
Abstract
Nanoparticles coated with cell membranes have been garnering growing attention due to their homologous binding capability of membrane molecules and consequent self-recognition by their source cells. In the present study, we report on the construction of doxorubicin and PD-L1 siRNA-loaded PLGA nanoparticles and their biological functionalization by cancer cell-derived membrane cloaking. The resulting cancer cell membrane-coated nanoparticles (CCMNPs) presented a core-shell nanostructure with highly specific self-recognition affinity to the homotypic cells, which can be attributed to the transference of cell adhesion molecules with homotypic binding properties. These findings facilitate the application of this bioinspired strategy for effective delivery of siRNA and precise tumour therapy.
Introduction
Nanomedicines based on targeting drug delivery systems have been intensively studied and developed for clinical cancer therapy [Citation1]. So far, enhanced permeability and retention (EPR) effect remained the main pattern for the uptake of tumour-specific delivery vehicles [Citation2–4]. However, most nanoparticles are recognized and eliminated as foreign substances by the immune system, thereby limiting the clinical applications of nanoparticles [Citation5]. There is an urgent demand for the seeking of a safer and more effective approach is urgently demanded. Nanodevices inspired and functionalized by natural biological materials have attracted surging interests due to the recent advancement in biomaterial engineering [Citation6–9]. Biomimetic nanoparticles modified with an active cell membrane have become one of the most attractive nanostructures [Citation1]. By cell membrane cloaking, biomimetic nanoparticles can be modified and functionalized with self-recognition and source-targeting capabilities, companied by long blood circulation and escape from immune capture.
Currently, cancer cell membranes (CCM) have drawn much attention for their membrane antigen-mediated homologous binding capability, which could be applied for the biomimetic functionalization of nanoparticles by offering the advantages of rich surface molecular diversity [Citation8,Citation10–13]. The CCM-based cancer cell functions such as extravasation, chemotaxis, and cancer cell adhesion, are inspiring researchers to explore the application prospect of CCMNPs for tumour-targeting drug delivery [Citation5,Citation14].
Overexpression of programmed death‐ligand 1 (PD-L1) on tumour cells is responsible for T cell hyporesponsiveness and blockade of PD-1/PD-L1 bindings by knockdown of PD-L1 using siRNA could resensitize cancer to T cell killing [Citation15]. For the co-delivery of chemical drug, doxorubicin (Dox) and siRNA targeting PD-L1 (si.PD-L1), we constructed PLGA nanocores loading Dox and si.PD-L1, camouflaged and function modified using CCM. The morphology, protein contents and self-recognition capability of homotypic cells of CCMNPs were characterized. Our results indicated that CCMNPs could homologously target to the source cells and hold great potential as an excellent nanoplatform guiding the delivery of homologous-targeting therapeutics.
Material and methods
Materials
Human MDA-MB-231 and Hela cell lines were obtained from Shanghai Institute of Cell Biology, Chinese Academy of Science (Shanghai, CN). SiRNA-cy5 of PD-L1 and control siRNA were purchased from GenePharma Co., LTD (Shanghai, CN). Lipofectamine 2000 (Invitrogen) was used for the transfection according to the manufacturer’s protocol. Anti-PD-L1 antibody (ab205921) and anti GAPDH antibody (60004–1-Ig) were acquired from Abcam and Proteintech respectively.
Preparation of Dox & si.PD-L1-loaded PLGA NPs (PLGA@[email protected])
To prepare PLGA nanoparticles, 200 μl of 20 μM siRNA and 10 mg/ml dox in RNAase free-deionized water was added under stirring, on a drop-by-drop basis, to 800 μl of dichloromethane (DCM) containing 25 mg of PLGA and the mixed solution was stirred magnetically for 2 min to obtain a primary W/O emulsion. Next, 2 ml of 2% PVA1500 (w/v) in ddH2O was added directly into the primary solution and further emulsified by sonication under 50 W for 1 min. After dilution using 6 ml of 2% PVA1500, the resulting product was further stirred magnetically for 3 h at room temperature to evaporate DCM. The precipitates were collected by centrifugation at 12,000 g for 30 min at 4 °C to obtain PLGA nanoparticles. Then, the PLGA NPs were washed twice with RNAase free-deionized water, resuspended in water and finally freeze-dried.
Preparation of cancer cell membrane fragments
Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Gibco) and penicillin-streptomycin (Hyclone) was used for the maintenance of Hela human cervix carcinoma cells and MDA-MB-231 human breast cancer cells. Hela cells were used to harvest membrane. In brief, cells were grown in cell culture dishes with 10 cm in diameter to full confluency and before detaching with 2 mM ethylenediaminetetraacetic acid (EDTA, USB Corporation) in phosphate buffered saline (PBS, Invitrogen). The collected cells were resuspended, washed twice using PBS and centrifuged at 500 × g for 5 min. Subsequently, the cell precipitates were suspended in a hypotonic lysing buffer consisting of 20 mM Tris-HCl (pH = 7.5, Mediatech), 10 mM KCl (Sigma Aldrich), 2 mM MgCl2 (Sigma Aldrich), and 1% phenylmethanesulfonyl fluoride (PMSF) (Beyotime Institute of Biotechnology) and lysed in ice bath for 15 min. The entire solution was freezed and thawed repeatedly, following centrifugation at 1000 × g for 10 min at 4 °C. The supernatants were centrifuged at 14,000 × g for 30 min at 4 °C to obtain the membrane fragments. The final fragments were collected in PBS for subsequent experiments.
Preparation and characterization of cancer cell membrane cloaked PLGA NPs (CCMNPs)
CCMNPs were synthesized as the extrusion approach previously reported. Briefly, the above cancer cell membrane fragments were physically extruded through a 400 nm polycarbonate membrane for 20 passes to obtain membrane vesicles. Anti-pan cadherin antibody (Alexa Fluor® 488) (Abcam) was used to characterize the cell membrane vesicles. Next, co-extrudation of nanocores and membrane vesicle through a 200 nm polycarbonate membrane were performed to coat the membrane vesicles onto PLGA cores for at least 5 cycles. Dynamic light scattering (DLS) measurements were applied to evaluate the particle size of PLGA NPs and CCMNPs. The morphology of final CCMNPs were obtained using transmission electron microscopy (TEM) under a JEM-2100 instrument. Briefly, PLGA NPs or CCMNPs were dropped onto carbon-coated 400 square mesh copper grids, following washing off with ddH2O. Grids were then negatively stained with 3% phosphotungstic acid (Yuanye Biotech) and excess solution was wicked away with absorbent paper before imaging. All assays were done in triplicate at room temperature.
Characterization of cancer cell membrane proteins
The protein content on the CCMNPs was analyzed by 10% SDS-PAGE. The cancer cell membrane vehicles and CCMNPs were heated to 70 °C for 10 min before loading to the gel. The gel was stained in Coomassie Blue solution (Beyotime Biotech.) for 2 h and then destained in water overnight following imaging.
Cell viability assays (MTT)
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) was used to evaluate the potential cytotoxicity of PLGA@[email protected] or CCMNPs. In brief, cells were seeded in 96-well plates with an average density of 103 cells per well and adherently cultured overnight. Then, the cells were incubated with medium containing PLGA@[email protected] or CCMNPs for 48 h. The DOX concentration of 1.5 µg/mL was used. MTT was added and co-incubated with the cells for 4 h, following crystals desolation with DMSO. Colour conversion was measured at 570 nm. All assays were repeated in triplicate and the values were represented as percentages.
Cellular uptake of Dox and si.PD-L1
Hela and MDA-MB-231 cells were plated at 50% confluency in tissue culture-treated 6-well plates. 24 h later, the cells were incubated with medium containing PLGA@[email protected] or CCMNPs at the same DOX concentration in triplicate for 1 h. Then, cells were harvested using 0.25% trypsin free of EDTA and washed twice with PBS. Final, flow cytometry data were collected using a Becton Dickinson FACSCanto II instrument and analyzed using Flowjo software (Treestar).
Homotypic targeting studies
For co-location imaging, Hela cells were plated at 50% confluency in confocal chambers overnight. Then, the cells were incubated with medium containing PLGA@[email protected] or CCMNPs at the DOX concentration of 1.5 µg/mL in triplicate for 1 h. The chambers were washed 3 times with PBS and imaging was done using a FLUOVIEW FV3000 Confocal Laser Scanning Microscope (Olympus).
Western blot analyses
Western blot assay was performed to measure the PD-L1 expression after si.PD-L1 transfection for 48 h as previous report [Citation16]. In brief, ice-cold PBS was used to wash the cells for twice before RIPA lysis buffer (Beyotime Biotechnology, CN) containing PMSF was added. After lysis on ice for 15 min, cell lysates collection was performed by centrifugation at 4 °C for 30 min and BCA protein assay kit (Beyotime Biotechnology, CN) was used to measure the protein contents. Then, equivalent proteins were loaded and separated by SDS-PAGE. 0.22 μm PVDF membranes (Merck Millipore, DE) were used for the transferation. After membrane blocking in 5% non-fat dry milk in PBST for 1 h, appropriate primary antibodies were used for incubation overnight at 4 °C. The PVDF membranes binding with primary antibodies were washed three times with PBST for 5 min each, following incubation with horse-radish peroxidase-coupled isotype-specific secondary antibodies for 1 h at room temperature. Finally, ChemiDoc MP imaging system (Bio-Rad) was used for the imaging of the immune complexes.
Statistical analysis
Significance analysis was performed using statistical package SPSS (version 20.0; IBM, New York, NY). All data were expressed as means ± SD. Differences between groups were assessed by one-way analysis of variance (ANOVA) and a p values <0.05 was considered as significant.
Results
Synthesis and characterization of CCMNPs
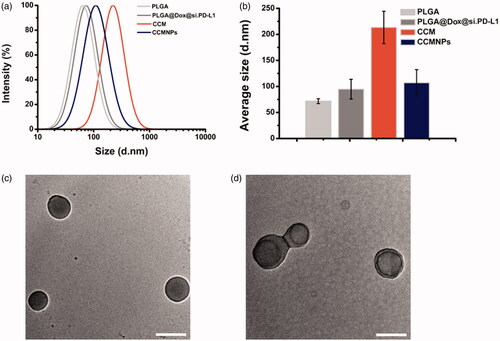
A well-studied nanoprecipitation process was used to prepare PLGA cores [Citation17]. Briefly, dox and siRNA dissolved in water was dropped into DCM containing PLGA. 2% PVA in water was added to form diblock copolymer. In order to cloak the bare PLGA NPs, Hela cells were chosen as a model cancer cell line to collect purified CCM and fabricate CCMNPs. Hypotonic lysis, mechanical membrane rupture and gradient centrifugation were performed sequentially to remove the intracellular contents of the harvested cells and gained purified membrane derivatives. Final, CCMNPs were obtained by physical co-extrusion of membrane vesicles and nanoparticle cores. Morphological changes in bare NP after complexing with Dox and siRNA were monitored using DLS. As measured using DLS, the bare PLGA NPs were approximately 70 nm in size. A slight size increase by ∼20 nm was observed in PLGA@[email protected] NPs formed by complexing bare PLGA with Dox and si.PD-L1. The membrane vesicles were about 200 nm. And the final CCMNPs with an average size approximate 110 nm was obtained upon fusion of the membrane vesicles with PLGA@[email protected] (). It has been reported that the size of bare cores could influence lipid bilayer coating on nanoscale substrates [Citation9]. These researches could help in interpreting the size decrease from membrane vesicles to CCMNPs. TEM visualization of PLGA@[email protected] NPs and CCMNPs showed that uniform membrane cloaks were coated around the polymeric cores to form a final spherical shape. A clear core-shell structure was observed in CCMNPs group upon negative staining with phosphotungstic acid ().
Figure 1. Characterization of CCMNPs. (a) Size intensity curves of bare PLGA cores, c, cancer cell membrane vesicles, and CCMNPs measured by dynamic light scattering (DLS). (b) Hydrodynamic size of bare PLGA cores, PLGA@[email protected], cancer cell membrane vesicles, and CCMNPs. Bars represent means ± SD (n = 3). (c,d) Transmission electron micrographs of (c) PLGA@[email protected] and (d) CCMNPs. Samples were negatively stained with phosphotungstic acid. All scale bars = 100 nm.
Characterization of cancer cell membrane proteins
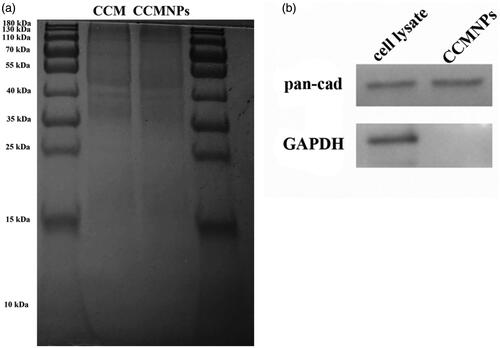
Protein ingredient analysis was carried out to validate the successful functionalization of PLGA@[email protected] with cancer cell membrane antigens. Based on the results of PAGE gel electrophoresis for protein content evaluation, we found similar results in CCM and CCMNPs lysates, which indicates that the membrane contents of the source cell membrane can be well retained during the CCM extracted and CCMNPs formulated progress (). To confirm the presence of specific membrane antigens onto the CCMNPs, expressions of cancer cell membrane protein marker, pan-cadherin, and intracellular marker, GAPDH were detected using Western blotting analysis (). A significant enrichment of cadherin and GAPDH was observed in cancer cell lysate, while CCMNPs have lost the intracellular biomarker and only the membrane retained. All the findings demonstrated the preferential retention of membrane proteins during the fabrication process.
Figure 2. Membrane antigen characterization of CCMNPs. (a) SDS-PAGE protein analysis of cancer cell membrane vesicles and CCMNPs. Samples were run at equal protein concentration and stained with Coomassie Blue. (b) Western blotting analysis for membrane-specific protein, pan-cadherin.
Homotypic targeting mediated by cancer cell surface antigens
Cancer cell membrane coating could provide self-recognition capability originating from homotypic cancer cells. To determine the specific targeted delivery of CCMNPs, MDA-MB-231 and Hela cells were incubated with CCMNPs for 1 h. The fluorescence intensities of Dox were monitored using FACS. No significant difference of fluorescence intensities in uptake of free dox or PLGA@[email protected] were found between MDA-MB-231 and Hela cells. But as for CCMNPs, the corresponding source cells uptook far more of those than heterotypic MDA-MB-231 cells (). Compared to blank controls, an approximating 100-fold in terms of the mean fluorescence intensity was observed in Hela cells while no obvious uptake of CCMNPs was found in MDA-MB-231 cells. As compared to PLGA@[email protected], the increased uptake of CCMNPs in corresponding homotypic cells and the decreased uptake of that in heterotypic cells further suggested the highly specific self-recognition affinity of CCMNPs to the homotypic cells.
Figure 3. Targeted delivery of CCMNPs based on self-recognition. (a) Flow cytometric analysis of Hela cells incubated with blank solution, free Dox, PLGA@[email protected] or CCMNPs. (b) Flow cytometric analysis of MDA-MB-231 cells incubated with blank solution, free Dox, PLGA@[email protected] or CCMNPs. (c) Cytotoxicity profiles in Hela and MDA-MB-231 cells obtained after 48 h incubation of PLGA, PLGA@[email protected], CCM or CCMNPs. *p < .05, and ***p < .001.
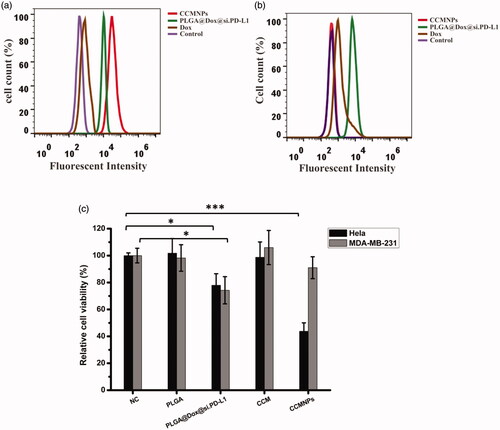
The in vitro cytotoxicity of CCMNPs was measured using MTT assay. As shown in , CCMNPs induced a much higher cytotoxicity in Hela cells while no significant effect was observed in MDA-MB-231 cells. The unique cytotoxicity of CCMNPs in Hela cells implicated the self-selective cellular uptake and biosecurity of CCM.
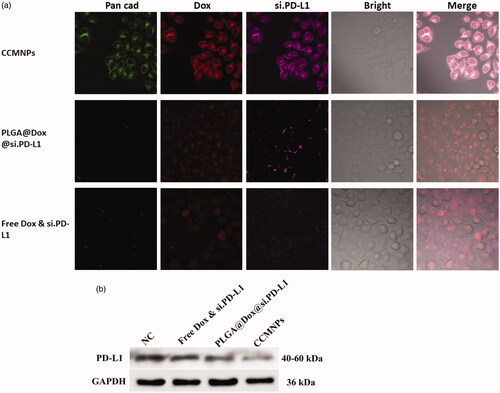
To further confirm the interactions between cells and particles originating from membrane functionalization, confocal fluorescence microscopy was applied to detect the specific uptake of CCMNPs. After 1 h incubation of CCMNPs or PLGA nanoparticles in cultured Hela cells, we observed a dramatically increased uptake as compared to both free drugs and drug-loaded PLGA cores (). We next examined the expression of PD-L1 protein to confirm the target delivery of CCMNPs. After treatment for 48 h, protein levels of PD-L1 were similar in NC and free Dox & si.PD-L1 treated groups. A most dramatical reduction was observed in CCMNPs group (), indicating the most uptake of si.PD-L1. These results indicate that coating of PLGA nanoparticles with cancer cell membranes can preferentially increase the affinity of the nanoparticles to the homotypic cancer cells, which can be attributed to the homotypic binding properties of cell adhesion molecules. These results indicate that coating of CCM onto PLGA nanoparticles can preferentially enhance the affinity of the particles to the homotypic cancer cells, which can be attributed to the homotypic binding properties of cell adhesion proteins.
Figure 4. CCMNPs serve as a homotypically targeted delivery vehicle. (a) Confocal fluorescent imaging of Hela cells incubated with CCMNPs, PLGA@[email protected] or free Dox & si.PD-L1. (b) PD-L1 knockdown was induced after CCMNPs incubation for 48 h.
Discussion
Construction of biomimetic core-shell nanoparticles by coating cellular membrane onto nanoparticles has been widely applied in drug delivery [Citation18–20], imaging [Citation21, Citation22], detoxification [Citation23], vaccination [Citation13], and phototherapy [Citation24]. Fusion of membrane extraction and bare nanoparticle core formed into a core-shell structure. Surface modification using cell membrane could function nanoparticles antigen camouflage, resulting escape from immune elimination and long circulation by innately mimicking the antigenic diversity of the source cells [Citation13]. Moreover, the remain of natural information of sourcing cells could endow the nanoparticles with biological functions for tumour targeting, while removing the nuclear components with serious safety concerns [Citation25]. The camouflage of cell membrane could benefit therapeutic nanoparticles by immune escape, homologous targeting abilities and specific adhesion to tumour sites [Citation1]. As for now, variety of cell membranes have been used for the surface modification of therapeutic nanoparticle cores, such as membrane derivates from RBCs, cancer cells, fibroblast cells, macrophages, stem cells, platelets and MDSCs [Citation1].
RBC membrane–camouflaged nanoparticles have been most widely discussed with many advantages based on their specific structure, surface proteins and functionalities [Citation12,Citation25]. However, no active targeting properties to tumours could be endowed by RBC membrane due to their heterology and it has been reported that the sourcing cells had a 20-fold higher uptake of cancer cell membrane-coated nanoparticles as compared to RBC membrane-coated nanoparticles as a result of homologous binding [Citation4]. These unique properties are firmly derived from their membrane biomarkers on the surface including N‐cadherin, galectin‐3, or epithelial cell adhesion molecule (EpCAM) [Citation26]. Therefore, it has been believed that homotypic cancer cell membranes could functionalize nanostructures with homologous targeting abilities, helping escape from immune elimination and prolong the blood circulation time, thereby promoting the development of highly specific and effective cancer therapy [Citation13].
In the present study, we observed the CCM was successfully coated onto the PLGA nanoparticle surfaces. The PLGA core and CCM shell mutually benefit each other with enhanced stability and superior targeting efficiency when combined into a core-shell structure as findings from previously reported RBCNPs [Citation27,Citation28]. The homologous-targeting and efficient delivery inspires the use of CCMNPs as potential new anticancer nanotherapeutics. Membrane coverage implicates the shield of nanoparticle cores from external exposure, which might minimize the foreign materials induced complement activation and immunological responses [Citation9]. The CCMNPs were also found to present cancer cell-specific proteins on the particle surface through the right-side-out oriented membrane coating [Citation4]. Combination of the cargo encapsulation within PLGA nanoparticles and membrane protein retention enable CCMNPs to serve as a multifunctional system integrating the steric stabilization of polysaccharides, the cargo loading capacity of polymeric cores, the biological immune-evasive activities of CCM, and the anchoring-insulating features of the lipid membranes, into a single, unified package [Citation9]. The introduction of tumour-related antigens could help elevating immune recognition or specifically inducing an immune response for immunotherapy [Citation5,Citation29,Citation30].
For now, receptor-ligand interactions aimed at surface antigens were the main strategy for cancer targeting [Citation31,Citation32]. In our study, CCMNPs exhibited a source targeting ability by the specific self-recognition of homologous cancer cells. It has been demonstrated that surface antigens with homophilic adhesion domains could be used as decoys for integrating to tumour cells with homophilic binding proteins, such as carcinoembryonic antigen and cadherins [Citation13,Citation33]. The integration of cancer cell membranes might endow the bare nanoparticles with cell-to-cell adhesive property to guide the targeted therapy for cancer. The homotypic binding mechanism of Hela cells have been well established in vivo previously [Citation4]. In the present study, we also used Hela cells to explore the cancer targeting possibilities of CCMNPs. Hela membrane cloaking PLGA nanoparticles presented a significant more powerful cellular internalization to the sourcing cells as compared to bare PLGA NPs while a reduced binding of nanoparticles was observed in MDA-MB-231 cells. These findings demonstrated that the enhanced particle-to-cell adhesion was contributed to homotypic cell membrane. The homologous adhesive tendencies implicate the potential of CCMNPs to target metastatic sites [Citation34]. Surface adhesion molecules play important roles in dissemination of cancer cells and their metastatic propensities [Citation34,Citation35]. Therefore, by functionalizing nanoparticles with specificities exhibited by cancer cell membrane, it holds great potential to prepare naturally targeted nanoformulations with cancer-cancer binding properties, thereby helping to deliver the therapeutics directly to primary cancers or distant sites with high efficacy.
Conclusions
Summarily, cancer cell membrane cloaking polymeric nanoparticles presents excellent self-recognition internalization by the source cancer cells, thereby inducing effective camouflage with multiple membrane antigens and surface functionalization. We have demonstrated successful extraction of cancer cell membranes, functionalization of PLGA nanoparticles and self-targeting via a homotypic binding mechanism for the drug delivery. Toward future translation, this nature-inspired nanotechnology with cell membrane-derived functionalization may facilitate the development of highly specific self-targeting nanosystems for personalized cancer therapy. Furthermore, the specific homo-recognition to the source cells may implicate the potentials of this nanoplatform directly tracing and attacking the cancer cells with highly invasive ability.
Related Research Data
References
- Zhen X, Cheng P, Pu K. Recent advances in cell membrane–camouflaged nanoparticles for cancer phototherapy. Small. 2019;15:1804105.
- Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71–79.
- Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6.
- Zhu JY, Zheng DW, Zhang MK, et al. Preferential cancer cell self-recognition and tumor self-targeting by coating nanoparticles with homotypic cancer cell membranes. Nano Lett. 2016;16:5895.
- Li R, He Y, Zhang S, et al. Cell membrane-based nanoparticles: a new biomimetic platform for tumor diagnosis and treatment. Acta Pharmaceutica Sinica B. 2018;8:14–22.
- Yoo JW, Irvine DJ, Discher DE, et al. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521–535.
- Balmert SC, Little SR. Biomimetic delivery with micro- and nanoparticles. Adv Mater. 2012;24:3757–3778.
- Hu CM, Fang RH, Luk BT, et al. 'Marker-of-self' functionalization of nanoscale particles through a top-down cellular membrane coating approach. Nanoscale. 2013;5:2664–2668.
- Luk BT, Hu CM, Fang RH, et al. Interfacial interactions between natural RBC membranes and synthetic polymeric nanoparticles. Nanoscale. 2013;6:2730–2737.
- Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–2127.
- Hamdy S, Molavi O, Ma Z, et al. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine. 2008;26:5046–5057.
- Hu CM, Zhang L, Aryal S, et al. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci USA. 2011;108:10980–10985.
- Fang RH, Hu CM, Luk BT, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181–2188.
- Iero M, Valenti R, Huber V, et al. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15:80–88.
- Teo PY, Yang C, Whilding LM, et al. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: strategies to enhance T cell killing. Adv Healthcare Mater. 2015;4:1180–1189.
- Yue C, Yang M, Tian Q, et al. IGFBP7 is associated to prognosis and could suppress cell survival in cholangiocarcinoma. Artif Cells Nanomed Biotechnol. 2018;46:817–825.
- Yang C, Vu-Quang H, Husum DMU, et al. Theranostic poly(lactic-co-glycolic acid) nanoparticle for magnetic resonance/infrared fluorescence bimodal imaging and efficient siRNA delivery to macrophages and its evaluation in a kidney injury model. Nanomed Nanotechnol Biol Med. 2017;13:2451–2462.
- Biagiotti S, Paoletti MF, Fraternale A, et al. Drug delivery by red blood cells. IUBMB Life. 2011;63:621–631.
- Aryal S, Hu CM, Fang RH, et al. Erythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and release. Nanomedicine (London, England). 2013;8:1271–1280.
- Hu CM, Fang RH, Wang KC, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118–121.
- Rao L, Bu LL, Cai B, et al. Cancer cell membrane-coated upconversion nanoprobes for highly specific tumor imaging. Adv Mater. 2016;28:3460–3466.
- Fang RH, Luk BT, Hu CM, et al. Engineered nanoparticles mimicking cell membranes for toxin neutralization. Adv Drug Deliv Rev. 2015;90:69–80.
- Hu CM, Fang RH, Copp J, et al. A biomimetic nanosponge that absorbs pore-forming toxins. Nat Nanotech. 2013;8:336–340.
- Fang RH, Kroll AV, Gao W, et al. Cell membrane coating nanotechnology. Adv Mater. 2018;30:1706759.
- Fang RH, Jiang Y, Fang JC, et al. Cell membrane-derived nanomaterials for biomedical applications. Biomaterials. 2017;128:69–83.
- Chen Z, Zhao P, Luo Z, et al. Cancer cell membrane-biomimetic nanoparticles for homologous-targeting dual-modal imaging and photothermal therapy. ACS Nano. 2016;10:10049–10057.
- Gao W, Hu CM, Fang RH, et al. Surface functionalization of gold nanoparticles with red blood cell membranes. Adv Mater. 2013;25:3549–3553.
- Hu CM, Fang RH, Zhang L. Erythrocyte-inspired delivery systems. Adv Healthcare Mater. 2012;1:537–547.
- Pulido J, Kottke T, Thompson J, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012;30:337–343.
- Castle JC, Kreiter S, Diekmann J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081–1091.
- Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782.
- Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768.
- Iurisci I, Cumashi A, Sherman AA, et al. Synthetic inhibitors of galectin-1 and -3 selectively modulate homotypic cell aggregation and tumor cell apoptosis. Anticancer Res. 2009;29:403–410.
- Khaldoyanidi SK, Glinsky VV, Sikora L, et al. MDA-MB-435 human breast carcinoma cell homo- and heterotypic adhesion under flow conditions is mediated in part by Thomsen-Friedenreich antigen-galectin-3 interactions. J Biol Chem. 2003;278:4127–4134.
- Glinsky VV, Glinsky GV, Glinskii OV, et al. Intravascular metastatic cancer cell homotypic aggregation at the sites of primary attachment to the endothelium. Cancer Res. 2003;63:3805–3811.