
Abstract
Excessive generation and accumulation of amyloid-β (Aβ) fragments by familial mutations of amyloid precursor protein (APP) and presenilin 1 (PS1) play a key role in causing oxidative stress, mitochondrial abnormalities and neuronal apoptosis in Alzheimer’s disease (AD). Anagliptin, a novel DPP-4 inhibitor, is a clinical drug for the management of type II diabetes approved for use in 2012. Little on the pharmacological function of anagliptin against Aβ-induced cytotoxicity in neuronal cells is known. Here, we examined the protective capacities of anagliptin against cytotoxicity in N2a neuronal cells overexpressing APP Swedish mutant and PS1 exon 9 deletion mutant (N2a/Swe.D9). Our results demonstrate that anagliptin reduced the production of ROS and the expression of NADPH oxidase 4 (NOX-4) in N2a/Swe.D9 cells. We also reported that anagliptin activates the antioxidant system by increasing the level of reduced glutathione (GSH) and glutathione peroxidase (GPx) activity. Notably, anagliptin is able to improve mitochondrial function by elevating mitochondrial membrane potential (MMP) and adenosine triphosphate (ATP) production. Additionally, our results demonstrate that anagliptin decreased the vulnerability of cells to hydrogen peroxide (H2O2)-induced secondary insult by increasing cell viability and reducing the secretion of lactate dehydrogenase (LDH) and high mobility group box 1 protein (HMGB-1). Importantly, we found that treatment with anagliptin suppressed the mitochondrial-dependent apoptosis pathway by preventing the translocation of cytochrome C, reducing cleavage of caspase-3, and the inhibiting expression of Bax. These results implicate that anagliptin may have potential as a therapeutic agent for AD treatment.
Introduction
Alzheimer’s disease (AD) is an important disease of dementia among the elderly. Excessive production of the amyloid-β peptide (Aβ) has been considered as an important pathological characteristic of AD [Citation1]. Aβ40 and Aβ42 fragments are produced by amyloid precursor protein (APP) through proteolytic processes involving β- and γ-secretases. Presenilin 1 (PS1) has been identified as the main component of γ-secretase. Mutations in APP and PS1 are linked with familial AD by altering the consequence of AD pathogenesis [Citation2]. The Swedish double mutation (K670N/M671 L) is one of the most famous familial APP missense mutations. Previous reports have demonstrated various PS1 mutations including exon 9 deletion [Citation3]. Overexpression of these mutations increases the generation of Aβ fractions, especially Aβ42 [Citation4]. Increased Aβ production is associated with oxidative stress. Mitochondrial abnormalities are considered as a central pathological mechanism in AD. Impairment of mitochondrial function including reduced mitochondrial membrane potential (MMP) and decreased adenosine triphosphate (ATP) production have been identified in AD brains. Importantly, mitochondrial-dependent apoptosis plays a key role in the pathological progression of AD [Citation5]. Amelioration of Aβ-induced cytotoxicity is, therefore, an important therapeutic strategy against AD.
Dipeptidyl peptidase (DPP)-4 inhibitors are a new family of incretin-based drugs, which were developed to treat type 2 diabetes mellitus (T2DM) [Citation6] by increasing the level of glucagon-like peptide 1 (GLP-1) and stimulating insulin secretion [Citation7]. Anagliptin is a novel DPP-4 inhibitor which was approved for the management of T2DM in 2012 [Citation8]. In addition to lowering blood glucose levels, anagliptin has demonstrated a wide range of pharmacological properties. For example, administration of anagliptin exerts a cholesterol-lowering effect in rodent models by suppressing intestinal cholesterol transport [Citation9]. Another study reported that anagliptin treatment could prevent intimal hyperplasia formation induced by balloon injury through elevating the activity of superoxide dismutase (SOD) and upregulating the expression of SOD-1 [Citation10]. Interestingly, T2DM patients are reported to have a two-fold higher risk of developing AD [Citation11]. Recently, DPP-4 inhibitors were proposed as a promising treatment for AD owing to their ability to suppress Aβ accumulation, tau hyperphosphorylation, and neuroinflammation [Citation12]. Concordantly, linagliptin, another DPP-4 family member, has been shown to play an important role in rescuing insulin signaling impairment caused by Aβ and ameliorating tau hyper-phosphorylation caused by Aβ [Citation13]. Administration of saxagliptin was also shown to reduce the production of Aβ and phosphorylated tau, attenuate neuroinflammation, and improve cognitive performance in the test animals [Citation14]. However, it is still not known whether anagliptin possesses a protective function in AD. In the current study, using an N2a neuronal cell model, we assessed the beneficial capacities of anagliptin against Aβ-induced neurotoxicity.
Materials and methods
Cell culture
Neuroblastoma 2a (N2a) stable cell line co-transfected with Swedish mutations of APP and ΔE9 deleted mutation of PS1 (N2a/Swe.D9) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)/Opti–MEM supplemented with 5% fetal bovine serum (FBS) and 0.1% penicillin/streptomycin [Citation15]. Cells were stimulated with 0.5, 1 μM anagliptin [Citation16] for 24 h.
Determination of Aβ
Secretion of Aβ40 and Aβ42 in the culture supernatant was measured by a sandwich enzyme-linked immunosorbent assay (S-ELISA) using commercial ELISA kits from R&D Systems, USA.
Real-time polymerase chain reaction (PCR) analysis
After the indicated stimulation, RNA was extracted from N2a cells using Qiazol (Qiagen, USA). cDNA was produced using a Bio-Rad cDNA synthesis kit. Expression of NADPH oxidase 4 (NOX-4) was assessed using SYBR Green-based real-time PCR. Quantification of NOX-4 mRNA was performed using the 2−ΔΔCt method. We used the following primers: Human NOX-4 primer (F: 5′-CTTTTGGAAGTCCATTTGAG-3′; R: 5′-GTCTGTTCTCTTGCCAAAAC-3′); Human GAPDH primer (F, 5′-CAAAATCAAGTGGGGCGATGCTG-3′, R, 5′-TGTGGTCATGAGTCCTTCCACGAT-3′).
Western blot
Cell lysates were prepared with cell lysis buffer. Equal amounts of cell lysates (20 μg) were run on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then electronically transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% skim dry milk. Samples were probed with primary antibodies in a cold room overnight. After 3 washes, the transferred membrane was probed with an HRP-linked secondary antibody for 1 h at room temperature (RT). Bands were visualized with enhanced chemiluminescence (ECL) (Bio-Rad, USA). We used the following antibodies in the current study: NOX-4 (#NB110-58849, Novus Biologicals, USA); β-actin (#MAB8929, R&D Systems, USA); cytochrome C (#MAB897, R&D Systems, USA); cleaved caspase-3 (#MAB835, R&D Systems, USA); Bax (#AF820, R&D Systems, USA); Bcl-2 (#MAB8272, R&D Systems, USA).
Measurement of reduced glutathione (GSH)
The levels of reduced GSH represent the antioxidant capacity of cells and tissues. The level of reduced GSH was assessed with a 5,5’-dithiobis-(2-nitrobenzoic acid)-glutathione disulfide reductase recycling assay [Citation17].
Determination of glutathione peroxidase (GPx) activity
GPx activity was measured to reflect the antioxidant capacity of cells and tissues. After treatment, cell lysate was mixed with reaction buffer: potassium phosphate buffer (50 mM, pH 7.0), EDTA (1 mM), GSH (1 mM), GSH reductase (0.2 U/mL), tertbutyl-hydroperoxide (0.2 mM), and NADPH (0.2 mM). OD at 340 nm was monitored to reflect GPx activity.
Measurement of lactate dehydrogenase (LDH) release
LDH release from N2a cells was determined to reflect cell death. After the indicated stimulation, leakage of LDH was determined using a kit. Briefly, 50 μL of supernatant was added to a 96-well plate and mixed with 50 μL reaction buffer for 30 min. OD at 490 nm was monitored to reflect LDH leakage.
Determination of reactive oxygen species (ROS)
The levels of ROS in N2a cells were assessed using the ROS-sensitive dye 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). After the necessary stimulation, cells were washed and probed with DCFH-DA (10 μM) for 30 min. Fluorescence in N2a cells was visualized using a fluorescence microscope.
Measurement of mitochondrial membrane potential (MMP)
Mitochondrial function was assayed by evaluating MMP. After the necessary stimulation, N2a cells were probed with tetramethylrhodamine (TMRM) (20 nmol/L) for 60 min at 37 °C in darkness. Fluorescence in N2a cells was recorded with a fluorescence microscope.
Measurement of adenosine triphosphate (ATP)
Production of ATP was assessed using a commercial bioluminescence kit (Thermo Fisher Scientific, USA). After the necessary stimulation, cells were lysed. After a brief centrifugation at 14,000 × g, 100 μL supernatant was collected and mixed with 100 μL reaction reagent to generate luciferin light. Luciferin light intensity was detected using a microplate luminometer.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Cell apoptosis was assessed using a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining assay. Cells were fixed by being treated with formaldehyde (4%) for 10 min and then permeabilized by being treated with Triton X-100 (0.1%) for 15 min. N2a cells were cultured with a terminal deoxynucleotidyl transferase enzyme in an incubator at 37 °C for 30 min. Nuclei of cells were stained with DAPI.
Dihydroethidium (DHE) staining
The dye dihydroethidium (DHE) purchased from Sigma-Aldrich, USA was dissolved in anhydrous dimethyl sulfoxide (DMSO). Cells were probed with 5 μmol/L DHE in Krebs buffer at 37 °C for 20 min in a dark room. Cells were then washed 3 times, and the fluorescence intensity was determined using a fluorescence microscope (Olympus, Japan).
Statistical analysis
Experimental values are shown as means ± standard error of measurement (SEM). A one-way analysis of variance (ANOVA) method was used to determine statistical significance. p < .05 was treated as statistical significance.
Results
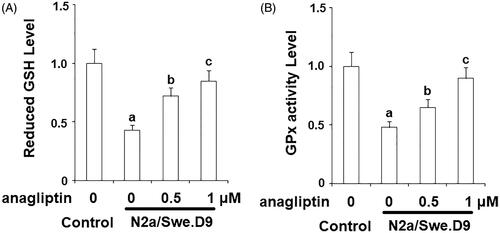
Firstly, we measured the level of secreted Aβ. The results of ELISA in indicate that secreted Aβ40 and secreted Aβ42 in N2a/Swe.D9 cells were obviously higher than the vehicle control. Aβ is associated with oxidative stress in neuronal cells. Here, DCFH-DA staining demonstrates that ROS in N2a/Swe.D9 cells was remarkably higher than in the vehicle control. However, the presence of anagliptin reduced the levels of ROS (). Consistently, treatment with anagliptin inhibited the fluorescence density of dihydroethidium (DHE), another ROS staining probe (). NOX-4 is responsible for the production of mitochondrial ROS. Interestingly, we found that the expression of NOX-4 was remarkably higher in N2a/Swe.D9 cells than in the vehicle control. As expected, anagliptin suppressed the expression of NOX-4 (). The GSH antioxidant system is essential for protecting cells against oxidative stress. Therefore, we then investigated whether treatment with anagliptin affected the GSH system. The results in and demonstrate that reduced GSH and GPx activity was significantly lower in N2a/Swe.D9 cells than in vehicle control cells. The presence of anagliptin ameliorated the decrease in reduced GSH and GPx activity.
Figure 1. Secretion of Amyloid β (Aβ) was increased in AD cell models. The levels of secreted Aβ40 and Aβ42 as determined by an ELISA assay (a, p < .01 vs. vehicle control).
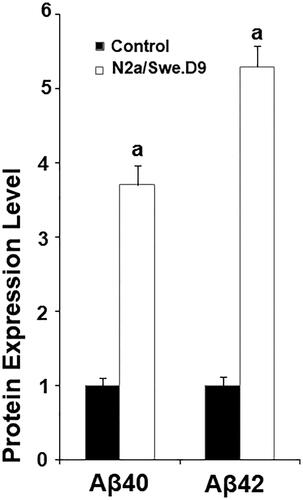
Figure 2. Anagliptin (0.5, 1 μM) ameliorates oxidative stress in N2a/Swe.D9 cells. Cells were incubated with 0.5, 1 μM anagliptin for 24 h. (A) ROS was measured through DCFH-DA staining; (B) Cells were stained with dihydroethidium (DHE) (a, b, c, p < .01 vs. the previous group).
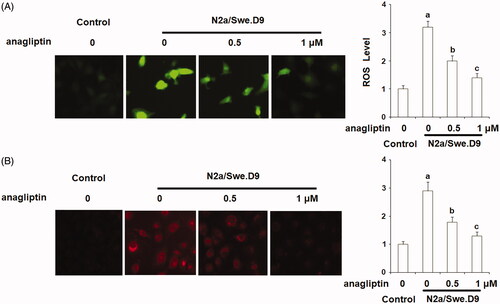
Figure 3. Anagliptin (0.5, 1 μM) suppresses the expression of NOX-4. Cells were incubated with 0.5, 1 μM anagliptin for 24 h. (A) mRNA of NOX-4; (B) Protein of NOX-4 (a, b, c, p < .01 vs. the previous group).
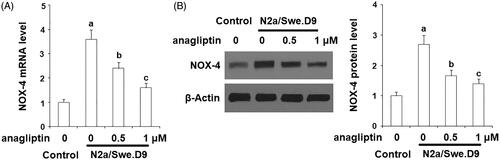
Figure 4. Anagliptin (0.5, 1 μM) restored the reduction of reduced glutathione (GSH) in AD cell models. N2a/Swe.D9 cells were stimulated with 0.5, 1 μM anagliptin for 24 h. (A) Reduced GSH was determined; (B) GPx activity was measured (a, b, c, p < .01 vs. the previous group).
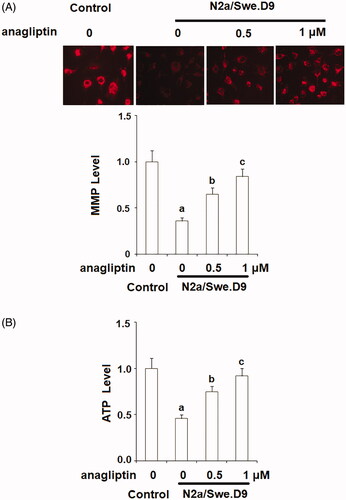
Mitochondrial dysfunction occurs in the brains of AD patients. Increased production and accumulation of Aβ have been associated with the deterioration of mitochondrial dysfunction. Here, we assessed the beneficial effects of anagliptin on mitochondrial function through examining the levels of MMP via TMRM staining and intracellular ATP using a commercial bioluminescence assay. The results in display that the presence of anagliptin could restore the reduction in MMP and ATP.
Figure 5. Anagliptin (0.5, 1 μM) improved mitochondrial dysfunction in an AD cell model. N2a/Swe.D9 cells were cultured with 0.5, 1 μM anagliptin for 24 h. (A) Intracellular levels of MMP were measured by TMRM staining; (B) Intracellular ATP was measured using a bioluminescence method (a, b, c, p < .01 vs. the previous group).
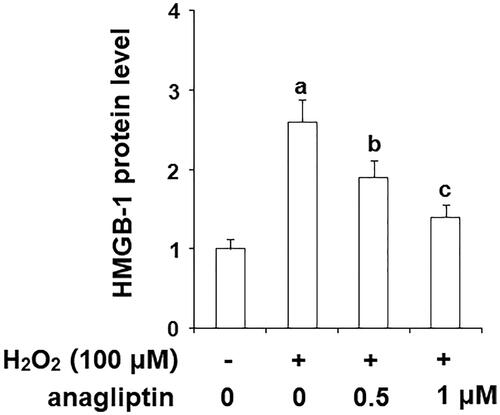
Oxygen free radicals are important factors involved in the pathogenetic cascade of AD. The generation of hydrogen peroxide (H2O2) is linked with the formation of insoluble Aβ. Notably, excessive generation of H2O2 may cause secondary damage in AD brains. The MTT assay results in indicate that stimulation with H2O2 markedly reduced the viability of N2a/Swe.D9 cells, which was ameliorated by the presence of anagliptin. The release of LDH is an important index of the integrity of the cell membrane and cell death. Consistently, anagliptin significantly suppressed H2O2-induced LDH release (). The secretion of high mobility group box 1 protein (HMGB-1) is associated with inflammation and cell death. The levels of secreted HMGB-1 were measured using an ELISA assay. The results in display that H2O2 stimulation obviously increased the secretion of HMGB-1, which was suppressed by anagliptin. These findings suggest that anagliptin possesses an important protective effect against H2O2-induced secondary insult in neuronal cells.
Figure 6. Anagliptin (0.5, 1 μM) protects N2a/Swe.D9 cells against secondary insult by H2O2. Cells were stimulated with H2O2 (100 μM) with or without anagliptin (0.5, 1 μM) for 24 h. (A). Cell viability as determined by MTT assay; (B). Leakage of LDH as examined using a kit (a, b, c, p < .01 vs. the previous group).
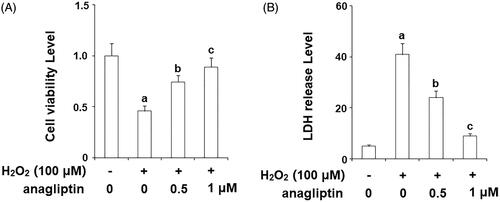
Figure 7. Anagliptin (0.5, 1 μM) protects against H2O2-induced HMGB-1 secretion in an AD cell model. N2a/Swe.D9 cells were stimulated with H2O2 (100 μM) with or without anagliptin (0.5, 1 μM) for 24 h. Secretion of HMGB-1 was measured by ELISA (a, b, c, p < .01 vs. the previous group).
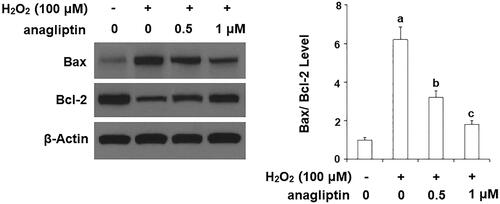
Excessive apoptosis of neuronal cells is an important pathological characteristic of AD. The results of TUNEL assay in demonstrate that stimulation with H2O2 led to a significant increase in apoptosis of N2a/Swe.D9 cells. However, the presence of anagliptin significantly ameliorated H2O2-induced apoptosis. MMP disruption results in leakage of cytochrome C from mitochondria into the cytoplasm and subsequent cleavage of caspase-3. Our results indicate that H2O2 stimulation significantly promoted leakage of cytochrome C from mitochondria into the cytoplasm, which was markedly attenuated by treatment with anagliptin (). Notably, H2O2 stimulation significantly upregulated the levels of cleaved caspase-3, which was significantly inhibited by treatment with anagliptin (). The Bax/Bcl2 family proteins play essential roles in mediating mitochondria-dependent apoptosis. Consistently, we found that anagliptin mitigated the elevation of Bax () and reduced expression of Bcl-2 () in response to H2O2. These findings implicate that anagliptin protects against H2O2-induced apoptosis via a mitochondria-dependent pathway.
Figure 8. Anagliptin protects against H2O2-induced apoptosis in an AD cell model. N2a/Swe.D9 cells were stimulated with H2O2 (100 μM) with or without anagliptin (0.5, 1 μM) for 24 h. Apoptosis of cells was assayed by TUNEL staining (a, b, c, p < .01 vs. the previous group).
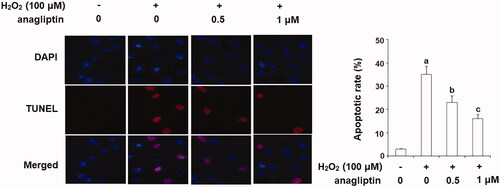
Figure 9. Anagliptin prevents the leakage of cytochrome C from mitochondria into cytosol and activation of caspase-3. N2a/Swe.D9 cells were stimulated with H2O2 (100 μM) with or without anagliptin (0.5, 1 μM) for 24 h. (A) Levels of cytochrome C in the cytosol. (B) Cleaved caspase-3 (a, b, c, p < .01 vs. the previous group).
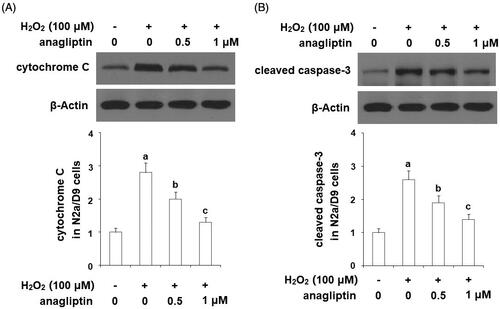
Figure 10. The effects of anagliptin on Bax/Bcl-2 expression. N2a/Swe.D9 cells were stimulated with H2O2 (100 μM) with or without anagliptin (0.5, 1 μM) for 24 h. (A) Representative bands of Bax/Bcl-2; (B) Quantification of Bax/Bcl-2 (a, b, c, p < .01 vs. previous column group).
Discussion
Excessive generation of Aβ fragments is one of the most crucial events in the pathological progression of AD [Citation18]. Familial-associated APP and PS1 mutations cause excessive secretion of Aβ and are intensively linked with cytotoxicity in AD brains [Citation19]. Transgenic (Tg) mouse models that overexpress familial AD-associated APP mutant or PS1 mutant have been widely used in preclinical studies of AD [Citation20]. Additionally, neuronal cell lines bearing APP Swedish mutant or PS1 mutant have been used to explore the underlying mechanisms of cytotoxicity in AD [Citation15]. A previous study demonstrated that N2a neuronal cells containing APP Swedish mutant and ΔE9 deleted PS1 mutant exhibited elevated production of ROS, nitrous oxide, deterioration of mitochondrial function, as well as apoptosis [Citation15]. Abnormal generation of Aβ results in impairment in the bioenergetics of neuronal mitochondria by impairing the mitochondrial electron transport chain and subsequent ATP generation [Citation21]. Also, overexpression of APP Swedish mutant in neuronal cells leads to reduced mitochondrial DNA replication and transcription as well as mitochondrial biogenesis [Citation22]. Importantly, mitochondrial dysfunction in neuronal cells results in mitochondria-associated apoptosis [Citation23]. Amelioration of oxidative stress and improvement of mitochondrial-dependent apoptosis have been considered as novel therapeutic targets for AD. Here, we characterized the intracellular protective functions of anagliptin against endogenous Aβ. These results demonstrate that anagliptin can mitigate oxidative stress, improve mitochondrial function, and reduce apoptosis in AD model cells.
Anagliptin, a newly developed selective inhibitor of DPP-4, has been clinically used for improving long-term glycemic control by increasing GLP-1 and subsequent insulin secretion [Citation24]. Interestingly, AD and T2DM have many common pathological characteristics, such as insulin resistance, amyloid accumulation and neurodegeneration in the brain [Citation25]. Intranasal administration of insulin is capable of protecting the cognitive performance of AD patients. Importantly, DPP-4 inhibitors have been recently reported to prevent pathological development of AD through several intracellular mechanisms. For example, linagliptin treatment is able to rescue Aβ-induced dysregulation of insulin signaling, tau hyperphosphorylation, and neuronal apoptosis [Citation13]. Administration of another DPP-4 inhibitor saxagliptin protected against streptozotocin-induced AD-like neurodegenerative progress by attenuating Aβ accumulation, tau phosphorylation, inflammatory responses and improving memory retention in rats [Citation14]. In this study, we proved that anagliptin could protect neuronal cells against H2O2-induced secondary insult. Consistently, a recent study demonstrated that pre-incubation with anagliptin prevented apoptosis in HUVECs by suppressing the generation of ROS, expression of NOX-4, activation of Bax, cleavage of caspase-3, and overexpression of cytochrome C [Citation10]. Oxidative stress, mitochondrial dysfunction, and related apoptosis have been considered as central pathological mechanisms of multiple neurodegenerative diseases. The protective effects of anagliptin against mitochondrial dysregulation found in this study suggest that anagliptin might exert a wide range of beneficial properties in all kinds of neurodegenerative disorders. It should be noted that anagliptin plays an important role in increasing GLP-1 levels by inhibiting DPP-4 activity. GLP-1 has been identified as a neurotrophic factor, which can protect against neuronal damage by enhancing long-term potentiation (LTP) and increasing synapse formation [Citation26]. Additionally, GLP-1 analogs including exendin-4 and liraglutide, which are available treatment drugs for T2DM, have displayed beneficial effects in AD by attenuating memory deficit and reducing Aβ plaques in the brain [Citation27]. We speculated that the neuroprotective effects of anagliptin against Aβ toxicity might be associated with the inhibition of DPP-4 and the resulting increase in GLP-1.
There are several limitations to this study. Firstly, we only examined the protective properties of anagliptin against Aβ-induced cytotoxicity in N2a cells. The pathological mechanisms of AD are complex. A variety of brain cells including astrocytes and microglia have been reported to participate in the pathogenesis of AD [Citation28]. Secondly, in addition to excessive production and accumulation of Aβ, a diverse range of risk factors including hyperphosphorylation of tau protein and cytokine-mediated neuroinflammation have been reported to contribute to the development of AD. Therefore, future in vivo investigations will help us to enhance our understanding of the pharmacological roles of anagliptin in AD.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Nalivaeva NN, Turner AJ. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br J Pharmacol [In press]. 2019.
- Li NM, Liu KF, Qiu YJ, et al. Mutations of beta-amyloid precursor protein alter the consequence of Alzheimer’s disease pathogenesis. Neural Regen Res. 2019;14:658–665.
- Cho YY, Kwon OH, Park MK, et al. Elevated cellular cholesterol in familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of β-amyloid precursor protein. PLoS One. 2019;14:e0210535.
- Aso E, Juvés S, Maldonado R, et al. CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AβPP/PS1 mice. J Alzheimers Dis. 2013;35:847–858.
- Area-Gomez E, de Groof A, Bonilla E, et al. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018;9:335.
- Wang X, Zheng P, Huang G, et al. Dipeptidyl peptidase-4(DPP-4) inhibitors: promising new agents for autoimmune diabetes. Clin Exp Med. 2018;18:473–480.
- Xie W, Song X, Liu Z. Impact of dipeptidyl-peptidase 4 inhibitors on cardiovascular diseases. Vascul Pharmacol. 2018;109:17–26.
- Nishio S, Abe M, Ito H. Anagliptin in the treatment of type 2 diabetes: safety, efficacy, and patient acceptability. Diabetes Metab Syndr Obes. 2015;8:163–171.
- Goto M, Furuta S, Yamashita S, et al. Dipeptidyl peptidase 4 inhibitor anagliptin ameliorates hypercholesterolemia in hypercholesterolemic mice through inhibition of intestinal cholesterol transport. J Diabetes Investig. 2018;9:1261–1269.
- Li Q, Li J, Liu Y, et al. Anagliptin prevents apoptosis of human umbilical vein endothelial cells by modulating NOX-4 signaling pathways. Biomed Pharmacother. 2018;103:1623–1631.
- Isik AT, Soysal P, Yay A, et al. The effects of sitagliptin, a DPP-4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer’s disease. Diabetes Res Clin Pract. 2017;123:192–198.
- Angelopoulou E, Piperi C. DPP-4 inhibitors: a promising therapeutic approach against Alzheimer’s disease. Ann Transl Med. 2018;6:255.
- Kornelius E, Lin CL, Chang HH, et al. DPP-4 Inhibitor linagliptin attenuates Abeta-induced cytotoxicity through activation of AMPK in neuronal Cells. CNS Neurosci Ther. 2015;21:549–557.
- Kosaraju J, Gali CC, Khatwal RB, et al. Saxagliptin: a dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 2013;72:291–300.
- Sheng B, Song B, Zheng Z, et al. Abnormal cleavage of APP impairs its functions in cell adhesion and migration. Neurosci Lett. 2009;450:327–331.
- Jiang T, Jiang D, Zhang L, et al. Anagliptin ameliorates high glucose- induced endothelial dysfunction via suppression of NLRP3 inflammasome activation mediated by SIRT1. Mol Immunol. 2019;107:54–60.
- Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165.
- Jan AT, Azam M, Rahman S, et al. Perspective insights into disease progression, diagnostics, and therapeutic approaches in Alzheimer’s disease: a judicious update. Front Aging Neurosci. 2017;9:356.
- Dai MH, Zheng H, Zeng LD, et al. The genes associated with early-onset Alzheimer’s disease. Oncotarget 2017;9:15132–15143.
- Sasaguri H, Nilsson P, Hashimoto S, et al. APP mouse models for Alzheimer’s disease preclinical studies. Embo J. 2017;36:2473–2487.
- Crouch PJ, Harding SME, White AR, et al. Mechanisms of A beta mediated neurodegeneration in Alzheimer’s disease. Int J Biochem Cell Biol. 2008;40:181–198.
- Sheng B, Wang X, Su B, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120:419–429.
- Cabezas-Opazo FA, Vergara-Pulgar K, Pérez MJ, et al. Mitochondrial dysfunction contributes to the pathogenesis of Alzheimer’s Disease. Oxid Med Cell Longev. 2015;2015:509654.
- Chiba Y, Yamakawa T, Tsuchiya H, et al. Effect of anagliptin on glycemic and lipid profile in patients with type 2 Diabetes Mellitus. J Clin Med Res. 2018;10:648–656.
- Carelli-Alinovi C, Misiti F. Erythrocytes as Potential Link between Diabetes and Alzheimer’s Disease. Front Aging Neurosci. 2017;9:276.
- Salcedo I, Tweedie D, Li Y, et al. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol. 2012;166:1586–1599.
- Hölscher C. Central effects of GLP-1: new opportunities for treatments of neurodegenerative diseases. J Endocrinol. 2014;221:T31–T41.
- Fernandez CG, Hamby ME, McReynolds ML, et al. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front Aging Neurosci. 2019;11:14.