
Abstract
Recently, we showed that parathyroid hormone-like hormone (PTHLH), a cytokine-like polyprotein, is critical for extracellular matrix (ECM) deposition through the activation of hepatic stellate cells (HSCs). Here, we show that N-terminal PTHLH is secreted into the supernatant of injured hepatocytes, its expression is positively correlated with liver fibrosis severity based on mice liver biopsies, and it is primarily expressed in the cytoplasm of hepatocytes along the fibrous septa of fibrotic livers. PTHLH overexpression in mice was achieved through adeno-associated virus-mediated gene delivery (AAV9-PTHLH), and liver fibrosis was induced with carbon tetrachloride (CCl4). We observed that AAV9-PTHLH induced spontaneous development of liver fibrosis and increased sensitivity to CCl4. PTHLH increased Hedgehog (Hh) pathway activation in a PTH1R-dependent manner, and the effect of PTHLH was primarily mediated by protein kinase C (PKC) θ. PTHLH-mediated PTH1R-PKC θ pathway activation is a key event in the profibrotic Hh-dependent activation of HSCs.
Introduction
Liver fibrosis resulting from chronic liver injury occurs through various aetiologies and triggers wound-healing that is characterized by excess deposition of extracellular matrix (ECM) [Citation1,Citation2], leading to the formation of precancerous hepatocellular carcinoma (HCC) [Citation3] lesions. Hepatic stellate cells (HSCs) are responsible for ECM deposition in liver fibrosis [Citation4,Citation5]. Damaged hepatocytes secrete various types of factors including cytokines and growth factors that can induce differentiation of HSCs into myofibroblast-like cells [Citation6]. These cells express α-smooth muscle actin (α-SMA) and synthesize fibrillar collagens, resulting in liver fibrosis [Citation4,Citation7].
Parathyroid hormone-like hormone (PTHLH), also referred to as parathyroid hormone-related protein (PTHrP) [Citation8], is widely distributed in many organs, including the liver [Citation8–10]. The secretory N-terminal PTHLH(1–40) fragment shares homology with parathyroid hormone (PTH) and is referred to as the common PTH/PTHLH type 1 receptor (PTH1R) ligand, and PTH1R usually participates in the protein kinase A (PKA) intracellular signalling pathway [Citation11]. Through an autocrine or paracrine mechanism, PTHLH suppresses the growth of HepG2 cells, and during endotoxaemia, PTHLH levels increase and can potentially cause an acute hepatic response [Citation12,Citation13]. Recently, PTHLH has been reported to be involved in renal fibrogenesis in cooperation with transforming growth factor-β1(TGF-β1) [Citation14–16]. Our previous results showed that PTHLH(1–40) activates HSCs and promotes ECM deposition [Citation17]. In addition, we found that PTHLH promotes intestinal fibrosis by activating the transcription factor Runx2 [Citation18]. These results suggest that PTHLH is involved in liver fibrosis progression; however, the exact effects of PTHLH on hepatic fibrosis and its molecular mechanism are poorly understood. The Hedgehog (Hh) signalling pathway plays a vital role in orchestrating wound healing responses [Citation19,Citation20]. HSCs are a type of Hh-responsive cell involved in liver fibrosis progression, and HSCs express the Hh-receptor Patched. Under normal circumstances, Hh ligands in cooperation with Patched liberate Smo signalling, resulting in a cascade that ultimately activates members of the Gli transcription factor family [Citation21–24]. Thus, we further reasoned that the liver repair process might be regulated by PTHLH and Hh pathways because there is obvious evidence that PTHLH and Hh pathways are important regulators of cartilage development [Citation25,Citation26].
In the current study, we found that damaged hepatocytes release an N-terminal PTHLH fragment and subsequently activate HSCs, and this activation was blocked by Hh signalling pathway inhibitors. In vivo, data showed that PTHLH levels increased in liver fibrosis, and PTHLH overexpression promoted not only the hepatic fibrosis process but also the spontaneous development of hepatic fibrosis. More importantly, the Hh pathway was activated during PTHLH-induced hepatic fibrosis, which was blocked by neutralizing antibodies targeting PTH1R and a protein kinase C (PKC) θ inhibitor. Together, these results suggest that the PTHLH-PTH1R-PKC θ-Hh axis plays an important role in the modulation of HSC activation.
Materials and methods
Chemicals
Mineral oil and carbon tetrachloride (CCl4) were obtained from Sigma-Aldrich (St. Louis, Missouri, USA). To induce hepatocyte damage, hepatocytes were treated with 16 mM CCl4 for different periods of time (0 h, 24 h or 48 h) [Citation27].
Animals and treatment
Adult male Sprague–Dawley rats (180–220 g) and adult male BALB/C mice (6–8 weeks old) were purchased from and housed in the Central Laboratory of Animal Science at Southern Medical University (Guangzhou, China). Experimental procedures were approved by the Animal Ethics Committee of Nanfang Hospital. Liver fibrosis was induced by CCl4 administered subcutaneously at a dose of 1 ml/kg (a 1:4 mixture with mineral oil) twice weekly for 3 or 7 consecutive weeks (n = 5 in each group). Liver tissues were collected at different weeks after the CCl4 injections [Citation28]. Serum was collected to determinate biochemical parameters, including AST and ALT.
PTHLH lentivirus and adeno-associated virus
Recombinant adeno-associated viruses carrying the green fluorescent protein (AAV9-GFP) or PTHLH gene (https://www.ncbi.nlm.nih.gov/gene/, accession number: NM_008970, last accessed July 1, 2018) were purchased from Hanbio, Shanghai, China. AAV9-GFP or AAV9-PTHLH was administered through the tail vein with a 28-gauge insulin syringe at a dose of 1 × 1011 plaque-forming units in 0.1 ml of saline under sevoflurane anaesthesia, and haemostasis was achieved by brief manual pressure. LV5-EF-1a-GFP + Puro vectors encoding the mouse PTHLH gene (GeneBank accession number: NM_008970) or human PTHLH gene (GeneBank accession number: NM_002820.2, lasted accessed December 23, 2018) were purchased from Genepharma (Suzhou, China). Target cells (1 × 105), including AML12 and LO2 cells, were infected with 1 × 106 recombinant lentivirus-transducing units in the presence of 5 μg/ml polybrene (GenePharma).
Cell isolation and culture
Primary hepatocytes were isolated from Sprague–Dawley rats, and primary HSCs were isolated from male BALB/C mice. The cells were assessed for purity and viability and then seeded at a density of 3 × 102 cells/mm in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum (FBS) and penicillin/streptomycin [Citation29,Citation30]. The clonally derived rat HSC line T6, the human HSC line LX-2, the normal human hepatocyte line LO2 and the normal mouse hepatocyte line AML12 were purchased from the Type Culture Collection of the Chinese Academy of Sciences in Shanghai, China. All cells were cultured in RPMI 1640 medium containing 10% FBS at 37 °C with 5% CO2. At 70% confluence, the cells were growth-arrested in serum-free medium overnight before the experiments. LX2 cells were treated with LV-PTHLH-LO2 or LV-NC-LO2 cell (4 days of culture) conditioned medium, and primary quiescent mouse HSCs were treated with LV-PTHLH-AML12 or LV-NC-AML12 cell conditioned medium (4 days of culture) for 24 h. Co-culture of LX2 and primary quiescent mouse HSCs with LV-PTHLH-AML12 or LV-PTHLH-LO2 cells was conducted using a non-contact co-culture transwell system (Corning, USA). Inserts containing LV-PTHLH or LV-NC hepatocyte cell lines were transferred to a 6-well plate seeded with LX2 or primary quiescent mouse HSCs. After 24 h of co-culture, LX2 or primary quiescent mouse HSCs were harvested for further analyses.
Reagents
Recombinant PTHLH(1–40) protein was purchased from Bachem (Bubendorf, Switzerland), and used at 10 nM. A broad-spectrum PKC inhibitor (Gö6983, 10 µM) and a PKC θ inhibitor (rottlerin, 10 µM) were purchased from Sigma-Aldrich. Cells were pretreated with neutralizing PTHLH antibody (5 µg/ml) and neutralizing PTH1R antibody (5 µg/ml) for 2 h and then incubated with PTHLH(1–40) or the supernatant of damaged LX2 cells for 48 h; both neutralizing antibodies obtained from Abcam (Cambridge, MA, USA).
Serum biochemistry
Serum ALT and AST levels were measured using standard enzymatic procedures according to the manufacturers’ instructions (Thermo Fisher, Pittsburgh, PA, USA) [Citation31].
Hydroxyproline assay
The concentration of liver hydroxyproline was measured with a Hydroxyproline Assay Kit (Sigma, USA) according to the manufacturer’s recommended protocol.
Histology and immunohistochemistry
Paraffin-embedded sections of liver samples were stained with either H&E or Sirius Red. Deparaffinized slides were immunostained for α-SMA (Abcam), Shh (Millipore, Billerica, MA, USA), Gli2 (Abcam), PTH1R (Abcam) and PTHLH (Santa Cruz, CA, USA). The immunostained sections were examined with an Olympus IX73 microscope (Olympus, Tokyo, Japan). Positive staining for α-SMA, Shh, PTH1R, PTHLH and GLI2 was semi-quantified according to a previously described method [Citation32].
Sirius red staining and morphometry
Sirius Red staining was performed on paraffin-embedded sections as previously described [Citation33]. Computer-aided quantification of the fibrotic areas was performed on Sirius Red-stained liver sections from different groups (n = 5 in each group), as indicated in the relevant figure legends. The fibrotic areas were calculated according to a previously described method [Citation32].
Trichloroacetic acid (TCA) precipitation of proteins
Supernatant proteins were extracted from the cell medium according to a reported protocol [Citation34].
Western blot analysis
Western blot analysis was performed according to general protocols. The following primary antibodies were used: anti-PTHLH antibody was from Santa Cruz, CA; anti-rabbit PTH1R, anti-rabbit α-SMA, anti-rabbit TGF-β1, anti-Smo and anti-rabbit collagen I antibodies were obtained from Abcam; antibodies targeting pPKC δ, pPKC α/β, pPKC ε, pPkc θ, pPKC ζ, total PKC δ, total PKC α/β, total PKC ε, total PKC θ and total PKC ζ were from Cell Signaling Technology (Beverly, MA, USA); anti-PKA C-α, anti-PKA RI-α/β, anti-P-CREB(Ser133) and anti-total CREB antibodies were also from Cell Signaling Technology. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was obtained from Zhonshanjinqiao (Wuhan, China). Acquisition of blot images was performed using a Gene5 image acquisition system (Syngene, Frederick, MD, USA).
Quantitative real-time PCR
Total RNA extraction, cDNA synthesis and real-time PCR were performed according to general protocols [Citation18,Citation35]. Data analysis was performed using the 2−△△CT method. GAPDH was used as an internal control. The primer pairs are listed in .
Table 1. Sequences of specific primer pairs.
Statistical analysis
The data are presented as mean ± SD. SPSS 13.0 software (SPSS Inc., Chicago, IL, USA) was used for analysis. Multiple comparisons were performed through one-way ANOVA. p < .05 was considered statistically significant.
Results
PTHLH was released into the supernatant of damaged hepatocytes and subsequently activated HSCs
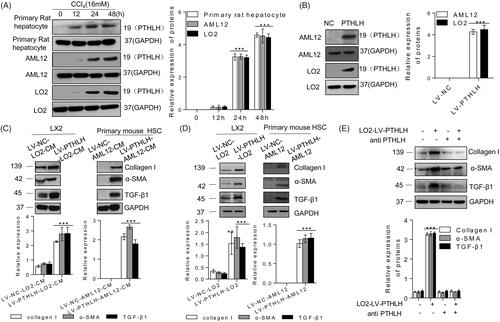
To confirm our previously reported results, we isolated primary rat hepatocytes and mouse HSCs and determined whether their viability and purity were sufficient for experimental use (Supplementary Figure 1(A)). Consistent with previous results, we found that PTHLH was expressed in hepatocytes from different species [Citation12,Citation13], including primary rat hepatocytes and AML12 and LO2 cell lines (Supplementary Figure 1(B)). To determine whether hepatocytes can secrete PTHLH, LO2 and AML12 cells were infected with recombinant lentivirus carrying PTHLH (LV-PTHLH) or lentivirus carrying NC (LV-NC) (Supplementary Figure 1(C)), and the three types of hepatocytes were induced by CCl4. After 12–48 h of culture, the N-terminal PTHLH level was detected in the supernatant using the TCA precipitation method. The results showed that the levels of N-terminal PTHLH were increased and easily detectable compared with the baseline levels in the untreated or LV-NC cell medium (). Subsequently, we tested whether N-terminal PTHLH in the culture medium could activate HSCs. Primary mouse HSCs or LX2 cells were stimulated with conditioned medium from hepatocytes (LV-NC-AML12-CM, LV-PTHLH-AML12-CM, LV-NV-LO2-CM and LV-PTHLH-LO2-CM), and α-SMA, collagen I, and TGF-β1 protein expression were found to be significantly upregulated in the LV-PTHLH group (). These results reminded us that overexpression of PTHLH itself is harmful to hepatocytes. To confirm whether PTHLH can promote HSC activation in vivo, LX2 cells or primary mouse HSCs were co-cultured with LV-PTHLH-LO2, LV-NC-LO2, LV-PTHLH-AML12 or LV-NV-AML12 cells in a non-contact transwell system that allowed the exchange of soluble factors but was impermeable to the cells. As shown in , the expression levels of α-SMA, collagen I, and TGF-β1 in HSCs and LX2 cells were upregulated. To further confirm the functional implication of PTHLH in HSCs, we treated LX2 cell cultures with neutralizing PTHLH antibody before conditioned medium from LV-PTHLH-LO2 or LV-NC-LO2 cells was added and then assessed the TGF-β1, collagen I and α-SMA levels. Neutralization of endogenous PTHLH activity significantly abrogated the increase in TGF-β1, collagen I and α-SMA proteins in LX-2 cells ().
Figure 1. Accumulation of N-terminal PTHLH in supernatants of damaged hepatocytes and PTHLH treatment activated HSCs. (A, B) Primary rat hepatocytes and AML12 and LO2 cells were incubated with 16 mM CCl4 for the time period indicated or transduced with LV-NC or LV-PTHLH, and the protein expression of N-terminal PTHLH in the growth medium of hepatocytes was determined using the TCA method. Equal loading was confirmed by comparison with GAPDH levels. (C) LX2 cells were treated with LV-PTHLH-LO2 or LV-NC-LO2 cell conditioned medium (LV-PTHLH-LO2-CM and LV-NC-LO2-CM) and primary quiescent mouse HSCs were treated with LV-PTHLH-AML12 or LV-NC-AML12 cell conditioned medium (LV-PTHLH-AML12-CM and LV-NC-AML12-CM) for 24 h. Collagen I, TGF-β1 and α-SMA protein levels were analysed through Western blotting. (D) Western blotting to determine Collagen I, TGF-β1 and α-SMA protein expression in LX2 cells or primary quiescent mouse HSCs co-cultured with LV-NC or LV-PTHLH hepatocytes (LO2 or AML12) for 24 h. (E) LX2 cells were pretreated with neutralizing PTHLH antibody (5 µg/ml) for 2 h and then recombinant PTHLH(1–40) for 48 h. Western blots show the decrease in collagen I, TGF-β1 and α-SMA protein expression in PTHLH(1–40)-treated cells in response to the neutralizing PTHLH antibody (5 µg/ml). The relative protein levels were normalized to the GAPDH level. The data are expressed as the mean ± SD of three independent experiments. *p < .05, **p < .01 and ***p < .001 versus controls.
PTHLH/PTH1R was upregulated in fibrotic livers
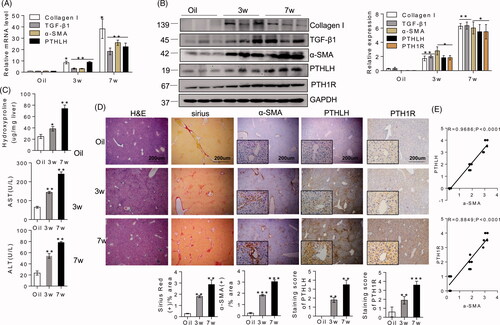
We initially examined the PTHLH levels in liver biopsies from a CCl4-induced mice model of liver fibrosis (at week 3 or 7), with olive oil treatment as the control. With respect to the involvement of PTHLH in liver fibrosis development, we found that the PTHLH mRNA levels at week 7 were twice as high as those at week 3 () and were correlated with the elevated mRNA levels of TGF-β1, collagen I and α-SMA. Western blot analysis showed that PTHLH expression in the livers was significantly elevated with the progression of CCl4-induced hepatic fibrosis (). Additionally, PTHLH and PTH1R protein upregulation was accompanied by increased expression of TGF-β1, collagen I, and α-SMA. These results suggest that PTHLH expression is positively correlated with the severity of liver fibrosis. Consistent with the hepatic hydroxyproline content (), the CCl4 induced mice model exhibited more severe liver fibrosis at week 7 than at week 3, based on H&E and Sirius Red staining of liver samples (). Serum ALT and AST levels () were significantly increased at week 7 compared with week 3. Furthermore, we detected the distribution of PTHLH in injured livers. PTHLH expression was detected through immunohistochemical staining of the CCl4-treated livers. The results showed that PTHLH was located in the parenchymal regions of normal livers; however, CCl4 treatment elevated PTHLH expression in the cytoplasm of parenchymal cells as well as in hepatocytes in portal tracts and along fibrous septa (). We also detected the expression of PTH1R, a specific receptor for PTHLH, and found that the PTH1R protein level was increased in the cell membrane of hepatocytes from CCl4 treated mice (). In addition, PTHLH and PTH1R upregulation were strongly correlated with α-SMA expression, which indicates HSC activation ().
Figure 2. In CCl4-induced liver fibrosis mice, PTHLH was highly expressed in fibrotic livers. BALB/C mice were administered CCl4 (1 ml/kg, 1:4 mixture with mineral oil) twice/week for 3 and 7 weeks. (A, B) Quantitative RT-PCR and immunoblot analyses showed upregulation of PTHLH mRNA and protein in liver fibrosis models mice accompanied by increased TGF-β1, collagen I, and α-SMA increased. (C) Hepatic hydroxyproline content and serum ALT and AST levels. (D) Representative histology of livers from oil- and CCl4-treated mice at 3 and 7 weeks after H&E and Sirius Red staining. The Sirius Red staining was quantified by morphometric analysis. Immunohistochemical staining showing localization of α-SMA, PTH1R and PTHLH. The data for the accompanying graphs were generated from morphometric analysis. (E) Pearson correlation and linear analysis revealed that the α-SMA area was positively associated with PTHrP and PTH1R accumulation in liver sections from the fibrosis model. The relative mRNA levels are expressed as the fold induction over the control group after normalization to GAPDH, and the relative protein levels were also normalized to the GAPDH level. The data are the mean ± SD fold values over the control. *p < .05, **p < .01.
AAV9-PTHLH mice were more susceptible to liver fibrosis development in response to CCl4
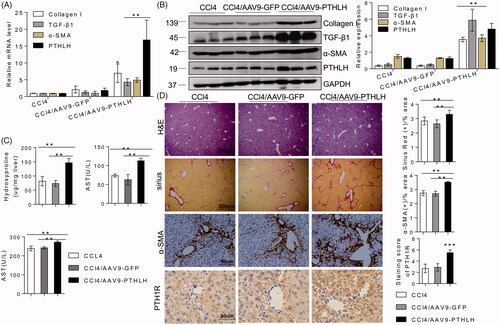
Because recombinant adeno-associated vectors exhibit a unique tropism to the liver, we generated AAV9-PTHLH to overexpress PTHLH in liver cells and generated AAV9-GFP as the control. As shown in , quantitative RT-PCR and Western blot analyses showed that PTHLH gene delivery significantly increased the expression levels of TGF-β1, collagen I and α-SMA compared with those in mice treated with CCl4 alone and in CCl4-treated AAV-GFP mice (). In addition, PTHLH gene overexpression increased liver injury and collagen bundle distribution assessed by H&E and Sirius Red staining, respectively (). Sirius Red staining showed increased fibrosis in livers infected with AAV9-PTHLH compared with the CCl4-treated alone and CCl4-treated AAV9-GFP livers. Consistently, higher hepatic α-SMA and PTH1R expression () was found in the CCl4-treated AAV9-PTHLH mice. Furthermore, the hepatic hydroxyproline content () and serum ALT and AST levels () were significantly elevated in the CCl4-treated AAV9-PTHLH mice.
Figure 3. PTHLH accelerated murine liver fibrosis induced by CCl4. Liver fibrosis was induced by CCl4 administration (1 ml/kg, 1:4 mixture with mineral oil) twice/week for 5 weeks. Mice were treated with AAV9-GFP or AAV9-PTHLH through the tail vein, at doses of 1 × 1011 plaque-forming units in 0.1 ml of saline for 2 weeks before the first CCl4 injection. (A, B) The relative mRNA and protein levels of collagen I, TGF-β1, α-SMA and PTHLH were normalized to GAPDH levels. (C) The liver hydroxyproline levels and serum ALT and AST levels. (D) Representative microphotographs of H&E-stained or Sirius Red-stained paraffin-embedded sections of liver tissues and immunohistochemical staining for α-SMA and PTH1R; the Sirius Red, PTH1R and α-SMA staining scores were determined by morphometric analysis. The relative mRNA levels are expressed as the fold induction over the level in the vehicle-treated group after normalization to GAPDH, and the relative protein levels were also normalized to the GAPDH level. The data are the mean ± SD fold values over the control. *p < .05, **p < .01.
AAV9-PTHLH induced the spontaneous development of liver fibrosis
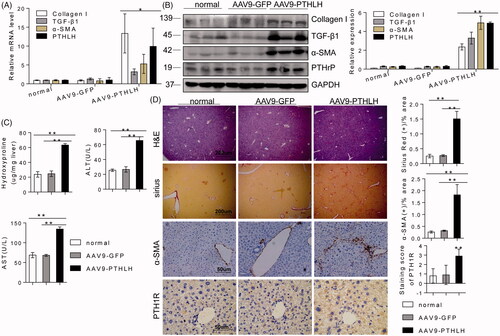
A previous study reported that mice with certain gene mutations or overexpression patterns could spontaneously develop liver fibrosis [Citation22,Citation33], and we observed that overexpression of PTHLH itself is harmful to hepatocytes. Therefore, we sought to confirm the results in vivo. In our study, interestingly, AAV9-PTHLH mice spontaneously exhibited hepatic fibrosis in our facility within 6 months. As shown in , the mRNA and protein levels of TGF-β1, collagen I and α-SMA were increased in the AAV9-PTHLH mice (). Histological examination using H&E staining revealed that 80% (4 of 5) of the AAV9-PTHLH-treated mice displayed cytoplasmic vacuolization, compared with 0% (0 of 5) of the mice in the normal and AAV9-GFP-treated groups (). Sirius Red staining showed significantly increased liver fibrosis in the AAV9-PTHLH mice (). Furthermore, immunochemical staining revealed that α-SMA and PTH1R expression () was significantly increased in the AAV9-PTHLH mice. The hepatic hydroxyproline content () and serum ALT and AST levels () were significantly increased in the AAV9-PTHLH mice.
Figure 4. AAV9-PTHLH in the liver induced hepatic fibrosis. Mice were divided into three groups, normal mice not subjected to any treatment, and an AAV9-GFP and AAV9-PTHLH group that were treated with AAV9-GFP or AAV9-PTHLH, respectively, through the tail vein at doses of 1 × 1011 plaque-forming units in 0.1 ml of saline, and these mice hosted for 6 months. (A, B) Western blot analysis and quantitative RT-PCR showing the expression levels of collagen I, TGF-β1, α-SMA and PTHLH in response to AAV9-PTHLH injection into mice. (C) The liver hydroxyproline and serum ALT and AST levels in different group. (D) Representative microphotograph of H&E and Sirius Red-stained paraffin-embedded sections of liver tissues and immunohistochemistry staining of α-SMA and PTH1R. Sirius Red, PTH1R and α-SMA staining scores were determined by morphometric analysis. The relative mRNA levels are expressed as fold induction over the level in the normal group after normalization to GAPDH, and the relative protein levels were also normalized to the GAPDH level. The data are the mean ± SD fold values over the control. *p < .05, **p < .01.
PTHLH(1–40) promoted HSC activation through the Hh signalling pathway
Recent studies have revealed that Hh signalling plays an important role in many types of fibrosis, including liver fibrosis [Citation19]. Both the Hh pathway and PTHLH are essential for regulating the proper development of digit structures [Citation36]. Therefore, we explored the relationship between PTHLH and the Hh pathway. First, we found that Shh, Gli2 and Ptch expression levels were increased in liver fibrosis (). Then, immunohistochemistry was used to characterize the cell type responsible for Shh and Gli2 upregulation. We found Shh and Gli2 positivity in portal tract cells, ballooned hepatocytes, and periportal hepatocytes (). We tested the effects of PTHLH (1–40) on Hh pathway activation. In response to PTHLH (1–40) at 1–100 nM for 12–48 h, Ptch, Shh and Gli2 protein expression levels were significantly increased in LX2 cells (). To determine the role of the Hh pathway in PTHLH-induced HSC activation, LX2 cells were exposed to cyclopamine (Smo inhibitor) and GANT61 (Gli inhibitor) before PTHLH (1–40) treatment. As shown in , inhibition of Smo and Gli activity significantly suppressed the PTHLH(1–40)-induced TGF-β1, collagen I and α-SMA production, which suggested that the Hh pathway is crucial for PTHLH (1–40)-induced HSC activation in an LX2 cell model.
Figure 5. PTHLH triggered HSC activation and ECM production through the Hh pathway. (A, B) Western blotting and quantitative RT-PCR analyses showing the expression levels of Shh, Gli2 and Ptch in liver fibrosis model mice (3 and 7 weeks). (C) Photomicrographs of immunohistochemical staining for Gli2 and Shh at 3 weeks or 7 weeks showing Gli2 and Shh accumulation in fibrotic septa of fibrotic livers from mice subjected to control treatment or the CCl4 treatment (to induce liver fibrosis). Shh and Gli2 staining scores were obtained through morphometric analysis. (D) LX2 cells were treated with various concentrations of PTHLH(1–40) for 48 h or 10 nM PTHLH(1–40) for various time periods. Western blot analysis was performed to determine the expression levels of Ptch, Gli2 and Shh. (E) LX2 cells were pretreated with the SMO inhibitor of cyclopamine (5 µM) and GLI inhibitor GANT61 (5 µM) before exposure to 10 nM PTHLH(1–40) for 48 h. Collagen I, TGF-β1, α-SMA, Smo and Gli2 expression levels were measured through Western blot analysis. The relative mRNA levels are expressed as fold induction over the level in the vehicle-treated group after normalization to GAPDH, and the relative protein levels were also normalized to the GAPDH level. The data are the mean ± SD fold values over the control. *p < .05, **p < .01, ***p < .001 versus controls.
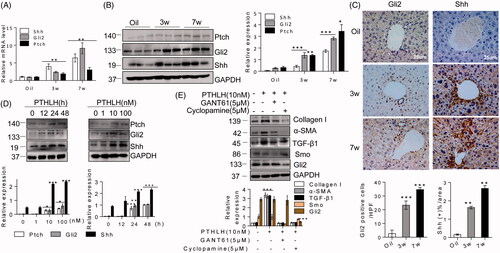
To confirm the role of the Hh pathway in PTHLH-induced liver fibrosis, we analysed Ptch, Shh and Gli2 expression in AAV9-PTHLH mouse liver fibrosis. Immunoblotting and qRT-PCR analyses showed that PTHLH gene delivery significantly increased the expressions levels of Ptch, Shh and Gli2 in CCl4 models compared with those in the vehicle controls (). Immunohistochemistry showed that Shh and Gli2 expression was increased in AAV-PTHLH mice ().
Figure 6. PTHLH treatment resulted in Shh and Gli2 accumulation in fibrotic livers. (A) Shh, Gli2 and Ptch mRNA and protein expression was observed in AAV9-GFP and AAV9-PTHLH mice after CCl4 injection for 5 weeks. (B) Quantitative RT-PCR and Western blot analyses were used to determine the mRNA and protein levels of Ptch, Gli2 and Shh in mice treated with AAV-GFP and AAV9-PTHLH and housed for 6 months. (C) Immunohistochemical staining for Shh and Gli2 and morphometric measurement of the Shh and Gli2 staining scores in AAV9-GFP and AAV9-PTHLH mice after CCl4 injection for 5 weeks. (D) Histomorphometric analysis to determine the percentage of Shh and Gli2 in mice treated with AAV-GFP and AAV9-PTHLH and housed for 6 months. The relative mRNA levels are expressed as fold induction over the level in the vehicle-treated group after normalization to GAPDH, and the relative protein levels were also normalized to the GAPDH level. The data are the mean ± SD fold values over the control. *p < .05, **p < .01, and ***p < .001 versus controls.
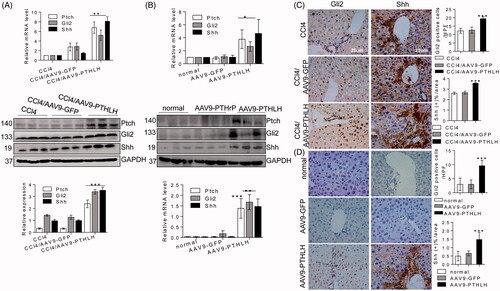
To determine whether PTHLH(1–40) could induce Hh signalling activation in vivo, mice were injected with an adeno-associated virus through the tail and hosted for 6 months. The Ptch, Shh and Gli2 mRNA and protein expression levels were significantly increased in the AAV9-PTHLH mice compared with the AAV9-GFP and vehicle control mice (). The immunohistochemistry results showed that Shh and Gli2 expression levels were upregulated in the AAV-PTHLH mice ().
PTHLH regulates the Hh signalling pathway through the PTH1R-PKC θ signalling axis
The N-terminal PTHLH(1–40) fragment usually shares homology with PTH1R ligand. Consistent with previous results [Citation37], we found that PTH1R was expressed in both HSCs and hepatocytes (). We had already found that an increase in PTHLH was associated with PTH1R upregulation in mouse liver fibrosis () and PTHLH(1–40) induced PTH1R production in a concentration- and time-dependent manner (). Therefore, to further address the functional implications of PTHLH in HSCs, we treated cells with neutralizing PTH1R antibody before PTHLH(1–40) treatment. Neutralization of endogenous PTH1R activity significantly abrogated the increases in TGF-β1, collagen I, α-SMA and Hh pathway-related proteins levels in LX-2 cells (). These results demonstrate that PTHLH triggers HSC activation through PTH1R.
Figure 7. PTHLH (1–40) induced LX2 cell activation through PTH1R-PKC θ. (A) Western blotting analysis of protein harvested from different cells was conducted to confirm PTH1R expression in HSCs and hepatocytes. (B) LX2 cells were treated with various concentrations of PTHLH (1–40) for 48 h or 10 nM PTHLH (1–40) for various time periods. Western blotting was used to examine the expression levels of PTH1R. (C) Cells were pretreated with a neutralizing antibody targeting PTH1R (5 µg/ml) prior to treatment with PTHLH(1–40), and Western blotting was used to show collagen I, TGF-β1, α-SMA, Shh, Gli2 and Ptch protein expression. (D) In PTHLH(1–40) treated LX2 cells, the expression levels of phosphorylated and total PKC isoforms (δ, α, β, ε, ζ) were analysed by Western blotting. (E) LX2 cells were pretreated with the broad-spectrum PKC inhibitor Gö6983 (10 µM) or the PKC-θ inhibitor rottlerin (10 µM) for 1 h and then incubated with 10 nM PTHLH(1–40) for 48 h. Collagen I, TGF-β1 and α-SMA expression was then measured by Western blotting. The relative protein levels were normalized to GAPDH, PKC δ, PKC α/β, PKC ε, t PKC θ, or PKC ζ levels as appropriate. The data are the mean ± SD fold values over the control. *p < .05, **p < .01, and ***p < .001 versus controls.
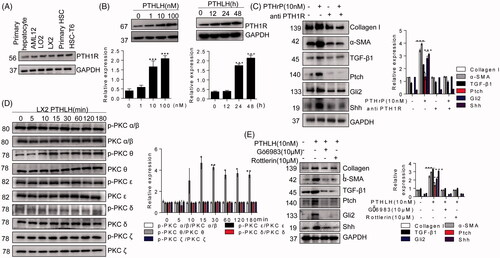
PTH1R signals typically couple with PKA, but also couple with members of the PKC pathway [Citation11]. PKC family members have been classified into three groups: the classical PKC α, β1, β2, and γ isoforms; the novel PKC δ, ε, η and θ isoforms; and the atypical PKC λ/ι and ζ isoforms. As shown in , exposure of LX2 cells to PTHLH(1–40) significantly induced PKC θ phosphorylation at 10 min. However, PTHLH(1–40) had no effect on the phosphorylation of PKC α, PKC β, PKC ε, PKC δ or PKC ζ in cultured LX2 cells. In addition, PTHLH(1–40) treatment had no effect on activation of PKA or CREB (cAMP-responsive element binding protein) (Supplementary Figure 2(A,B)). To confirm the role of PKC θ in HSC activation in response to PTHLH(1–40), LX2 cells were preincubated with Gö6983 or rottlerin, and Figure shows that PTHLH(1–40)-induced HSC activation was suppressed by Gö6983 and rottlerin to a similar extent. Meanwhile, pretreatment of LX2 cells with Gö6983 and rottlerin suppressed PTHLH(1–40)-induced Hh pathway activation ().
Discussion
Previous reports have largely focused on the role of PTHLH in bone formation and tumour progression through its binding to PTH1R [Citation38]. Synthetic human PTHLH(1–40) has been shown to increase lumbar spine bone density and has also been investigated as a potential anabolic therapy for osteoporosis [Citation39]. For example, a PTHLH(1–40) analogue (abaloparatide-SC) that increased bone mineral density and lowered the risk of osteoporosis-related fractures is in a phase 3 ACTIVE trial [Citation40]. However, PTHLH can also promote tumour development and fibrosis processes, and we cannot ignore such side effects.
Researchers showed that interaction of PTHLH with TGF-β1 promoted fibrogenesis in obstructed mouse kidneys and chronic pancreatitis [Citation41,Citation42]. Some studies have shown that in a well-differentiated human hepatoma cell line (HepG2), growth medium demonstrated N-terminal PTHLH biological activity [Citation12]. In addition, PTHLH(1–34) was induced in adult livers during endotoxaemia and was capable of stimulating the acute phase response [Citation13]. Moreover, our previous research revealed that PTHLH(1–40) can activate HSCs [Citation17]. Therefore, we predicted that PTHLH might have a similar effect in promoting hepatic fibrosis. However, the function and mechanism of PTHLH in liver fibrosis is not known. The present study clearly demonstrated that AAV9-PTHLH accelerated liver fibrosis induced in mouse livers by CCl4 treatment. Notably, AAV9 system mouse models maintained for 6 months spontaneously exhibited liver fibrosis, suggesting a causative role for PTHLH signalling in liver fibrosis.
PTHLH is involved in a wide range of physiological and developmental processes. In fact, PTHLH not only serves as an autocrine and intracrine signalling factor but also demonstrates paracrine activities. A significant correlation between PTHLH expression and hepatic fibrogenesis was found, and we observed that N-terminal PTHLH was expressed in the supernatant of damaged hepatocytes. At the same time, conditioned medium from LO2-LV-PTHLH and AML12-LV-PTHLH cells activated and promoted the fibrosis process in HSCs. We propose that while PTHLH is expressed at a low level in the parenchyma of normal adult livers, its release can be induced upon exposure to other pathogenetic factors. Our study found that hepatocytes are one of the major cell types that express PTHLH with a predominantly cytoplasmic localization. Pilot studies have shown that lentivirus-mediated PTHLH overexpression results in its extracellular release. Exogenously supplied recombinant PTHLH (1–40) showed the same impact on HSCs, raising the possibility that PTHLH released from injured hepatocytes might enhance trans-differentiation of HSCs in a paracrine manner. To the best of our knowledge, the present study is the first to demonstrate that injured hepatocytes can secrete PTHLH and promote fibrosis.
A better understanding of the mechanism responsible for PTHLH-induced HSC activation is crucial to provide novel therapies that may attenuate liver fibrosis. A series of studies have suggested that the Hh signalling pathway is a critical factor in HSC activation and liver fibrogenesis [Citation19,Citation21,Citation22,Citation43]. A relationship between the PTHLH and Hh pathways was observed in digit structures [Citation36]. In our animal model, the effect of PTHLH-triggered HSC activation and collagen fibre deposition was mediated through Hh signalling activation. As expected, the stimulatory effect of PTHLH on HSCs was significantly inhibited by the SMO inhibitor cyclopamine and the Gli-mediated transcriptional activity inhibitor GANT61, supporting the hypothesis that activation of Hh signalling is involved in PTHLH-induced HSC activation. Surprisingly, spontaneous liver fibrosis occurred in mice without exogenous inducers, and activation of Hedgehog signalling was also observed [Citation21,Citation22]. At the same time, previous research has shown that hepatic expression of Sonic hedgehog in mice can induce liver fibrosis [Citation22]. Commonly, PTHLH exerts its pathological effects through PTH1R to participate in biological responses in a paracrine manner [Citation11]. At the same time, we found PTH1R expressed in hepatocytes, and PTHLH promoted the increased PTH1R expression in a time-dependent manner. Therefore, we questioned whether PTHLH activated HSCs and the Hh pathway through a paracrine mechanism in hepatocytes. We found that PTHLH induced HSC activation and that Hh pathway activation was blocked by a neutralizing antibody targeting PTH1R. Binding of PTHLH(1–40) to PTH1R activates PKA; however, this result could also occur through PKC10. In our study, we found that PTHLH(1–40)-induced phosphorylation of PKC θ, promoted Hh pathway-related protein generation and resulted in ECM deposition.
In summary, we conclude that increased PTHLH deposition is correlated with liver fibrosis. Accumulation of PTHLH induced HSC activation and collagen fibre production, which were mainly mediated by the PTH1R-PKC θ-Hh axis. Thus, we speculate that inhibiting PTHLH in the liver might serve as an approach for blocking liver fibrosis progression.
| Abbreviations | ||
| PTHLH | = | parathyroid hormone-like hormone |
| PTHrP | = | parathyroid hormone-related protein |
| PTH1R | = | PTH/PTHLH type 1 receptor |
| PTH | = | Parathyroid hormone |
| α-SMA | = | α-smooth muscle actin |
| CCl4 | = | carbon tetrachloride |
| ECM | = | extracellular matrix |
| HSCs | = | hepatic stellate cells |
| TGF-β1 | = | transforming growth factor-β1 |
| Hh | = | Hedgehog |
| Shh | = | Sonic hedgehog |
| Gli | = | glioblastoma |
| Ihh | = | Indian hedgehog |
| HCC | = | hepatocellular carcinoma |
| AAV | = | adeno-associated virus |
| ALT | = | alanine aminotransferase |
| AST | = | aspartate aminotransferase |
| qRT-PCR | = | quantitative reverse-transcription polymerase chain reaction |
| HRP | = | horseradish peroxidase |
| TCA | = | trichloroacetic acid |
| Smo | = | Smoothened |
| PKC | = | protein kinase C |
| PKA | = | protein kinase A. |
Supplementary_materials_and_methods.docx
Download ()Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64:830–841.
- Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38:38–53.
- Bosch FX, Ribes J, Borràs J. Epidemiology of primary liver cancer. Semin Liver Dis. 1999;19:271–285.
- Yin C, Evason KJ, Asahina K, et al. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123:1902–1910.
- Shackel NA, Vadas MA, Gamble JR, et al. Beyond liver fibrosis: hepatic stellate cell senescence links obesity to liver cancer by way of the microbiome. Hepatology 2014;59:2413–2415.
- Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456.
- Ray K. Liver: hepatic stellate cells hold the key to liver fibrosis. Nat Rev Gastroenterol Hepatol. 2014;11:74.
- McCauley LK, Martin TJ. Twenty-five years of PTHrP progress: from cancer hormone to multifunctional cytokine. J Bone Miner Res. 2012;27:1231–1239.
- Martin TJ, Moseley JM, Gillespie MT. Parathyroid hormone-related protein: biochemistry and molecular biology. Crit Rev Biochem Mol.1991;26:377–395.
- Wysolmerski JJ. Parathyroid hormone-related protein: an update. J Clin Endocrinol Metab. 2012;97:2947–2956.
- Cheloha RW, Gellman SH, Vilardaga J-P, et al. PTH receptor-1 signalling-mechanistic insights and therapeutic prospects. Nat Rev Endocrinol. 2015;11:712–724.
- Li H, Seitz PK, Selvanayagam P, et al. Effect of endogenously produced parathyroid hormone-related peptide on growth of a human hepatoma cell line (Hep G2). Endocrinology 1996;137:2367–2374.
- Funk JL, Moser AH, Grunfeld C, et al. Parathyroid hormone-related protein is induced in the adult liver during endotoxemia and stimulates the hepatic acute phase response. Endocrinology 1997;138:2665–2673.
- Ortega A, Ramila D, Ardura JA, et al. Role of parathyroid hormone-related protein in tubulointerstitial apoptosis and fibrosis after folic acid-induced nephrotoxicity. Jasn. 2006;17:1594–1603.
- Ardura JA, Rayego-Mateos S, Ramila D, et al. Parathyroid hormone-related protein promotes epithelial-mesenchymal transition. JASN. 2010;21:237–248.
- Lorenzo O. Angiotensin II increases parathyroid hormone-related protein (PTHrP) and the type 1 PTH/PTHrP receptor in the kidney. J Am Soc Nephrol. 2002;13:1595–1607.
- Liang FFL, Li LX, Xue MM, et al. Activated effects of parathyroid hormone-related protein on human hepatic stellate cells. PLoS One. 2013;8:e76517.
- He S, Xue M, Liu C, et al. Parathyroid hormone-like hormone induces epithelial-to-mesenchymal transition of intestinal epithelial cells by activating the runt-related transcription factor 2. Am J Pathol. 2018;188:1374–1388.
- Omenetti A, Choi S, Michelotti G, et al. Hedgehog signaling in the liver. J Hepatol. 2011;54:366–373.
- Hu L, Lin X, Lu H, et al. An overview of hedgehog signaling in fibrosis. Mol Pharmacol. 2015;87:174–182.
- Yang L, Wang Y, Mao H, et al. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J Hepatol. 2008;48:98–106.
- Chung SI, Moon H, Ju HL, et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J Hepatol. 2016;64:618–627.
- Witek RP, Yang L, Liu R, et al. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology 2009;136:320–330.
- Chen Y, Choi SS, Michelotti GA, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012;143:1319–1329.
- Kobayashi T, Soegiarto DW, Yang Y, et al. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J Clin Invest. 2005;115:1734–1742.
- Zhang X, Cheng Q, Wang Y, et al. Hedgehog signaling in bone regulates whole-body energy metabolism through a bone-adipose endocrine relay mediated by PTHrP and adiponectin. Cell Death Differ. 2017;24:225–237.
- Anuja GI, Shine VJ, Latha PG, et al. Protective effect of ethyl acetate fraction of Drynaria quercifolia Against CCl4 induced rat liver fibrosis via Nrf2/ARE and NFkappaB signalling pathway. J Ethnopharmacol. 2018;216:79–88.
- Chang J, Lan T, Li C, et al. Activation of Slit2-Robo1 signaling promotes liver fibrosis. J Hepatol. 2015;63:1413–1420.
- Wu X, Wu X, Ma Y, et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat Commun. 2016;7:13498.
- Paik YH, Iwaisako K, Seki E, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011;53:1730–1741.
- Zhang L-L, Huang S, Ma X-X, et al. Angiotensin(1–7) attenuated Angiotensin II-induced hepatocyte EMT by inhibiting NOX-derived H2O2-activated NLRP3 inflammasome/IL-1β/Smad circuit. Free Rad Biol Med. 2016;97:531–543.
- He S, Guo W, Deng F, et al. Targeted delivery of microRNA 146b mimic to hepatocytes by lactosylated PDMAEMA nanoparticles for the treatment of NAFLD. Artif Cells Nanomed Biotechnol. 2018;46:217–228.
- Kao YCC, Jawan B, Chung YH, et al. Upregulation of hepatoma-derived growth factor is involved in murine hepatic fibrogenesis. J Hepatol. 2010;52:96–105.
- Koontz L. TCA precipitation. Meth Enzymol. 2014;541:3–10.
- Xu X, Sun S, Xie F, et al. Advanced oxidation protein products induce epithelial-mesenchymal transition of intestinal epithelial cells via a PKC delta-mediated, redox-dependent signaling pathway. Antioxid Redox Signal. 2017;27:37–56.
- Amano K, Densmore M, Fan Y, et al. Ihh and PTH1R signaling in limb mesenchyme is required for proper segmentation and subsequent formation and growth of digit bones. Bone 2016;83:256–266.
- Xie G, Choi SS, Syn WK, et al. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut 2013;62:299–309.
- Li JK, Huang DC, Siegel PM, et al. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J Clin Invest. 2011;121:4655–4669.
- Augustine M, Horwitz MJ. Parathyroid hormone and parathyroid hormone-related protein analogs as therapies for osteoporosis. Curr Osteoporos Rep. 2013;11:400–406.
- Esbrit P, Herrera S, Portal-Núñez S, et al. Parathyroid hormone-related protein analogs as osteoporosis therapies. Calcif Tissue Int. 2016;98:359–369.
- Ardura JA, Berruguete R, Ramila D, et al. Parathyroid hormone-related protein interacts with vascular endothelial growth factor to promote fibrogenesis in the obstructed mouse kidney. Am J Physiol Renal Physiol. 2008;295:F415–25.
- Bhatia V, Cao Y, Ko TC, et al. Parathyroid hormone-related protein interacts with the transforming growth factor-β/bone morphogenetic protein-2/gremlin signaling pathway to regulate proinflammatory and profibrotic mediators in pancreatic acinar and stellate cells. Pancreas 2016;45:659–670.
- Guy CD, Suzuki A, Zdanowicz M, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012;55:1711–1721.