
Abstract
Ropivacaine is a commonly used local anaesthetic, but its side effects remain largely unknown. In the present study, we investigated the side effects of ropivacaine in human neuronal SH-5Y5Y cells. We show that 0.5% and 1% ropivacaine could cause fission-like mitochondrial morphological changes. Ropivacaine exclusively induces mitochondrial fission protein DRP1, generation of ROS and causes mitochondrial dysfunction including decreasing mitochondrial membrane potential, the activity of cytochrome C oxidase and ATP production. The side effects of ropivacaine appear to be dependent on DRP1 expression as silencing of DRP1 in neuronal cells abolishes ropivacaine-induced morphological changes and mitochondrial dysfunction. Silencing of DRP1 prevents ropivacaine-induced cellular LDH release and cell death. Moreover, DRP1-deficient neuronal cells are resistant to ropivacaine-induced apoptosis and silencing of DRP1 rescues the activity of cytochrome C oxidase and cellular ATP production. Collectively, our data indicate that imbalances in mitochondrial dynamics, mitochondrial dysfunction and cell death resulting from ropivacaine are all dependent on DRP1 expression. Our study provides valuable data to assess the safety of ropivacaine.
Introduction
Anaesthetics are used to prevent pain during medical and dental procedures. Anaesthetics are categorized into two classes: general anaesthetics (GA) and local anaesthetics (LA). The former causes a reversible loss of consciousness, and the latter causes a reversible loss of sensation in a limited region of the body and while maintaining consciousness [Citation1]. Local anaesthetics act by interrupting neural conduction by inhibiting the influx of sodium ions. Generally, neuronal sodium channels exist in a resting state. When the neuron is stimulated, the channels assume an activated or open state and the ions diffuse into the cell where they initiate depolarization. Local anaesthetics (LAs) have a greater affinity for receptors of sodium channels during their activated and inactivated states than when they are in their resting state [Citation2]. Based on the linked chemical group, LAs can be divided into two types: amide and ester [Citation3]. Amide LAs are most commonly used clinically, and ester LAs are mostly used in topical and mucous formulations. The effects of amide LAs can last for 2 to 3 h and toxicity is more likely to occur in patients with impaired liver function [Citation4]. Meanwhile, ester LAs last only a few minutes.
Although they are widely used and benefit the majority of patients, LAs have been reported to cause neurotoxicity and cardiac toxicity [Citation5,Citation6]. These studies demonstrate that commonly used LAs including bupivacaine and ropivacaine could induce apoptosis in neuronal cells and have similar cytotoxicity in clinically used concentrations [Citation7,Citation8]. Both bupivacaine and ropivacaine are long-acting amide LAs. Administration of bupivacaine has been shown to induce neurological damage in a preclinical experimental rodent study [Citation9,Citation10]. Ropivacaine was developed after bupivacaine was noted to be associated with cardiac toxicity [Citation11]. Ropivacaine, a drug that can penetrate the blood–brain barrier is used routinely and effectively [Citation12]. However, many such drugs are known to activate neurotoxic pathways.
Mitochondria, the principal progenitors of ATP through the oxidative phosphorylation (OXPHOS) system, have been identified as the powerhouses of the cell [Citation13]. Mitochondria have been reported to participate in a variety of biological processes, including steroid and heme biosynthesis, calcium homeostasis, redox signaling, and cell apoptosis [Citation14]. The half-life of mitochondria varies in different types of cells and tissues. Cells of all types have developed numerous ways to perform mitochondrial quality control, including mitochondrial fission and mitochondrial fusion. Mitochondrial fission is important for mitochondrial replication and the removal of damaged organelles by selective autophagy [Citation15]. On the other hand, mitochondrial fusion is important for facilitating the exchange of material between mitochondria and may compensate for functional defects. The recruitment of the large GTPase DRP1 plays an important role in mitochondrial fission. Mitofusin 1 and Mitofusin 2 (Mfn1 and Mfn2) play key roles in mediating mitochondrial fusion in mammals.
In our current study, we investigated the effect of ropivacaine on cellular mitochondrial function in cultured neuronal cells line.
Methods and materials
Cell culture and ropivacaine treatment
The human neuronal cell line SH-SY5Y was purchased from ATCC (CRL-2266). Cells were cultured in complete media of Eagle’s Minimum Essential Medium with 10% fetal bovine serum [Citation16]. We purchased ropivacaine from Sigma-Aldrich. To test the effect of ropivacaine on neuronal cells, ropivacaine solution was mixed with cell culture media to make 0.5% and 1% concentrations. The cells were treated with ropivacaine for 72 h before the functional assessment.
Real time PCR analysis
We extracted total RNA from cultured cells using a High Pure RNA kit from Roche (#12033674001). The RNA concentration was quantified by Nanodrop. A total of 1 μg RNA was used to synthesize cDNA using iScript Supermix from Invitrogen [Citation17]. SYBR-based real-time PCR experiments were run to detect the mRNA transcripts of DRP1, MFN1 and MFN2 using an ABI 7500 platform.
Western blot analysis
SH-SY5Y cells treated under different conditions were lysed by RIPA buffer with a protease inhibitor. A total of 20 μg cell lysates were loaded onto 4–20% precasted PAGE gel to separate the proteins by size [Citation18]. The separated protein mix was transferred to PVDF membranes to detect the corresponding protein levels by specific antibodies. The following list of antibodies was used: DRP1 (1:3000, Cell Signaling Technology, USA), MFN1 (1:3000, Cell Signaling technology, USA), MFN2 (1:2000, Abcam, USA), cleaved caspase 3 (1:1000, Cell Signaling Technology, USA), cytochrome C (1:1000, Cell Signaling Technology, USA), β-actin (1:5000, Cell Signaling Technology, USA), anti-mouse IgG, HRP-linked antibody (1:2000, Cell Signaling Technology, USA), anti-rabbit IgG, HRP-linked antibody (1:2000, Cell Signaling Technology, USA).
Mitochondrial staining
We purchased MitoTracker Red dye from Thermo Fisher (#M7512) [Citation19]. Briefly, cells on cover slides were fixed with 4% PFA and permeabilized with 0.3% Tween 20. MitoTracker red dye was added to the fixed cells to stain mitochondria. Cells with stained mitochondria were counterstained with DAPI to label the nuclei. Mitochondrial morphology was visualized using a confocal microscopy system (LSM510 Zeiss, Germany).
Mitochondrial membrane potential assay
Mitochondrial membrane potential (MMP) assay was based on measurements from TMRM staining (tetramethyl rhodamine methyl and ethyl esters) [Citation20]. Based on the concentration of accumulated TREM in the mitochondria, aggregated fluorescent counts were measured to determine the depolarization of mitochondria and cell health status. We purchased a TMRM kit (Sigma-Aldrich, USA) and MMP activity was measured by fluorescent density and quantified to present the data.
Measurement of cellular ATP
To measure the source of cellular metabolic energy, we measured the total ATP levels of cells under different conditions. A bioluminescence assay kit was purchased from Thermo Fisher Scientific, USA [Citation21]. Cellular ATP concentrations were quantified according to a standard curve.
SiRNA transfection
Two siRNA duplexes that specifically target human DRP1 were designed and obtained from Dharmacon (Lafayette, CO). A non-sense siRNA was used as a control. To deliver the siRNA oligos, SH-SY5Y cells were seeded at the subconfluent density and transfected with siRNA with Oligofectamine (Invitrogen, USA) in accordance with the manufacturer’s guidelines [Citation22]. We also transfected cells with non-sense siRNA as a negative control. Cells were further incubated for 48 h at 37 °C after transfection. To ensure siRNA effectiveness, cells were processed for the evaluation of changes in COX subunit protein expression and mRNA by real-time RT-PCR.
Measurement of cytochrome C oxidase activity
Cytochrome c oxidase enzyme activity was measured in SH-SY5Y cells under the different conditions as previously described [Citation23]. Briefly, frozen cell pellets (∼5 million cells) were suspended in phosphate buffered saline and sonicated for 10 s on ice. Cytochrome C oxidase was measured after reacting with 1% reduced cytochrome C at 550 nm at 30 °C. Reduced cytochrome C was prepared by adding 5 μl/ml of 5% sodium hydrosulfite to cytochrome C extract from bovine heart (Sigma-Aldrich, USA) in 0.01 M KPO4 at pH7.
MTT cell survival and LDH cell toxicity experiment
An MTT assay was used to measure cell viability [Citation20]. Briefly, cells were incubated for 4 h with 0.8 mg/ml MTT in serum-free medium and followed by the addition of DMSO. The stabilized cell-MTT reaction mixture was transferred into 96-well plates and absorbance was recorded at 560 nm using a microplate spectrophotometer system. LDH cytotoxicity was assessed by leakage of lactate dehydrogenase (LDH) into the culture medium. The culture medium was collected to obtain cell-free supernatants under different conditions. The activity of LDH in the medium was determined using a commercially available kit from Thermo Fisher Scientific, USA. The data are presented as percentages of the control values.
ROS assay
Cellular ROS was measured by staining the cells under different conditions with 2′,7′–dichlorofluorescin diacetate dye (DCFH-DA) [Citation20]. The fluorescent image density was captured by a laser microscope compound which can be detected. The fluorescent density of images was quantified by the Image J software.
Flow cytometry
Flow cytometry analyses [Citation24] were performed using a MACSQuant analyzer (Miltenyi Biotec, Bergisch-Gladbach, Germany). The apoptotic population of cells was stained with a combination of annexin V–FITC and PI. The non-apoptotic cells were annexin V–FITC-negative and PI-negative, and the apoptotic cells were annexin V–FITC-positive and PI-negative.
Determination of GTPase DRP1 enzymatic activity
The enzymatic activity of GTPase DRP1 in human SH-SY5Y neuronal cells was assessed using a commercial GTPase Assay Kit from Innova Bioscience, USA as previously reported [Citation21]. GTPase can hydrolyze GTP to GDP and inorganic Pi. GTPase activity was measured based on the amount of Pi that the GTP produces. After stimulation with 0.5% and 1% for 72 h, cells were lysed and DRP1 was immunoprecipitated with anti-DRP1 antibody. ColorLock Gold (orange) substrate was added to the Pi generated from GTP, and GTP activity was measured based on the inorganic complex solution (green). Absorbance was recorded at the wavelength range of 650 nm.
Results
Ropivacaine induces fission-like changes in mitochondrial morphology in neuronal cells
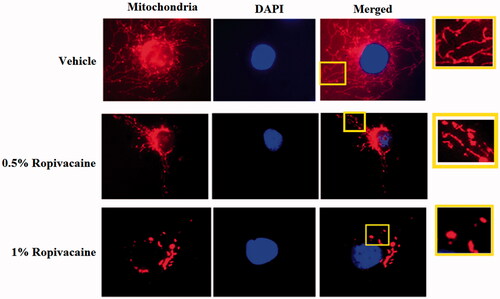
To examine the role of ropivacaine on mitochondrial dynamics, we treated human SH-5Y5Y neuronal cells with two doses of ropivacaine (0.5% and 1%). In non-treated cells, mitochondria formed a tightly packed network (). However, when cells were exposed to 0.5% and 1% ropivacaine, the mitochondria took on a fragmented and condensed structure with the higher dose (1%) having a more potent influence, suggesting that the balance of the mitochondrial network leaned toward fission (). These data suggest that ropivacaine treatment results in mitochondrial fission in neuronal cells.
Figure 1. Ropivacaine induces fission-like changes in mitochondrial morphology. Human SH-SY5Y neuronal cells were treated with 0.5% and 1% ropivacaine for 72 h. Mitochondrial morphology was measured by MitoTracker red staining; nuclei were stained with DAPI. The overlay pictures were shown to overall mitochondrial morphology. The insets were amplified mitochondrial structure.
Ropivacaine treatment induces expression of the mitochondrial fission protein DRP1
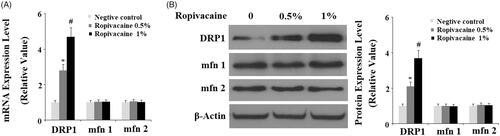
Since ropivacaine causes fission-like changes in mitochondrial morphology, we assessed the influence of ropivacaine on fission-related proteins. At the transcriptional level, 0.5% and 1% ropivacaine significantly increased DRP1 expression as compared to non-treated cells. However, ropivacaine did not have any effect on Mfn1 and Mfn2 expression (). We confirmed this finding by assessing their protein expression. Compared to non-treated cells, 0.5% and 1% ropivacaine induced roughly 2- and 4-fold higher DRP1 protein expression, respectively, but had no effect on Mfn1 and Mfn2 expression (). These data indicate that ropivacaine causes exclusive induction of DRP1 expression. Consistently, the results in supplementary Figure 1 demonstrate that the presence of ropivacaine significantly increased GTPase DRP1 activity.
Figure 2. Ropivacaine treatment increases the expression of mitochondrial fission protein DRP1. Human SH-SY5Y neuronal cells were treated with 0.5% and 1% ropivacaine for 72 h. (A) Ropivacaine increases the expression of DRP1, but not Mfn 1 and Mfn2 at the mRNA level; (B) Ropivacaine increases the expression of DRP1, but not Mfn 1 and Mfn 2 at the protein level (*, #, P < .01 vs. previous column group).
Ropivacaine treatment causes neuronal mitochondrial dysfunction
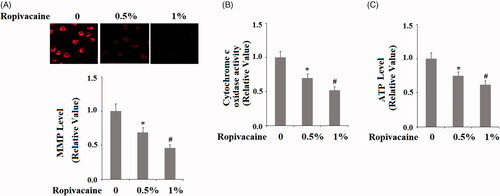
Next, we examined mitochondrial function through a series of experiments. Firstly, we measured mitochondrial membrane potential. Compared to non-treated cells, 0.5% and 1% ropivacaine treatment resulted in roughly 30% and 55% reductions in mitochondrial membrane potential, respectively (). Secondly, we measured the activity of the mitochondrial protein cytochrome C oxidase. Compared to non-treated cells, 0.5% and 1% ropivacaine treatment induced ∼25% and ∼50% reduction of cytochrome C oxidase activity (). Thirdly, we measured the influence of ropivacaine on cellular ATP production. Compared to non-treated cells, 0.5% and 1% ropivacaine treatment resulted roughly 25% and 45% reductions in ATP, respectively (). Together, these data demonstrate that ropivacaine treatment disturbs neuronal mitochondrial function.
Figure 3. Ropivacaine induced mitochondrial dysfunction in human SH-SY5Y neuronal cells. Human SH-SY5Y neuronal cells were treated with 0.5% and 1% ropivacaine for 72 h. (A) Mitochondrial membrane potential measured by TMRM; (B) Cytochrome c oxidase activity; (C) Intracellular ATP levels (*, #, P < .01 vs. previous column group).
Ropivacaine induces neuronal ROS production
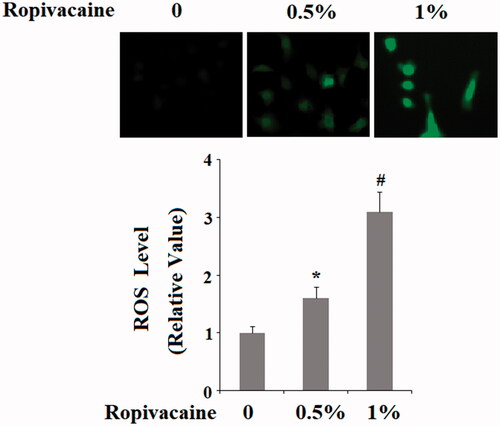
Ropivacaine-induced neuronal dysfunction suggests that the drug might influence the production of neuronal reactive oxidative species (ROS). Indeed, our data confirm this hypothesis. Compared to non-treated cells, 0.5% and 1% ropivacaine treatment gave rise roughly 1.7- and 3-fold higher ROS production ().
Figure 4. Ropivacaine-induced generation of intracellular ROS. Human SH-SY5Y neuronal cells were treated with 0.5% and 1% ropivacaine for 72 h. Intracellular ROS was determined by DCFH-DA assay (*, #, P < .01 vs. previous column group).
DRP1 deficiency ameliorates ropivacaine-induced mitochondrial fission
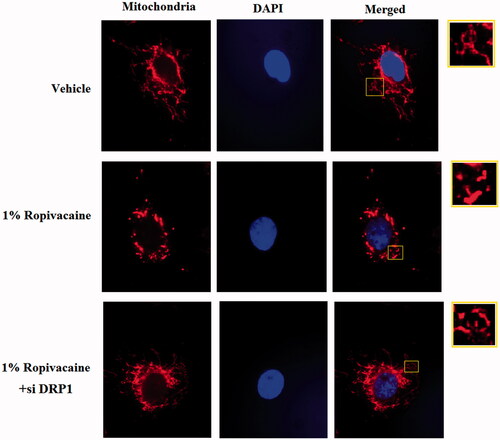
DRP1 controls the final process of mitochondrial fission and facilitates fission between two daughter mitochondria. We aimed to evaluate whether DRP1 is required for the process of ropivacaine-induced mitochondrial fission. To clarify this issue, we used an oligo-based gene silencing strategy to knockdown DRP1 in neuronal cells and subjected them to ropivacaine treatment. Our data show that DRP1-deficient neuronal cells are resistant to 1% ropivacaine treatment and under this condition, mitochondrial morphology consisted of a condensed network. However, DRP1-expressing control cells are susceptible to ropivacaine and exhibit a fission-like fragmented mitochondrial structure (). Thus, we conclude that DRP1 is indispensable for ropivacaine-induced mitochondrial fission.
Figure 5. Silencing of DRP1 ameliorated ropivacaine-induced mitochondrial fission. Human SH-SY5Y neuronal cells were infected with DRP1 siRNA Ad-virus for 12 h, followed by treatment with 0.5% and 1% ropivacaine for 72 h. Mitochondrial morphology was measured by MitoTracker red staining.
DRP1 deficiency ameliorates ropivacaine-induced mitochondrial dysfunction
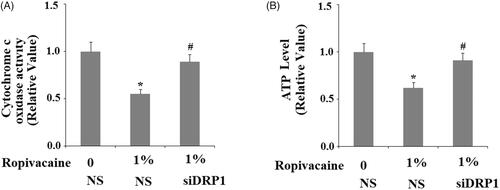
We tested the effect of ropivacaine on DRP1-deficient cells. Again, the DRP1-deficient cells showed higher resistance to ropivacaine treatment. The dose of 1% ropivacaine resulted in ∼50% cytochrome C oxidase activity in DRP1-expressing cells, while it barely had an effect on DRP1-deficient cells (). When measuring cellular ATP levels, treatment with the same concentration of ropivacaine caused 40% lower ATP production in DRP1-expressing cell while it had no obvious effect on ATP production in DRP1-deficient cells (). We came to the conclusion that DRP1 deficiency cells are resistant to Ropivacaine- caused mitochondrial damage.
Figure 6. Silencing of DRP1 ameliorated ropivacaine-induced mitochondrial dysfunction. Human SH-SY5Y neuronal cells were infected with DRP1 siRNA Ad-virus for 12 h, followed by treatment with 0.5% and 1% ropivacaine for 72 h. (A) Cytochrome C oxidase activity; (B) Intracellular ATP levels (*, #, P < .01 vs. previous column group).
Silencing of DRP1 ameliorates ropivacaine-induced LDH release and promotes neuronal viability
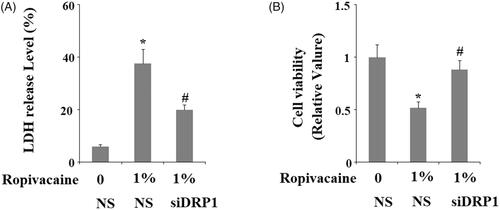
The loss of DRP1 appears to be protective against ropivacaine-induced cellular stress, so we tested cell viability in DRP1-deficient cells. The measurement of lactic dehydrogenase (LDH) release showed that 1% ropivacaine could induce about 40% LDH release in DRP1-expressing neuronal cells, while it only caused about 20% LDH release in DRP1-deficient cells (). MTT assay showed that the same concentration of ropivacaine caused roughly 50% neuronal death in DRP1-expressing cells while 95% of DRP1-deficient cells were viable (). Thus, DRP1-deficient cells are resistant to insult from ropivacaine.
Figure 7. Silencing of DRP1 ameliorated ropivacaine-induced release of LDH and reduction of cell viability. Human SH-SY5Y neuronal cells were infected with DRP1 siRNA Ad-virus for 12 h, followed by treatment with 0.5% and 1% ropivacaine for 72 h. (A) LDH release; (B) Cell viability was determined by MTT assay (*, #, P < .01 vs. previous column group).
DRP1 deficiency ameliorates ropivacaine-induced apoptosis
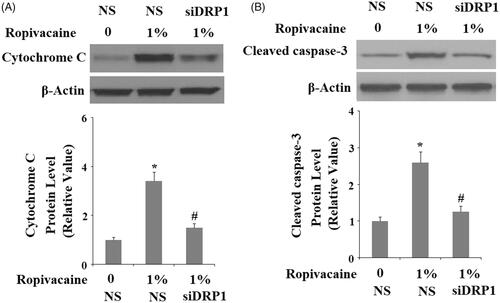
We next assessed the role of DRP1 in ropivacaine-induced apoptosis. For non-treated cells, roughly 7% of the neuronal population was detected to have undergone apoptosis via flow cytometry, while 1% ropivacaine caused apoptosis of roughly 50% of the population of DRP1-expressing cells, and it only caused ∼20% of cells to be apoptotic in DRP1-deficient cells (). Finally, we assessed two apoptotic signals in these DRP1-deficient cells: release of cytochrome C from mitochondria to cytosol and caspase 3 cleavage. Under non-treated conditions, the level of cytochrome C was extremely low and could barely be detected in cytosol. Upon treatment with 1% ropivacaine, DRP1-expressing cells had at least 3-fold higher release of cytochrome C into cytoplasm, while DRP1-deficient cells had an equivalent amount of cytochrome C (). Also, we found that 1% ropivacaine treatment significantly increased the level of cleaved caspase-3, which was prevented by silencing of DRP1.
Figure 8. Silencing of DRP1 ameliorated ropivacaine-induced apoptosis. Human SH-SY5Y neuronal cells were infected with DRP1 siRNA Ad-virus for 12 h, followed by treatment with 1% ropivacaine for 72 h. Apoptosis was determined by flow cytometry (*, #, P < .01 vs. previous column group).
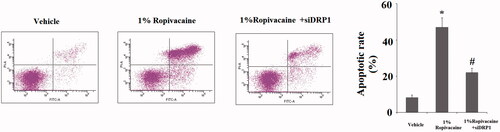
Figure 9. Silencing of DRP1 ameliorated ropivacaine-induced cytochrome C release from mitochondria to cytosol and activation of caspase-3. Human SH-SY5Y neuronal cells were infected with DRP1 siRNA Ad-virus for 12 h, followed by treatment with 1% ropivacaine for 72 h. (A) Cytochrome C in cytosol; (B) Cleaved caspase-3 (*, #, P < .01 vs. previous column group).
Discussion
Our study confirmed that ropivacaine, one of the most commonly used local anaesthetics, is cytotoxic in cultured SH-SY5Y neuronal cells. SH-SY5Y is a human neuroblastoma cell line widely used in cytotoxic research as it has a cytotoxic response similar to that of human primary neuronal cultures [Citation25]. The neurotoxic effect of ropivacaine can be reflected by its influence on mitochondrial hemostasis and cell viability. Our data demonstrate that clinically relevant doses of ropivacaine (0.5% and 1%) promote mitochondrial fragmentation, induce fission regulator DRP1 and lead to mitochondrial dysfunction. These data suggest that ropivacaine causes an imbalance in mitochondrial homeostasis and impairs neuronal cell function. Mechanistically, the mitochondrial fission protein DRP1 is indispensable for the cytotoxicity of ropivacaine. During mitochondrial fission, the dynamin-related GTPase DRP1 is recruited from the cytosol through the mitochondrial outer membrane [Citation21]. The impaired balance between mitochondrial fission and fusion has been associated with the pathogenesis of a variety of diseases, such as cancer, diabetes, cardiovascular disease and neurodegenerative disorders [Citation26]. Excessive expression and increased activity of DRP1 have been associated with neuronal death as well as the pathogenesis and progression of Alzheimer’s disease (AD) [Citation27]. Reduced DRP1 displays a protective effect against amyloid β (Aβ)-induced mitochondrial dysfunction and synaptic damage in AD [Citation28], which is consistent with our findings showing that silencing of DRP1 ameliorates ropivacaine-induced neuronal insult in human SH-SY5Y cells. As the loss of cellular DRP1 shows strong resistance to ropivacaine-induced mitochondrial disturbance and rescues the viability of neuronal cells under this stress, we conclude that expression of DRP1 is critical for ropivacaine-induced neuronal stress. We also examined whether treatment with ropivacaine has an impact on mitochondrial dynamics in non-neuronal cells. The liver hepatocellular cells Hep G2 were used. Interestingly, our results indicate that ropivacaine does not have a significant influence on mitochondrial fission nor on the expression of DRP1 (Data not shown). These findings suggest that the effects of ropivacaine on mitochondrial fission may be restricted to neuronal cells.
Ropivacaine is a long-acting, amide LA with an efficacy similar to that of bupivacaine. Ropivacaine was developed to replace bupivacaine and was shown to have a lesser cardiotoxic effect [Citation29]. Based on the findings of our study, the neurotoxic effect of ropivacaine appears to be significant and its side effects could disturb the normal function of mitochondria. This disruptive effect on mitochondria may be associated with numerous clinical and preclinical observations, as the majority of neurodegenerative disorders are tightly correlated with mitochondrial dysfunction [Citation30]. Dozens of clinical case reports and observational studies have shown that ropivacaine could induce severe neurotoxic side effects after intravenous injection or repeated use [Citation31–38].
According to an early dog experiment study, ropivacaine could induce neurological disorders similar to those seen in humans [Citation39]. In a snail experiment, ropivacaine was shown to activate phospholipase C activity induced by bursts of potential in the central snail neuron [Citation40]. In rat experiments, repeated intrathecal administration of ropivacaine caused neural damage [Citation41]. Another study in rats showed that ropivacaine injection is associated with peripheral nerve damage [Citation42]. Separate studies suggest that 1% ropivacaine treatment leads to neurotoxicity which can be revealed by ultrastructural and proteomic changes in the rat spinal cord [Citation43]. In more recent publications, clinically relevant concentrations of bupivacaine were shown to impair astrocytic mitochondrial function [Citation44]. Since bupivacaine and ropivacaine have similar chemical structures, we assume these data are consistent with our findings. In previous publications, ropivacaine has been shown to impair mitochondrial function in myocytes [Citation45], chondrocytes [Citation46], fibrous cells [Citation47] and other cell types.
Taken together, our findings support the notion that clinically relevant doses of ropivacaine could have significant neurotoxicity, and overdose-induced impaired neuronal mitochondrial homeostasis and cellular stress could be the mechanism behind this dangerous side effect. Therefore, careful evaluation and management of its use could be of clinical benefit for reducing its side effects.
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Bodenham AR, Howell SJ. General anaesthesia vs local anaesthesia: an ongoing story. Br J Anaesth. 2009;103:785–789.
- Becker DE, Reed KL. Local anesthetics: review of pharmacological considerations. Anesth Prog. 2012;59:90–102.
- Shipton EA. New formulations of local anaesthetics-part I. Anesthesiol Res Pract. 2012;2012:546409.
- Lirk P, Picardi S, Hollmann MW. Local anaesthetics: 10 Essentials. Eur J Anaesthesiol. 2014;31:575–585.
- Verlinde M, Hollmann MW, Stevens MF, et al. Local anesthetic-induced neurotoxicity. Int J Mol Sci. 2016;17:339.
- Sekimoto K, Tobe M, Saito S. Local anesthetic toxicity: acute and chronic management. Acute Med Surg. 2017;4:152–160.
- Werdehausen R, Fazeli S, Braun S, et al. Apoptosis induction by different local anaesthetics in a neuroblastoma cell line. Br J Anaesth. 2009;103:711–718.
- Cereda CM, Tofoli GR, Maturana LG, et al. Local neurotoxicity and myotoxicity evaluation of cyclodextrin complexes of bupivacaine and ropivacaine. Local neurotoxicity and myotoxicity evaluation of cyclodextrin complexes of bupivacaine and ropivacaine. Anesth Analg. 2012;115:1234–1241.
- Zhang K, Yang S, Luo C. TNF-alpha and TNF-R1 regulate bupivacaine-induced apoptosis in spinal cord dorsal root ganglion neuron. Eur J Pharmacol. 2018;833:63–68.
- Zheng J, Chen J, Wu G, et al. Inhibiting EZH2 rescued bupivacaine-induced neuronal apoptosis in spinal cord dorsal root ganglia in mice. J Anesth. 2018;32:524–530.
- Casati A, Putzu M. levobupivacaine and ropivacaine: are they clinically different? Best Pract Res Clin Anaesthesiol. 2005;19:247–268.
- Piegeler T, Votta-Velis EG, Bakhshi FR, et al. Endothelial barrier protection by local anesthetics: ropivacaine and lidocaine block tumor necrosis factor-α-induced endothelial cell Src activation. Anesthesiology 2014;120:1414–1428.
- Siekevitz P. Powerhouse of the Cell. Sci Am. 1957;197:131–144.
- Picard M, Wallace DC, Burelle Y. The rise of mitochondria in medicine. Mitochondrion 2016;30:105–116.
- Tandler B, Hoppel CL, Mears JA. Morphological pathways of mitochondrial division. Antioxidants (Basel). 2018;7:30.
- Gong Q, Wen X, Li H, et al. Up-regulation of Cav3.1 expression in SH-SY5Y cells induced by lidocaine hydrochloride. Artif Cells Nanomed Biotechnol. 2018;12:1–8.
- Askari S, Salehi R, Zarghami N, et al. The anticancer effects of biodegradable nanomagnetic dual natural components on the leptin gene expression in lung cancer. Artif Cells Nanomed Biotechnol. 2016;44:1753–1763.
- Sheng B, Wang X, Su B, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120:419–429.
- Wang J, Liu X, Qiu Y, et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J Hematol Oncol 2018;11:11.
- Xie X, Xu X, Sun C, et al. Protective effects of cilostazol on ethanol-induced damage in primary cultured hepatocytes. Cell Stress Chaperones. 2018;23:203–211.
- Manczak M, Kandimalla R, Yin X, et al. Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum Mol Genet. 2019;28:177–199.
- Lutz AK, Exner N, Fett ME, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem. 2009;284:22938–22951.
- da Silva EP, Jr, Nachbar RT, Levada-Pires AC, et al. Omega-3 fatty acids differentially modulate enzymatic anti-oxidant systems in skeletal muscle cells. Cell Stress Chaper. 2016;21:87–95.
- Ben Salem I, Prola A, Boussabbeh M, et al. Crocin and Quercetin protect HCT116 and HEK293 cells from Zearalenone-induced apoptosis by reducing endoplasmic reticulum stress. Cell Stress Chaper. 2015;20:927–938.
- Park CJ, Park SA, Yoon TG, et al. Bupivacaine induces apoptosis via ROS in the Schwann cell line. J Dent Res. 2005;84:852–857.
- Kandimalla R, Reddy PH. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim Biophys Acta. 2016;1862:814–828.
- Kandimalla R, Manczak M, Fry D, et al. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet. 2016;25:4881–4897.
- Manczak M, Kandimalla R, Fry D, et al. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet 2016;25:5148–5166.
- Graf BM, Abraham I, Eberbach N, et al. Differences in cardiotoxicity of bupivacaine and ropivacaine are the result of physicochemical and stereoselective properties. Anesthesiology 2002;96:1427–1434.
- de Castro IP, Martins LM, Tufi R. Mitochondrial quality control and neurological disease: an emerging connection. Expert Rev Mol Med. 2010;12:e12.
- Selander D, Sjovall J, Waldenlind L. Accidental i.v. injections of ropivacaine: clinical experiences of six cases. Reg Anesth. 1997;22:70.
- Ruetsch YA, Fattinger KE, Borgeat A. Ropivacaine-induced convulsions and severe cardiac dysrhythmia after sciatic block. Anesthesiology 1999;90:1784–1786.
- Klein SM, Benveniste H. Anxiety, vocalization, and agitation following peripheral nerve block with ropivacaine. Reg Anesth Pain Med. 1999;24:175–178.
- Ganapathy S, Sandhu HB, Stockall CA, et al. Transient neurologic symptom (TNS) following intrathecal ropivacaine. Anesthesiology 2000;93:1537–1539.
- Muller M, Litz RJ, Huler M. Grand mal convulsion and plasma concentrations after intravascular injection of ropivacaine for axillary brachial plexus blockade. Br J Anaesth. 2001;87:784–787.
- Al-Nasser B. Toxic effects of epidural analgesia with ropivacaine 0.2% in a diabetic patient. J Clin Anesth. 2004;16:220–223.
- Dhir S, Ganapathy S, Lindsay P, et al. Case report: ropivacaine neurotoxicity at clinical doses in interscalene brachial plexus block. Can J Anesth/J Can Anesth. 2007;54:912–916.
- Rodolà F, Anastasi F, Vergari A. Ropivacaine induced acute neurotoxicity after epidural injection. Eur Rev Med Pharmacol Sci. 2007;11:133–135.
- Bagdure DN, Reiter PD, Bhoite GR, et al. Persistent hiccups associated with epidural ropivacaine in a newborn. Ann Pharmacother. 2011;45:e35.
- Feldman HS, Arthur GR, Covino BG. Comparative systemic toxicity of convulsant and supraconvulsant doses of intravenous ropivacaine, bupivacaine, and lidocaine in the conscious dog. Anesth Analg. 1989;69:794–801.
- Zhong Z, Qulian G, Yuan Z, et al. Repeated intrathecal administration of ropivacaine causes neurotoxicity in rats. Anaesth Intensive Care. 2009;37:929–936.
- Whitlock EL, Brenner MJ, Fox IK, et al. Ropivacaine-induced peripheral nerve injection injury in the rodent model. Anesth Analg. 2010;111:214–220.
- Li L, Zhang T, Diao Y, et al. Injection of ropivacaine into the subarachnoid changes the ultrastructure and proteome of the rat spinal cord. Xenobiotica. 2013;43:908–914.
- Xing Y, Zhang N, Zhang W, et al. Bupivacaine indirectly potentiates glutamate-induced intracellular calcium signaling in rat hippocampal neurons by impairing mitochondrial function in cocultured astrocytes. Anesthesiology 2018;128:539–554.
- Sztark F, Malgat M, Dabadie P, et al. Comparison of the effects of bupivacaine and ropivacaine on heart cell mitochondrial bioenergetics. Anesthesiology 1998;88:1340–1349.
- Grishko V, Xu M, Wilson G, et al. Apoptosis and mitochondrial dysfunction in human chondrocytes following exposure to lidocaine, bupivacaine, and ropivacaine. J Bone Joint Surg Am. 2010;92:609–618.
- Cai XY, Xia Y, Yang SH, et al. Ropivacaine- and bupivacaine-induced death of rabbit annulus fibrosus cells in vitro: involvement of the mitochondrial apoptotic pathway. Osteoarthritis Cartilage. 2015;23:1763–1775.