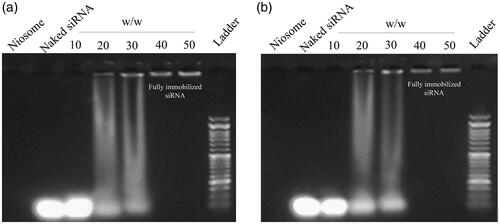
Complex formation of cationic PEGylated niosome with siRNA and leakage stability
As shown in , cationic PEGylated niosomes were able to bind siRNA in a dose-dependent manner. The weight ratio of 40:1 for cationic PEGylated niosomes to siRNA was an optimal condition. To analyze the effects of long-term storage on the leakage of siRNA from the Nio-siRNA, the complexes were stored at 4 °C for 4 months. The stability was measured using gel electrophoresis as shown in . The results demonstrated the complexes were even stable after 4 months with the lowest leakage.
Figure 6. (a) Agarose gel electrophoresis of free siRNA (1 µg) and siRNA loaded in niosome vesicles to determine the most effective ratio of niosome: siRNA. (b) Monitoring of siRNA loading stability after 4 months storage at 4 °C.
Cellular uptake assay
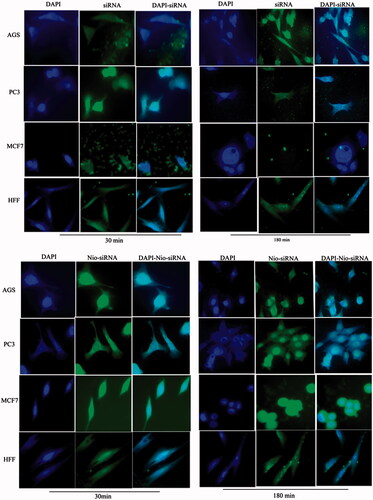
show the cellular uptake of free drugs (DOX, QC and siRNA) and drug-loaded niosome in AGS, PC3, MCF7 and HFF cell lines after 30 min and 180 min that were monitored by fluorescence microscopy. illustrates the successful delivery of DIL-labeled niosome to cells. According to , the number of cancer cells treated with Nio-DOX and Nio-QC were depicted as red and green fluorescence, respectively was higher than cells treated with free drugs. The results showed that DOX is accumulated in the nucleus region whereas the QC has a tendency to be hoarded in cytoplasm. According to the obtained data, the cationic niosomes were efficiently uptaked by the cancer cells. Seemingly, the endocytosis plays a key role in penetrating the drug-loaded niosomes into the cells. It also plays a crucial role in delivering DOX, QC and siRNA into the cells in comparison to free drugs passed through the cell membrane by a diffusion mechanism. On the other hand, nano-niosomes increase the solubility of QC considered a hydrophobic drug. The nano-niosomes can also increase the contacting surface area leading to a higher toxic effect in comparison to free QC [Citation35]. Niosomes-containing DOX can also be continuously released resulted in higher degrees of cytotoxicity as compared to free DOX. The intensity of fluorescent in HFF as normal human cells was much less than the cancer cells. These results demonstrate that the entry of Nio-DOX and Nio-QC into the normal cell was much lower with respect to cancer cells. As previously mentioned, our formulation is thermo- and pH-sensitive possessing a lower toxic effect on normal cells. These results confirmed the cellular uptake of Nio-DOX and Nio-QC by HFF showed a fewer purple and turquoise blue color intensity and lower amount of drugs in comparison to free drugs that was consistent with the results of cell viability exhibiting free drugs have a higher toxicity with respect to niosome-containing drugs. and illustrate the concurrent cellular uptake of DOX and QC at the both of free and niosomal forms that exhibit cancer cells successfully uptaked DOX and QC. displays the transfection and localization of free FAM-labeled siRNA and Nio-FAM siRNA in AGS, PC3, MCF7 and HFF cell lines after 30 and 180 min. The comparison of the free siRNA accumulation in the cytoplasm and Nio siRNA shows that transfection niosomal siRNA delivery system was far more efficient. The most fluorescence intensity for the drugs was found in the cells after 180 min of incubation. In another word, 180 min incubation had a higher fluorescence intensity for drugs with respect to incubation in 30 min.
Figure 7. Cellular uptake images of AGS, PC3, MCF7 and HFF cells, incubated with DIL-labeled Niosome (DIL-Nio) for 30 min and 180 min. DAPI (blue) was used for nucleus staining and DIL dye (green) was used for phospholipid staining.
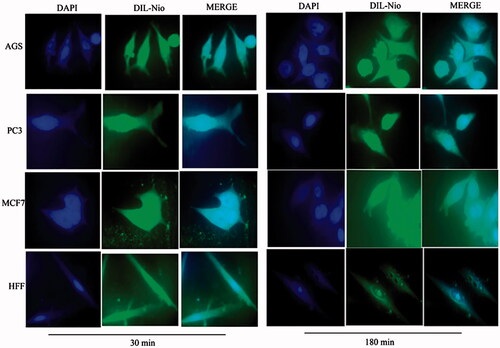
Figure 8. Cellular uptake images of AGS, PC3, MCF7 and HFF cells, incubated with free DOX for 30 min and 180 min. DAPI (blue) and DOX (red).
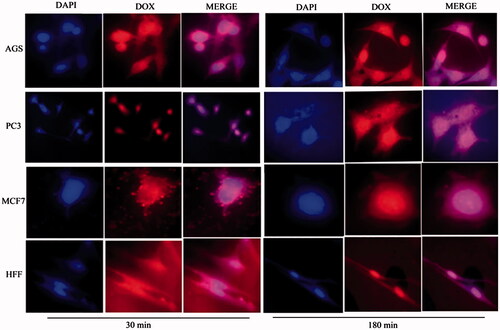
Figure 9. Cellular uptake images of AGS, PC3, MCF7 and HFF cells, incubated with DIL-Nio-DOX for 30 min and 180 min.
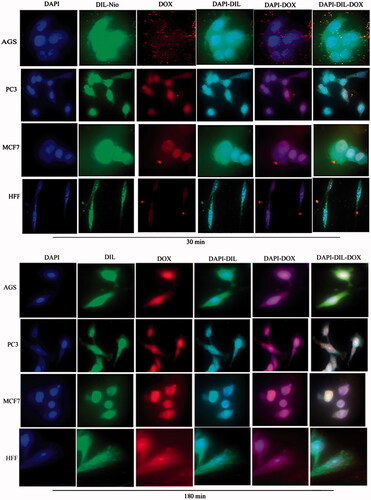
Figure 10. Cellular uptake images of AGS, PC3, MCF7 and HFF cells, incubated with (a) free QC and (b) DIL-Nio-QC for 30 min and 180 min. DAPI (blue) and QC (green).
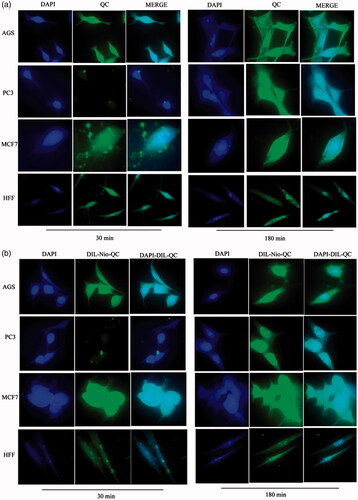
Figure 11. Cellular uptake images of AGS, PC3, MCF7 and HFF cells, incubated with free DOX and free QC for 30 min and 180 min.
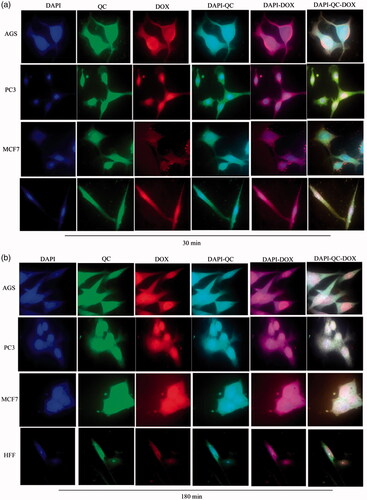
Figure 12. Cellular uptake images of AGS, PC3, MCF7 and HFF cells, incubated with Nio- DOX and Nio-QC for 30 min and 180 min.
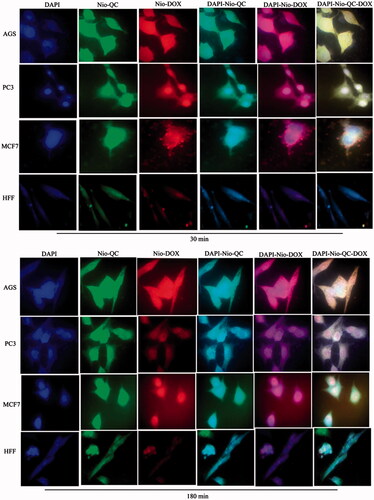
Figure 13. Cellular uptake images of AGS, PC3, MCF7 and HFF cells, incubated with (a) free siRNA and (b) Nio-siRNA for 30 min and 180 min. DAPI (blue) and FAM-labelled siRNA (green).
Cytotoxicity study
One of the classical approaches for the treatment of cancer is the use of chemotherapeutic agents to fight the cancer. Nowadays, many novel chemotherapeutic agents are applied for combating the cancer cells but the emergence of the drug resistance is one of the main obstacles that physician may encounter during the therapy. Cancer cells usually serve several pathways to get over the chemotherapy. So targeting several important pathways simultaneously in tumorigenesis by a combination therapy could overcome the drug resistance and improve the therapeutic index. In order to use of combination therapy in the treatment of cancer, we improved multifunctional niosomes that loaded DOX, QC and siRNA. Triple combination therapy would restrain multiple pathways of cancer cells and enhance a chance of recovery. To determine the inhibitory effects of both forms of drugs (DOX, QC) as the free and niosomes on AGS, PC3, MCF7 and HFF cells, a dose-response experiment was carried out. As indicated in , individual treatment with the free and the niosomal forms resulted in the growth inhibition of AGS, PC3, MCF7 and HFF cells in a dose-dependent manner. As shown in , blank niosome and free siRNA had little cytotoxicity and Nio-siRNA had more toxicity than the free siRNA indicating the improved action of cationic niosome formulations to deliver the siRNA. Summary of IC50 values of agents evaluated in . Given the values of IC50 for 72 h, revealed that HFF cells needed at least a 1.57, 2.01 and 1.78-fold higher concentration of free DOX and a 2.42, 4.17 and 3.34-fold higher concentration of free QC to attain IC50 compared to AGS, PC3 and MCF7, respectively. Notwithstanding, in HFF considered a normal cell line, QC and DOX either in free or in a niosomal form had a less toxicity when compared to other cancer cells (). As depicted in and , nano-niosomes efficiently delivered the DOX and QC to cancer cells, in fact, A 1.31, 1.34 and 1.32-fold reduction in DOX concentration and a 1.47, 1.45 and 1.55-fold reduction in QC concentration were measured, when the DOX and QC were administered in nano-niosomes compared to free DOX and free QC in AGS, PC3 and MCF7 cells, respectively. Literally, in all cancer cells, Nio-DOX and Nio-QC had the higher cytotoxicity as compared to free forms of DOX and QC. Contrariwise, the DOX and QC delivered in free form to HFF cells demonstrated a 1.33- and 1.27-fold lowered concentration, respectively. Herein, we surveyed the interactions between QC and DOX in cancer cells to find the effective combination doses of drugs. Co-administration of Nio-DOX and Nio-QC with a serial increasing dose of QC and constant dose of DOX (0.156 µg ml−1) demonstrated that high concentrations of QC increased the cytotoxicity of DOX, whereas the cytotoxicity of DOX was decreased when co-administrate with low concentrations of Quercetin (). The combination index (CI) calculated by the Chou and Talalay’s theorem to assess the antagonistic/synergistic effect of the drugs combination [Citation36]. represented that the low doses of QC (10 µg ml−1–30 µg ml−1) antagonize the DOX, while high doses of QC (above 40 µg ml−1) showed a synergism effect with DOX in cancer cells that consistent with the findings of Na Li et al. [Citation37] that exhibited a low concentration of QC decreased the intracellular levels of ROS via the induction of endogenous antioxidant enzymes thus exerting the anti-neoplastic properties of the drug in cancer cells and QC is able to reduce the DOX-induced oxidative stress insults. Samuel et al. also reported that there was a dose-dependent behavior for QC to show either antagonism or synergism with anti-neoplastic agents which is in agreement with our results [Citation38]. We also evaluated the co-administration of different concentrations of Nio-DOX with fixed concentration of Nio-QC (20 µg ml−1) which showed that low concentrations of QC antagonize the high concentrations of DOX in varying degrees compared to the low concentrations of DOX (). According to these results, we applied a combination therapy to increase the chemotherapeutic index to lower the side effects. showed the cytotoxic effect of the co-delivery of Nio-siRNA-DOX with Nio-QC was higher than other formulations (p < .05). In this study, we indicated that co-delivery of Nio-siRNA-DOX with co-administration of high dose of Nio-QC (triple combination therapy) decreased the cell survival when compared to other formulations and demonstrated a superior therapeutic efficacy.
Figure 14. Survival analysis (a) Cytotoxicity of blank niosome, free siRNA, Nio-siRNA for AGS, PC3, MCF7 and HFF after 72 h; (b) comparison between toxicity of free DOX and Nio-DOX in various concentrations for AGS, PC3, MCF7 and HFF after 24, 48 and 72 h; (c) comparison between toxicity of free QC and Nio-QC in various concentrations for AGS, PC3, MCF7 and HFF after 24, 48 and 72 h.
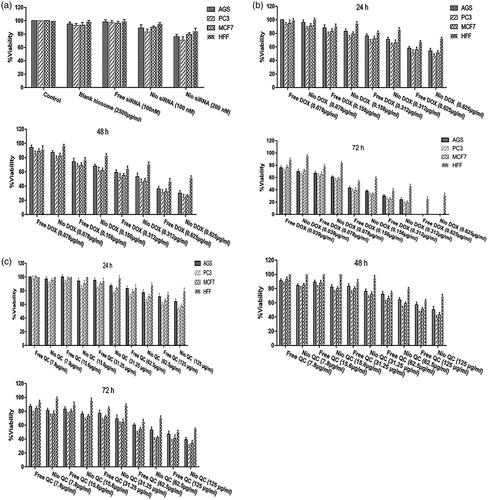
Figure 15. Comparison between cytotoxicity of 0.156 µg ml−1 Nio-DOX alone and combined with different concentrations of Nio-QC for 72 h (a) AGS; (b) PC3; (c) MCF7; and (d) HFF.
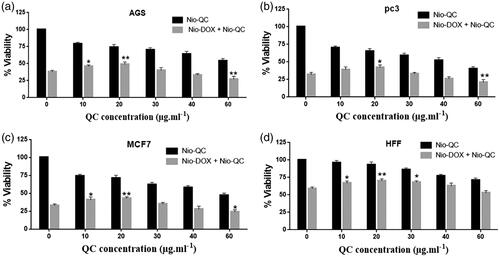
Figure 16. Comparison between cytotoxicity of different concentrations of Nio-DOX alone and combined with 20 µg ml−1 Nio-QC for 72 h (a) AGS; (b) PC3; (c) MCF7; and (d) HFF.
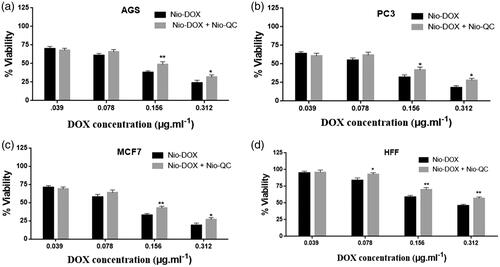
Figure 17. Comparison between cytotoxicity of Nio-DOX (0.156 µg ml−1) alone, combined with Nio-QC (40 µg ml−1) and co-delivery of Nio-siRNA (100 nM)-DOX (0.156 µg ml−1) with Nio-QC (40 µg ml−1) (triple combination therapy) for AGS; PC3; MCF7; HFF after 72 h.
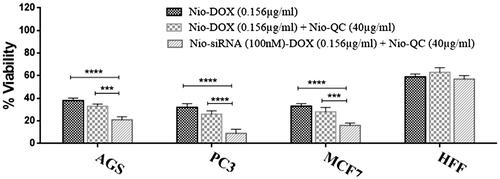
Table 4. The IC50 values of DOX and QC in the forms of free drug and drug niosomal form on AGS, PC3, MCF-7 and HFF cells after 72 h.
Table 5. The combination index of different concentration of QC in combinations with DOX.
Conclusions
In summary, we have successfully developed a novel optimized formulation of cationic PEGylated niosome for siRNA-drug delivery. The rounded shape, nanoscale size, high positively charge, good stability were corroborated by different characterization methods. In this delivery system, hydrophilic DOX was loaded within nano-carrier and hydrophobic QC was encapsulated within the lipid layer. The siRNA was also loaded on cationic PEGylated niosomes. Such bona fide delivery system demonstrates the high EE% with a contentious release profile. Cytotoxicity assay for this optimal formula with three different cancer cell lines showed that the IC50s of drugs loaded on PEGylated cationic niosomes were significantly lower than the free form of drugs. Contrarily, in normal cells the IC50s of drugs loaded in cationic niosomes were statistically higher than the un-encapsulated drug. These findings are consistent with thermo- and pH-sensitive drug release profile which confirms that an optimized formulation can act as a passive targeting which is dependent on difference of temperature and pH between the normal and cancer cells. It should be noted that the release of drugs is enhanced with high temperature and low pH. Generally, these newly developed cationic PEGylated niosomes in terms of siRNA-drug delivery system was effective to improve the therapeutic efficacy due to concurrent delivery of high amounts of siRNA and hydrophobic/hydrophilic drugs to augment the synergistic effects of triple-combination therapy.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Saadat S, Yousefifard M, Asady H, et al. The most important causes of death in Iranian population; a retrospective cohort study. Emergency. 2015;3:16–21.
- Dry JR, Yang M, Saez-Rodriguez J. Looking beyond the cancer cell for effective drug combinations. Genome Med. 2016;8:125.
- Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release. 2014;194:238–256.
- Xu B, Jin Q, Zeng J, et al. Combined tumor- and neovascular-“dual targeting” gene/chemo-therapy suppresses tumor growth and angiogenesis. ACS Appl Mater Interfaces. 2016;8:25753–25769.
- Shah N, Chaudhari K, Dantuluri P, et al. Paclitaxel-loaded PLGA nanoparticles surface modified with transferrin and Pluronic® P85, an in vitro cell line and in vivo biodistribution studies on rat model. J Drug Target. 2009;17:533–542.
- Refolo MG, D'Alessandro R, Malerba N, et al. Anti proliferative and pro apoptotic effects of flavonoid quercetin are mediated by CB1 receptor in human colon cancer cell lines. J Cell Physiol. 2015;230:2973–2980.
- Srivastava S, Somasagara RR, Hegde M, et al. Quercetin, a natural flavonoid interacts with DNA, arrests cell cycle and causes tumor regression by activating mitochondrial pathway of apoptosis. Sci Rep. 2016;6:24049.
- Dong Q, Chen L, Lu Q, et al. Quercetin attenuates doxorubicin cardiotoxicity by modulating Bmi-1 expression. Br J Pharmacol. 2014;171:4440–4454.
- Pappou EP, Ahuja N. The Role of oncogenes in gastrointestinal cancer. J Gastrointest Dig Syst. 2013(Suppl 1):S2–S15.
- Weinstein J, Jacobsen FW, Hsu-Chen J, et al. A novel mammalian protein, p55CDC, present in dividing cells is associated with protein kinase activity and has homology to the Saccharomyces cerevisiae cell division cycle proteins Cdc20 and Cdc4. Mol Cell Biol. 1994;14:3350–3363.
- Kim JM, Sohn HY, Yoon SY, et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin Cancer Res. 2005;11:3149–3482.
- Ding ZY, Wu HR, Zhang JM, et al. Expression characteristics of CDC20 in gastric cancer and its correlation with poor prognosis. Int J Clin Exp Pathol. 2014;7:722–727.
- Kanasty RL, Whitehead KA, Vegas AJ, et al. Action and reaction: the biological response to siRNA and its delivery vehicles. Mol Ther. 2012;20:513–524.
- Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138.
- Nematollahi MH, Torkzadeh-Mahanai M, Pardakhty A, et al. Ternary complex of plasmid DNA with NLS-Mu-Mu protein and cationic niosome for biocompatible and efficient gene delivery: a comparative study with protamine and lipofectamine. Artif Cells Nanomed Biotechnol. 2017;1–11.
- Haghiralsadat F, Amoabediny G, Helder MN, et al. A comprehensive mathematical model of drug release kinetics from nano-liposomes, derived from optimization studies of cationic PEGylated liposomal doxorubicin formulations for drug-gene delivery. Artif Cells Nanomed Biotechnol. 2018;46:169–177.
- Mokhtar M, Sammour OA, Hammad MA, et al. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm. 2008;361:104–111.
- Junyaprasert VB, Singhsa P, Suksiriworapong J, et al. Physicochemical properties and skin permeation of Span 60/Tween 60 niosomes of ellagic acid. Int J Pharm. 2012;423:303–311.
- Ruckmani K, Sankar V. Formulation and optimization of Zidovudine niosomes. AAPS PharmSciTech. 2010;11:1119–1127.
- Arzani G, Haeri A, Daeihamed M, et al. Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge. Int J Nanomed. 2015;10:4797–4813.
- El-Samaligy MS, Afifi NN, Mahmoud EA. Increasing bioavailability of silymarin using a buccal liposomal delivery system: preparation and experimental design investigation. Int J Pharm. 2006;308:140–148.
- Patel HM. Liposomes: a practical approach. FEBS Lett. 1990;275:242–243.
- Pawlikowska-Pawlęga B, Dziubińska H, Król E, et al. Characteristics of quercetin interactions with liposomal and vacuolar membranes. Biochim Biophys Acta. 2014;1838:254–265.
- Li J, Wang X, Zhang T, et al. A review on phospholipids and their main applications in drug delivery systems. Asian J Pharm Sci. 2015;10:81–98.
- Li Y, Pei Y, Zhang X, et al. PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. J Control Release. 2001;71:203–211.
- Kim JY, Kim JK, Park JS, et al. The use of PEGylated liposomes to prolong circulation lifetimes of tissue plasminogen activator. Biomaterials. 2009;30:5751–5756.
- Wang AZ, Gu F, Zhang L, et al. Biofunctionalized targeted nanoparticles for therapeutic applications. Expert Opin Biol Ther. 2008;8:1063–1070.
- Gang W, Jie WJ, Ping ZL, et al. Liposomal quercetin: evaluating drug delivery in vitro and biodistribution in vivo. Expert Opin Drug Deliv. 2012;9:599–613.
- Verhoef JJF, Anchordoquy TJ. Questioning the use of PEGylation for drug delivery. Drug Deliv Transl Res. 2013;3:499–503.
- Yang BB, Lum PK, Hayashi MM, et al. Polyethylene glycol modification of filgrastim results in decreased renal clearance of the protein in rats. J Pharm Sci. 2004;93:1367–1373.
- Swietach P, Vaughan-Jones RD, Harris AL, et al. The chemistry, physiology and pathology of pH in cancer. Philos Trans R Soc B Biol Sci. 2014;369:20130099.
- Lee RJ, Wang S, Turk MJ, et al. The effects of pH and intraliposomal buffer strength on the rate of liposome content release and intracellular drug delivery. Biosci Rep. 1998;18:69–78.
- Naderinezhad S, Amoabediny G, Haghiralsadat F. Co-delivery of hydrophilic and hydrophobic anticancer drugs using biocompatible pH-sensitive lipid-based nano-carriers for multidrug-resistant cancers [10.1039/C7RA01736G]. RSC Adv. 2017;7:30008–30019.
- Sezgin-Bayindir Z, Onay-Besikci A, Vural N, et al. Niosomes encapsulating paclitaxel for oral bioavailability enhancement: preparation, characterization, pharmacokinetics and biodistribution. J Microencapsul. 2013;30:796–804.
- Tavano L, Muzzalupo R, Picci N, et al. Co-encapsulation of antioxidants into niosomal carriers: gastrointestinal release studies for nutraceutical applications. Colloids Surf B Biointerfaces. 2014;114:82–88.
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446.
- Li N, Sun C, Zhou B, et al. Low concentration of quercetin antagonizes the cytotoxic effects of anti-neoplastic drugs in ovarian cancer. PLoS One. 2014;9:e100314
- Samuel T, Fadlalla K, Mosley L, et al. Dual-mode interaction between quercetin and DNA-damaging drugs in cancer cells. Anticancer Res. 2012;32:61–71.