
Abstract
Atherosclerosis is a chronic inflammatory disease of the blood vasculature. Endothelial dysfunction is an early event in the development of atherosclerosis and the endothelium plays an important role in the innate immune defense in the pathology of cardiovascular diseases. New therapies are being developed based on the involvement of the immune system in atherosclerosis. In this study, we demonstrate that a commonly used anti-rheumatic drug, tofacitinib, possesses vascular protective properties in cultured primary human aortic endothelial cells (HAECs). Tofacitinib ameliorates oxidized low-density lipoprotein (ox-LDL)-induced adhesion of THP-1 cells to HAECs, suppresses the expression of vascular adhesion molecules and production of cytokines, including vascular cell adhesion molecule 1 (VCAM-1), intercellular cell adhesion molecule-1 (ICAM-1), tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β). Moreover, tofacitinib inhibits elevation of endothelial lectin-like ox-LDL receptor-1 (LOX-1) and production of reactive oxygen species (ROS) triggered by ox-LDL. As a result, the presence of tofacitinib reduces ox-LDL-induced cytotoxicity and improves endothelial viability. Mechanistically, we demonstrate that tofacitinib suppresses ox-LDL-mediated activation of NF-κB inhibitor α (IκB-α), accumulation of nuclear p65 and activation of nuclear factor κB (NF-κB) promoter, indicating that tofacitinib inhibits NF-κB activation. Collectively, our data support that tofacitinib possesses a novel protective function in endothelial cells, implying that tofacitinib could have the therapeutic potential to modulate inflammation in cardiovascular diseases.
Introduction
Cardiovascular diseases are among the leading causes of death in many countries. Atherosclerosis is the major cause of numerous cardiovascular disorders. Atherosclerosis is not solely a disease of lipid deposition on the vessel wall, but also involves chronic inflammation of the blood vessels. Studies have been developing new therapies based on the concept of the involvement of the immune system in atherosclerosis [Citation1]. Inflammation is the immune system's response to harmful stimuli, such as pathogens, damaged cells, deleterious substances, and other stimuli. The inflammatory response acts to remove harmful stimuli and initiate the healing process, thus it is one of our body’s most powerful defense mechanisms [Citation2]. The inflammatory response in atherosclerosis involves both the innate and adaptive arms of the immune response [Citation3]. Adhesion and infiltration of immune cells mediated by endothelial cells is an important mechanism of vascular inflammation [Citation4].
In healthy conditions, the immune system recognizes the difference between self and non-self. However, an overactive and misguided immune response against foreign substances and tissues could lead the immune system to inadvertently attack the tissues or cells of the body itself. This can result in autoimmune disease [Citation5]. Cytokines play pivotal roles in maintaining an appropriate immune response. Pro-inflammatory cytokines contribute to the initiation and propagation of autoimmune inflammation, whereas antiinflammatory cytokines facilitate the regression of inflammation and recovery from the acute phase of the disease [Citation6]. Many cytokine pathway inhibitor-based therapies have been developed based on the results of studies of cytokines’ intracellular signaling pathways. The targeting of Janus kinase-signal transducers and activators of transcription (JAK-STAT) signaling led to the discovery of two small molecules in clinical therapy: tofacitinib and ruxolitinib [Citation7]. Tofacitinib is the first JAK inhibitor tested clinically and has been used to treat rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis. Ruxolitinib is another JAK inhibitor that has been used for the treatment of myelofibrosis. Clinical trials have demonstrated that targeting interleukin-1β (IL-1β) can reduce the risk of cardiovascular disease, suggesting the therapeutic importance of cytokine-mediated inflammation [Citation8]. In recent years, in light of an increased understanding of the roles of JAK-STAT signals in the vascular system, targeting JAK-associated pathways has been proposed as a potential therapeutic target for the treatment of atherosclerosis [Citation9]. In our current study, we applied tofacitinib to cultured vascular endothelial cells and investigated its effect on endothelial function upon stimulation with oxidized LDL (ox-LDL).
Materials and methods
Cell culture and treatment experiment
Primary human coronary endothelial cells (HAECs) were purchased from Lonza (Basel, Switzerland). HAECs were grown in 2% serum endothelial growth media (EGM2) in low-passage numbers (less than 10). Human monocyte cell line THP-1 was from American Type Culture Collection (ATCC) stock (TIB-202™) and grown in 10% fetal serum containing in Dulbecco's Modified Eagle Medium (DMEM). All cell cultures were maintained in a 5% (v/v) CO2/95% (v/v) nitrogen incubator at 37 °C. HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 24 h or 48 h.
Cellular adhesion assay
HAECs were grown to 70% confluence in 6-well plates and stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 24 h. THP-1 monocytes at a density of 2 × 105 were labeled with calcein acetoxymethyl ester (calcein AM, Invitrogen, USA) for 30 min and incubated with HAECs for 2 h. Each well was rinsed three times with PBS containing 1% BSA to remove unbound cells. Plates were fixed with 4% paraformaldehyde and calcein-labeled THP-1 cells bound to HAECs were visualized with a fluorescent microscope at 10× magnification.
Quantitative real-time PCR analysis
The total RNA from HAECs was extracted using a micro RNeasy Micro Kit from Qiagen (Duesseldorf, Germany) (Cat.74004) in accordance with the manufacturer’s manual. RNA concentrations were quantified by Nanodrop. A total of 1 μg RNA was used to synthesize cDNA using iScript™ Reverse Transcription Supermix (Bio-Rad, #1708840) for RT-qPCR from Invitrogen (Waltham, MA USA). SYBR-based real-time PCR experiments were performed to detect the total mRNA transcripts of human LOX-1, ICAM-1, VCAM-1, TNF-α and IL-1β on an ABI 7500 platform. Primer sequences are shown in .
Table 1. Primers used in this study.
Western blot analysis
HAECs under the different conditions were lysed by radioimmunoprecipitation assay (RIPA) buffer with protease and phosphatase inhibitors. The nuclear extracts were obtained by lysing the cells with hypotonic buffer to remove the cytoplasm. A total of 20 μg cell lysates or nuclear extracts were immobilized by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The separated protein mixes were then transferred to polyvinylidene fluoride (PVDF) membranes and blotted against their specific antibodies and corresponding secondary antibody. The immunoblots were visualized by Pierce™ (Dallas, Texas, USA) ECL Plus western blotting substrate (Catalog # 32132). The following antibodies were used in this study: mouse anti-VCAM-1 monoclonal antibody mab (1:1000, #643-VM, R&D Systems), mouse anti-E-selectin (1:1000, #BBA16, R&D Systems (Dresden, Germany)), rabbit anti-p65 mab (1:1000, #4764, Cell Signaling Technology, USA), mouse anti-LOX-1 mab (1:1000, #AF1564, R&D Systems, USA), rabbit anti-p-IκBα mab (1:1000, # 2859, Cell Signaling Technology, USA), IκBα (1:3000, #4812, Cell Signaling Technology, Danvers, MA, USA), rabbit Lamin B mab (1:3000, #13435, Cell Signaling Technology, USA) and β-actin (1:5000, #4970, Cell Signaling Technology, USA); HRP-linked anti-rabbit IgG antibody (1:2000, #7074, Cell Signaling Technology, USA); HRP-linked anti-mouse IgG antibody (1:2000, #7076, Cell Signaling Technology, USA).
Enzyme-linked immunosorbent assay (ELISA) assay
To measure the secreted levels of TNF-α and IL-1β, HAECs culture media was collected for analysis. Two ELISA kits were purchased from R&D Systems: a human TNF-α Quantikine ELISA Kit (#DTA00C, R&D Systems), and a human IL-1β Quantikine ELISA Kit (#DLB50, R&D Systems). The procedure was performed in accordance with the manufacturer’s instructions. The data were collected by 96-plate reader spectrometry.
3 –(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide, thiazolyl blue tetrazolium bromide (MTT) assay
The viability of HAECs was measured by MTT assay. Briefly, cells were incubated for 4 h with 0.8 mg/ml MTT in serum-free medium followed by the addition of dimethyl sulfoxide (DMSO). The stabilized cell-MTT reaction mixture was transferred into 96-well plates and absorbance was recorded at 560 nm using a microplate spectrophotometer system.
Measurement of lactate dehydrogenase (LDH) release
LDH cytotoxicity was assessed by measuring leakage of LDH into the culture medium. The culture medium was collected to obtain cell-free supernatants in different conditions. The activity of LDH in the medium was determined using a commercially available kit from Thermo Fisher Scientific, Waltham, MA, USA. The data are presented as percentages of the control values.
Measurement of ROS
Cellular reactive oxygen species (ROS) production was measured by quick staining of the cells under different conditions with 2′, 7′–dichlorofluorescein diacetate dye (DCFH-DA). After the indicated treatment, DCFH-DA was added to the culture medium at a final concentration of 5 μM and incubated for 1 h at 37 °C in darkness. After three gentle washes, the fluorescent image density was captured by a laser microscope. Image Pro Plus software (Media Cybernetics, Inc. (Rockville, MD, USA)) was used to analyze the average ROS levels. Six fields were randomly selected from each group. Regions of interest (ROI) in each field were characterized and the integrated density value (IDV) of the ROI was calculated. Average level of ROS = IDV/cell number. The relative value of intracellular ROS was normalized to the untreated group.
Measurement of luciferase activity
The transcriptional activity of NF-κB was measured using a luciferase assay. Cells were co-transfected with NF-κB promoter and a firefly luciferase promoter using Lipofectamine 2000 reagent from Invitrogen (Cat 11668027). At 24 h post transfection, the cells were treated with 100 mg/L ox-LDL in the presence or absence of 100 and 500 nM tofacitinib for an additional 24 h. The total cell lysates were collected to measure the dual luciferase activity of Renilla and firefly luciferase. Relative luciferase activity was calculated by normalizing the activity of firefly luciferase to Renilla luciferase.
Statistical analysis
Data are shown as means ± standard derivation (SD). Statistical significance of differences was assessed by one-way analysis of variance (ANOVA) followed by Bonferroni post-test comparisons. p Values less than .05 were determined to be statistically significant.
Results
Tofacitinib suppresses ox-LDL-induced adhesion of immune cells to endothelial cells
To test the effect of tofacitinib on endothelial function, we performed an immune-endothelial adhesion experiment upon ox-LDL stimulation. Endothelial cells treated with ox-LDL is known to induce the inflammatory response and trigger adhesion of immune cells. In our experiment, we prelabeled THP-1 cells and added them to HAECs in the combined condition of ox-LDL and tofacitinib. As shown in and referenced to non-treated cells, ox-LDL treatment induced adhesion of approximately 3.3-fold more THP-1 cells to HAECs. However, there were only 2.1- and 1.5-fold more THP-1 cells bound to endothelial cells when 100 and 500 nM tofacitinib were added to the medium, respectively. Clearly, tofacitinib significantly attenuated ox-LDL-induced adhesion of immune cells to endothelial cells.
Figure 1. Tofacitinib suppresses ox-LDL-induced attachment of human monocyte THP-1 cells to HAECs. HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 24 h. Representative fluorescence microscopic images of THP-1 cells attached to HAECs and quantitative analysis. Scale bar, 50 μM. (*, p < .01 vs. vehicle control; #, p < .01 vs. ox-LDL group, $, p < .01 vs. ox-LDL + 100 nM tofacitinib group, ANOVA, n = 5–6).
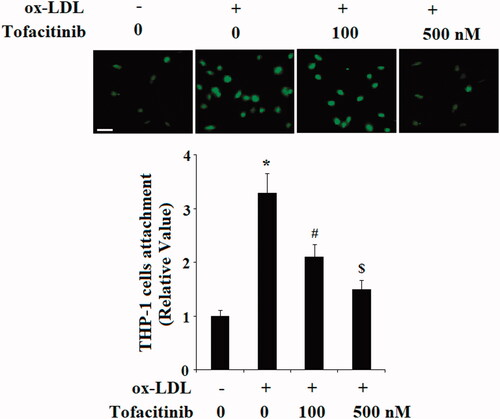
Tofacitinib inhibits ox-LDL-induced vascular adhesion molecules in endothelial cells
The capacity of tofacitinib to inhibit adhesion of immune cells to endothelial cells prompted our investigation of the expression of vascular adhesion molecules in these conditions. We measured the expression of two major molecules, which are known to trigger the adhesion of immune cells to endothelial cells: VCAM-1 and E-selectin. As shown in and compared to the control group, ox-LDL caused approximately 12-fold higher induction of VCAM-1 mRNA. However, there was only approximately 7- and 3-fold higher induction of VCAM-1 in the presence of 100 and 500 nM of tofacitinib, respectively. At the same time, ox-LDL caused approximately 9-fold higher induction of E-selectin mRNA. However, induction of E-selectin was only 5- and 3-fold higher when two doses of tofacitinib were added, respectively. Meanwhile, we verified the action of tofacitinib by measuring the protein levels of these molecules under same conditions. As shown in , VCAM-1 and E-selectin were barely detectable at the basal level and treatment with ox-LDL resulted in induction of a large amount of these proteins. However, ox-LDL had much lower induction of these proteins in the presence of the two doses of tofacitinib in a dose-dependent manner.
Figure 2. Tofacitinib inhibits ox-LDL-induced expression of VCAM-1 and E-selectin. (A) Real-time PCR analysis of VCAM-1 and E-selectin; (B) Western blot analysis of VCAM-1 and E-selectin (*, p < .01 vs. vehicle control; #, p < .01 vs. ox-LDL group, $, p < .01 vs. ox-LDL + 100 nM tofacitinib group, ANOVA, n = 5–6). HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 24 h.
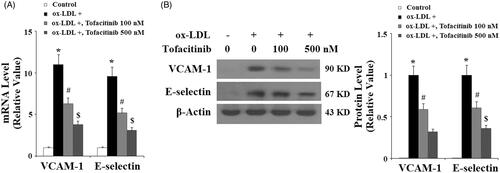
Tofacitinib inhibits ox-LDL-induced LOX-1 expression in endothelial cells
Next, we assessed the influence of tofacitinib on the endothelial receptor of ox-LDL, LOX-1. LOX-1 is a major receptor of ox-LDL which disturbs endothelial function. We assumed that tofacitinib would have an inhibitory effect on the expression of LOX-1. Indeed, both mRNA and protein experiments confirmed this action. As shown in and compared to the control, ox-LDL caused approximately 5-fold higher induction of LOX-1 mRNA. However, it induced only approximately 3.5- and 2.5-fold expression of LOX-1 in the presence of 100 and 500 nM tofacitinib, respectively. As shown in , ox-LDL treatment caused approximately 3.5-fold higher induction of LOX-1 protein at the protein level. However, it only induced approximately 2.5- and 1.5-fold higher expression of LOX-1 protein in the presence of the two doses of tofacitinib, respectively.
Figure 3. Tofacitinib decreases ox-LDL-induced expression of LOX-1. HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 24 h. (A) Real-time PCR analysis of LOX-1; (B) Western blot analysis of LOX-1 (*, p < .01 vs. vehicle control; #, p < .01 vs. ox-LDL group, $, p < .01 vs. ox-LDL + 100 nM tofacitinib group, ANOVA, n = 5–6).
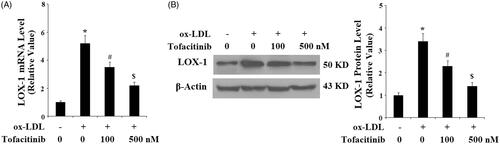
Tofacitinib inhibits ox-LDL-induced pro-inflammatory cytokines in endothelial cells
We also assessed cytokine levels in our treatment experiment. As shown in and referenced to the control, ox-LDL resulted in approximately 5.5-fold higher TNF-α mRNA, but it only caused approximately 3.5- and 2-fold higher TNF-α mRNA in the presence of 100 and 500 nM tofacitinib, respectively. Similarly, ox-LDL resulted in 5.2-fold expression of IL-1β mRNA, but only approximately 3- and 2-fold IL-1β expression when two doses of tofacitinib were added, respectively. We obtained consistent results by measuring the expression of proteins secreted into the culture media. As shown in and referenced to the control, ox-LDL induced approximately 3.5-fold higher TNF-α secretion but induced only approximately 2.3- and 1.2-fold higher TNF-α when 100 and 500 nM tofacitinib were added, respectively. Again, ox-LDL induced approximately 3.7-fold higher IL-1β secretion, but only approximately 2.2- and 1.2-fold IL-1β expression in the presence of the two doses of tofacitinib, respectively.
Figure 4. Tofacitinib suppresses ox-LDL-induced production of the pro-inflammatory cytokines TNF-α and IL-1β. HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 24 h. (A) Real-time PCR analysis of TNF-α and IL-1β; (B). ELISA analysis of TNF-α and IL-1β (*, p < .01 vs. vehicle control; #, p < .01 vs. ox-LDL group, $, p < .01 vs. ox-LDL + 100 nM tofacitinib group, ANOVA, n = 5–6).
Tofacitinib ameliorates ox-LDL-induced generation of reactive oxygen species (ROS)
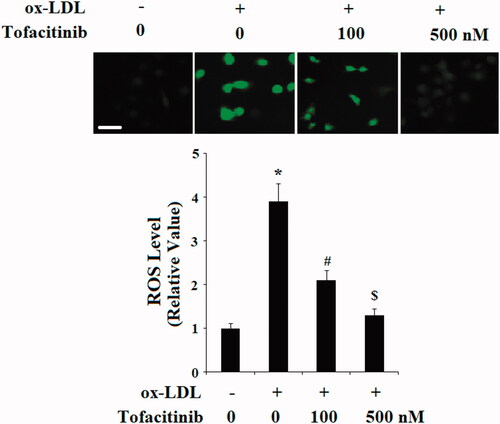
The presence of ox-LDL in endothelial cells can cause generation of reactive oxygen species (ROS) which leads to endothelial injury. We then assessed the influence of tofacitinib on ox-LDL-induced endothelial ROS generation. As shown in and referenced to the control, the addition of ox-LDL to HAECs led to approximately 4-fold higher generation of ROS, but it only resulted in approximately 2.2- and 1.3-fold higher ROS production in the presence of 100 and 500 nM tofacitinib. Therefore, tofacitinib inhibits generation of ROS in a dose-dependent manner.
Figure 5. Tofacitinib ameliorates ox-LDL-induced generation of reactive oxygen species (ROS). HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 24 h. Intracellular ROS was determined by the DCFH-DA assay. Scale bar, 100 μM. (*, p < .01 vs. vehicle control; #, p < .01 vs. ox-LDL group, $, p < .01 vs. ox-LDL + 100 nM Tofacitinib group, ANOVA, n = 5–6).
Tofacitinib ameliorates ox-LDL-induced LDH release and reduction of cell viability
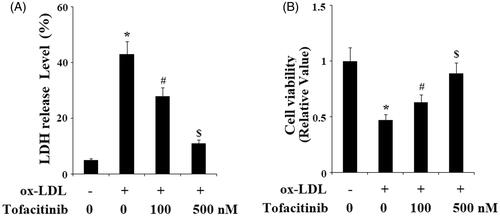
Next, we assessed whether tofacitinib could influence ox-LDL-mediated endothelial cytotoxicity and viability. Based on the above experiments, we assumed that tofacitinib may exert a protective effect on endothelial cells. As shown in the results of our LDH release experiment in , only 5% LDH was detected at the basal level, however, treatment with ox-LDL caused more than 40% LDH release. The release of ox-LDL was only approximately 30 and 10% in the presence of 100 and 500 nM tofacitinib, respectively. Meanwhile, we obtained meaningful results regarding cell viability under these conditions. As shown in and referenced to the control, ox-LDL caused a reduction in cell viability of more than half. However, the same ox-LDL treatment resulted in only approximately 35 and 10% cell death in the presence of the two doses of tofacitinib, respectively.
Figure 6. Tofacitinib ameliorates ox-LDL-induced LDH release and reduction of cell viability. HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 48 h. (A). LDH release was determined using a commercial kit; (B) Cell viability was determined by MTT assay (*, p < .01 vs. vehicle control; #, p < .01 vs. ox-LDL group, $, p < .01 vs. ox-LDL + 100 nM tofacitinib group, ANOVA, n = 5–6).
Tofacitinib inhibits ox-LDL-induced endothelial NF-κB activation
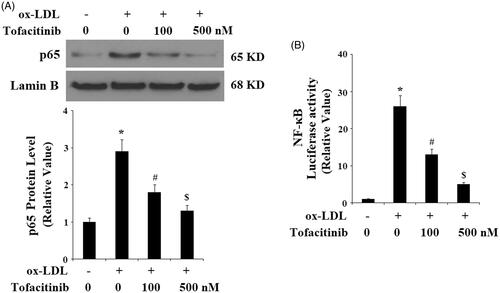
Finally, we explored the molecular mechanism through which tofacitinib could influence endothelial cells. We found that tofacitinib had a profound influence on the NF-κB pathway. Firstly, we examined the activation and degradation of the key kinase in NF-κB, IκBα. Phosphorylation of IκBα kinase leads to its degradation and subsequent activation of NF-κB. As shown in and compared to the control, ox-LDL caused approximately 3-fold higher activation of IκBα, however, there was only approximately 2- and 1.5-fold induction of the same kinase in the presence of 100 and 500 tofacitinib, respectively. Secondly, we examined nuclear translocation of the NF-κB subunit p65 and transfected NF-κB promoter activity. As shown in and compared with the control, ox-LDL gave rise to approximately 3-fold higher accumulation of p65 in the nucleus. However, it only allowed approximately 1.7- and 1.4-fold higher accumulation of p65 in the presence of 100 and 500 nM tofacitinib, respectively. As shown in , when NF-κB was transfected into the cells, ox-LDL treatment induced approximately 25-fold higher promoter activity than the control, but there was only approximately 13- and 5-fold higher promoter activity in the presence of the two doses of tofacitinib, respectively.
Figure 7. Tofacitinib mitigates ox-LDL-induced phosphorylation and degradation of IκBα. HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (500 nM) for 6 h. Phosphorylated and total levels of IκBα were determined by Western blot analysis (*, p < .01 vs. vehicle control; #, p < .01 vs. ox-LDL group, ANOVA, n = 5–6).
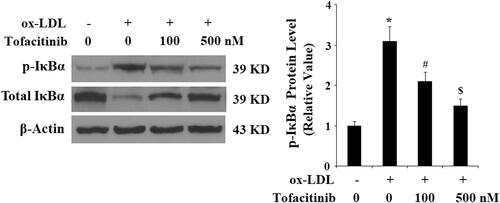
Figure 8. Tofacitinib inhibits ox-LDL-induced activation of NF-κB. HAECs were stimulated with 100 mg/L ox-LDL in the presence or absence of tofacitinib (100, 500 nM) for 24 h. (A). Nuclear level of p65; (B). Luciferase activity of NF-κB promoter (*, p < .01 vs. vehicle control; #, p < .01 vs. ox-LDL group, ANOVA, n = 5–6).
Discussion
The concept of atherosclerosis as an immune disease has been proposed for many years. Endothelial dysfunction is an early event in the development of atherosclerosis [Citation10]. We aimed to test the capacity of commonly available anti-inflammation agents to modulate inflammation in endothelial cells. The Janus kinase (JAK) and signal transducer and activator of transcription (STAT) pathway has been implicated in the down-regulation of many cytokines [Citation11]. Pharmacological inhibition of JAKs blocks the actions of type I/II cytokines. Tofacitinib is targeted to inhibit JAK1, JAK3, and to a lesser extent, JAK2 [Citation12]. Tofacitinib has been evaluated to be effective for various immune-mediated diseases, especially in rheumatoid arthritis [Citation13].
In vascular cells, the JAK/STAT signaling pathway is a stress responsive signaling cascade that transduces signals from cell surface receptors to the nucleus. Various stress factors are reported to activate the JAK/STAT signaling pathway to induce agents of vascular inflammation such as angiotensin II, mechanical stress, oxidative stress, and various cytokines [Citation14]. It also plays a role in pathogen infection of the host’s vascular system [Citation15,Citation16]. The JAK/STAT-mediated molecular mechanism has been shown to be involved in the anti-atherosclerotic effect of angiotensin-converting enzyme 2 (ACE 2) [Citation17]. Another study has shown that the lipid-reducing agent statin suppresses the adhesion of monocytes to endothelial cells by inhibiting JAK/STAT in endothelial cells [Citation18].
In our study, we applied ox-LDL, the most pathophysiologically relevant molecule, to mimic the hyperlipidemia environment in atherosclerosis and found that the inhibition of the JAK/STAT pathway by tofacitinib could provide protection against ox-LDL-induced cellular injury. We provide evidence from four experiments to support the beneficial roles of tofacitinib. First and foremost, tofacitinib mitigates ox-LDL-induced cellular LDH release and increases endothelial viability. Second, our data indicate that tofacitinib potently inhibits induction of vascular adhesion molecules and cytokines. We tested the expressions of four major pro-inflammatory molecules including VCAM-1, ICAM-1, TNF-α, and IL-1β, all of which were inhibited by tofacitinib. Notably, the high expression of VCAM-1 has been shown to be closely associated with the development of atherosclerosis [Citation19]. The inhibitory role of tofacitinib on VCAM-1 elevation indicates its potential protective role in atherosclerosis development. Third, the presence of tofacitinib decreases ox-LDL-induced adhesion of monocytes to endothelial cells, which can be explained as a result of its inhibition of the expression of vascular adhesion molecules and cytokines. Fourth, our data suggest that tofacitinib possesses an anti-ROS property as it reduces the ox-LDL-induced generation of cellular ROS. Mechanistically, we report that there are two possible molecular signaling pathways that are influenced by tofacitinib: NF-κB and the LOX-1 receptor. Inhibition on NF-κB can be reflected by the action of tofacitinib on IκBα activation/degradation and nuclear deposition of p65 as well as inhibition of NF-κB promoter. Meanwhile, tofacitinib appears to suppress the ox-LDL receptor on endothelial cells, LOX-1, indicating that receptor-mediated signals could be the mechanism by Tofacitinib too. We hypothesize that the inhibition of LOX-1 expression induced by tofacitinib could be the cause of its subsequent action on NF-κB, as it is known that ox-LDL activates NF-kB by binding to LOX-1 on the cell surface [Citation20].
Previous studies have shown that the JAK-STAT pathway plays an important role in the pathological process of atherosclerosis. Altogether, findings regarding inhibition of the JAK-STAT pathway by tofacitinib both in vitro and in atherosclerosis models have contributed to our knowledge about inflammation and cardiovascular disease. Our data present ample evidence supporting the notion that the anti-rheumatic drug, tofacitinib has a beneficial role in vascular endothelial cells. Thus, tofacitinib could have the potential implication of modulating vascular inflammation in the development of atherosclerosis. Future clinical trials of tofacitinib with cardiovascular outcomes would shed more light on its therapeutic premise in cardiovascular diseases.
Disclosure statement
None of the authors of this article declare that they have any conflicts of interest that need to be disclosed.
Additional information
Funding
References
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212.
- Chen L, Deng H, Cui H, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9:7204–7218.
- Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Rev. 2007;65:S140–S146.
- Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009;31:5–22.
- Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015;125:2228–2233.
- Moudgil KD, Choubey D. Cytokines in autoimmunity: role in induction, regulation, and treatment. J Interferon Cytokine Res. 2011;1:695–703.
- Gadina M. Janus Kinases: an ideal target for the treatment of autoimmune diseases. J Investig Dermatol Symp Proc. 2013;16:S70–S72.
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131.
- Schwartz DM, Kanno Y, Villarino A, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16:843–862.
- Widlansky ME, Gokce N, Keaney JF Jr, et al. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–1160.
- Rawlings JS, Rasler KM, Douglas A, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283.
- Furumoto Y, Gadina M. The arrival of JAK inhibitors: advancing the treatment of immune and hematologic disorders. Biodrugs. 2013;27:431–438.
- Dhillon S. Tofacitinib: a review in rheumatoid arthritis. Drugs. 2017;77:1987–2001.
- Grote K, Luchtefeld M, Schieffer B. JANUS under stress-role of JAK/STAT signaling pathway in vascular diseases. Vascul Pharmacol. 2005;43:357–363.
- Miller DM, Rahill BM, Boss JM, et al. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J Exp Med. 1998;187:675–683.
- Chaudhuri A, Yang B, Gendelman HE, et al. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood 2008;111:2062–2072.
- Zhang C, Zhao YX, Zhang YH, et al. Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells. Proc Natl Acad Sci USA. 2010;107:15886–15891.
- Jougasaki M, Ichiki T, Takenoshita Y, et al. Statins suppress interleukin-6-induced monocyte chemo-attractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br J Pharmacol. 2010;159:1294–1303.
- Cybulsky MI, Iiyama K, Li H, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262.
- Matsunaga T, Hokari S, Koyama I, et al. NF-kappa B activation in endothelial cells treated with oxidized high-density lipoprotein. Biochem Biophys Res Commun. 2003;303:313–319.