
Abstract
Background
The anti-inflammatory function of microRNA-124 (miR-124) has been a matter of extensive studies in the last few years. Although LINC00305 regulates biological activities by acting as a miR sponge, it is still unexplored whether LINC00305 is involved in inflammation by regulating miR-124.
Methods
Cell viability and apoptosis were evaluated with commercial kits, cell counting kit-8 (CCK-8) and Annexin V-fluorescein isothiocyanate (FITC) kit, respectively. LINC00305, miR-124 and mRNA levels were quantified by quantitative reverse transcription PCR (qRT-PCR). Protein level was assessed with Western blot assay and enzyme-linked immunosorbent assay (ELISA).
Results
The expression of LINC00305 was up-regulated by lipopolysaccharide (LPS). LINC00305 overexpression further suppressed the cell viability, promoted apoptosis and induced inflammation in LPS-treated ATDC5 cells while its silence enhanced the cell viability, inhibited apoptosis and ameliorated inflammation. miR-124 was negatively regulated by LINC00305 and its overexpression abolished the effects of LINC00305 in the presence of LPS. LINC00305 further triggered the Notch/nuclear factor kappa B (NF-κB) signalling pathway in LPS-treated ATDC5 cells and this activation was abrogated when ATDC5 cells overexpressed miR-124.
Conclusion
LINC00305 might emerge as a novel suppressor for miR-124. LINC00305-caused miR-124 silence compromises ATDC5 cell viability and ultimately results in inflammatory insults by activating Notch/NF-κB pathway.
Introduction
Osteoarthritis (OA) has been broadly viewed as a prevalent joint disease and characterizes pain and degenerative lesions. OA is classically considered as a multifactorial disease. Particularly, ageing [Citation1] and trauma [Citation2] are the main risk factors. Other factors, including obesity, inflammation, gender, hormones, metabolic syndrome and high blood pressure [Citation3,Citation4], are devoted to OA development and cause a more severe outcome. As a consequence, there remains an enormous need to explore innovative therapeutic interventions. Accumulating evidence indicates that inflammation drives many pathologic changes [Citation5]. Not only the local inflammation in the joint but also a systemic inflammation might be associated with OA pathogenesis [Citation6,Citation7]. Noncoding RNAs, for instance, long non-coding RNAs (lncRNAs) and microRNAs (miRs), are crucial mediators in the progression and management of the inflammation [Citation8].
A recent investigation proposed that miscellaneous characteristics of posttraumatic OA apparently vary with genetic composition, suggesting that phenotypic modulation is associated with genetic variability [Citation9]. A pivotal role of noncoding RNA has been recognized in OA, such as lncRNAs and miRs [Citation10]. It has been verified that lncRNAs act as a sponge for miRs to mediate biological progresses in OA [Citation11,Citation12]. Mechanically, miRs serve as the important regulator of gene expression at a post-transcriptional level [Citation13,Citation14]. Of particular interest is a crosstalk between lncRNAs and miRs in the physiologic and pathologic processes [Citation15]. The sponge/decoy function of lncRNAs on miRs plays a central role in regulating the expression of transcription factors [Citation16,Citation17].
Most recently, the up-regulated expression of LncRNA LINC00305 (LINC00305) was observed in atherosclerosis and LINC00305 is primarily overexpressed by monocytes, suggesting that LINC00305 participates in monocyte-mediated inflammation [Citation18]. Bioinformatics analysis revealed that LINC00305 mediates apoptosis by acting as a miR sponge [Citation19]. Furthermore, the anti-inflammatory role of miR-124 has been confirmed in animal models [Citation20–22]. Besides, in the progress of fracture healing, miR-124 was reported to be sponged by lncRNA and then exhibits a functional role [Citation23]. However, it is still unexplored whether LINC00305 involves in the progression of inflammation via mediating miR-124 in OA.
In this study, we demonstrated a vital function of LINC00305 in regulating miR-124 response to lipopolysaccharide (LPS) stimulus. Through triggering inflammation in ATDC5 cells, we detected an increase of LINC00305, which exerted a pro-inflammatory effect in the presence of LPS. Moreover, the down-regulation of miR-124 by LINC00305 might contribute to the activation of Notch/nuclear factor kappa B (NF-κB) signalling cascade.
Materials and methods
Cell culture and stimulation
ATDC5 cells were purchased from Riken Bio Resource Center Cell Bank (RIKEN) (Cell No: RCB0565) (Tsukuba, Japan). ATDC5 cells were cultured according to the supplier’s protocol. In short, ATDC5 cells were maintained in Dulbecco’s modified Eagle medium/nutrient mixture F-12 medium (DMEM/HamF12) (Thermo Fisher Scientific, Waltham, MA, USA) containing 5% fetal bovine serum (FBS) (Gibco, Gaithersburg, MD, USA), 100 U/mL penicillin (Sigma, St. Louis, MO, USA), and 100 μg/mL streptomycin (Sigma) in a humidified incubator containing 5% CO2 at 37 °C. ATDC5 cells were used in the fifth or tenth passage of culture. For LPS stimulation, cells were seeded in a 75 cm2 flask and cultured in media containing LPS (Sigma) at different concentrations (0, 1, 2, 5, and 10 μg/mL) for 12 h.
CCK-8 assay
Cell viability was assessed with a cell counting kit-8 (CCK-8) (Dojindo Molecular Technologies, Gaithersburg, MD, USA). In brief, cells were seeded in 96-well plate with a density of 5000 cells per well. After stimulation with LPS for 12 h, ATDC5 cells were incubated for 1 h with CCK-8 solution. The absorbance was evaluated at 450 nm with a Microplate Reader (Bio-Rad, Hercules, CA). ATDC5 cells cultured without LPS were applied to normalize cell viability.
Apoptosis examination
An Annexin V-fluorescein isothiocyanate (FITC) apoptosis analysis kit (Item No: ab14085; Abcam, Cambridge, MA, USA) was applied to analyze apoptosis referring to manufacturer’s description. Briefly, 5 × 105 cells were collected, trypsinized with trypsin (Pierce, Appleton, WI, USA) and washed with phosphate buffered saline (PBS) (Sigma). Subsequently, ATDC5 cells were stained with Annexin V-FITC and propidium iodide (PI) in the presence of RNase A (Sigma) for 5 min after stimulation with LPS. Finally, ATDC5 cells were visualized with a FACScan flow cytometer (Beckman Coulter, Fullerton, CA, USA).
RNA isolation and quantitative reverse transcription PCR (qRT-PCR)
Mouse interleukin-6 (IL-6) and tumour necrosis factor alpha (TNF-α) mRNA levels, as well as LINC00305 RNA level, were determined using SYBR Green-based quantitative PCR. Total RNA was extracted with TRIzol reagent kit (Invitrogen, Carlsbad, CA, USA) according to the user’s guide. We performed a qRT-PCR in a thermal cycler using a One Step SYBR PrimeScript RT-PCR Kit (TaKaRa Biotechnology, Kyoto, Japan) and specific primers. OLIGO Primer Analysis Software (Molecular Biology Insights, Cascade, CO, USA) was applied to design primers. Results of comparative qRT-PCR were normalized with β-actin. As for the expression of miR-124, RNA was reversely transcribed using random hexamer and a Taqman MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, PCR was conducted with Taqman Universal Master Mix II (Thermo Fisher Scientific). The expression of miR-124 was normalized to U6 with 2−△△Ct method.
Transfection and selection of stable transfected cells
To silence LINC00305 gene, the short hairpin (sh)-RNA was ligated into pGPU6/Neo plasmid (GenePharma, Shanghai, China) (sh-LINC00305) and transfected into ATDC5 cells with Lipofectamine 3000 reagent (Invitrogen, Waltham, MA, USA) to directly target LINC00305 according to the supplier’s recommendations. sh-LINC00305 and sh-RNA negative control (sh-NC) were all synthesized by GenePharma. In order to up-regulate LINC00305, the sequence of LINC00305 cDNA was ligated into pcDNATM3.1/V5-His-TOPO (Invitrogen) and transfected into ATDC5 cells with Lipofectamine 3000. Cells were cultured in media containing geneticin selective antibiotic (Invitrogen) for selection. In order to up-regulate miR-124, we used miR-124 mimic (GenePharma) to simulate endogenous miR-124. After transfection with Lipofectamine 3000 for 48 h, ATDC5 cells were collected.
Enzyme-linked immunosorbent assay (ELISA) for protein expression
To investigate the expression of IL-6 and TNF-α at protein level, we applied Mouse IL-6 and TNF-α Quantikine ELISA kits (R&D Systems, Abingdon, UK) with reference to the supplier’s description.
Western blot
After LPS treatment, the cells were washed with PBS and lysed by RIPA lysis buffer (Beyotime, Shanghai, China) in the presence of protease inhibitor (Roche Applied Science, Indianapolis, IN, USA) for protein extraction. The extracted protein was quantified with BCATM protein assay kit (Pierce). A Bio-Rad Bis-Tris Gel system was established with reference to the manufacturer’s instructions (Bio-Rad, Hercules, CA, USA). Protein was transferred onto polyvinylidene difluoride (PVDF) membrane (Millipore, MA, USA). The appropriate antibodies (anti-poly(ADP-ribose) polymerase [PARP] diluted 1:1000, anti-cleaved PARP diluted at 1:1000, anti-IL-6 diluted at 1:1000, anti-TNF-α diluted at 1:1000, all purchased from Abcam; anti-caspase-3 diluted at 1:1000, anti-caspase-9 diluted at 1:1000, anti-β-actin diluted at 1:1000, anti-Notch1 diluted at 1:1000, anti-Notch2 diluted at 1:1000, anti-total (t)-inhibitor of nuclear factor kappa-B alpha [IκBα] diluted at 1:1000, anti-phospho (p)-IκBα diluted at 1:1000, anti-t-p65 diluted at 1:1000, anti-p-p65 diluted at 1:1000, all purchased from Cell Signaling Technology, Danvers, MA, USA) were incubated with proteins at 4 °C overnight and then incubated with rabbit anti-mouse IgG conjugated by horseradish peroxidase (Cell Signaling Technology) diluted at 1:5000. The signals were captured with Bio-Rad ChemiDocTM XRS system and analyzed with Image software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data was the mean of three independent experiments, each conducted in triplicate. The data was reported as mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism 8.0 software (GraphPad, San Diego, CA, USA). Two-tailed Student’s t-test was performed. A one-way analysis of variance (ANOVA) was conducted followed by the Bonferroni’s test to compare the difference between multiple groups. The p values < .05 were considered to indicate a statistical significance.
Results
LINC00305 was elevated in LPS-treated ATDC5 cells
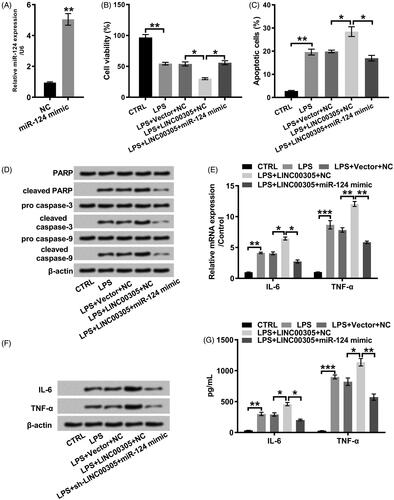
After stimulation with LPS, an obvious inflammation lesion was noticed in ATDC5 cells. Specifically, the viability of ATDC5 cells was attenuated by LPS in a dose-dependent relationship (2, 5, and 10 μg/mL) (p < .005, p < .01 or p < 0.001) (). We stimulated ATDC5 cells with LPS at a concentration of 5 μg/mL because the percentage of cell viability was half-repressed. Besides, the proportion of apoptotic cells was visibly elevated by LPS (p < .01) (). The cleavage of PARP, caspase-3 and caspase-9 further suggested LPS induced the significant insults in ATDC5 (). Additionally, inflammatory progress caused by LPS was evidenced by the overexpression of IL-6 (p < .01) and TNF-α (p < .001) at mRNA () and protein (,F)) levels. Of note, LINC00305 was enhanced by LPS in a concentration-dependent manner (p < .05 or p < .01) (), suggesting that the LPS-caused inflammatory insults might be ascribed to the up-regulation of LINC00305.
Figure 1. LPS promoted inflammatory insults in ATDC5 cells. (A) ATDC5 cells were stimulated with LPS at the indicated concentrations (0, 1, 2, 5, and 10 μg/mL) for 12 h. The cell viability was analyzed with a CCK-8 assay. ATDC5 cells were treated with 5 μg/mL LPS for 12 h in the downstream experiments. (B) Apoptotic cells were stained with Annexin-FITC/PI and then observed with a flow cytometry. (C) Immunoblot assay was conducted to evaluate the expression of apoptosis-related proteins. (D) Inflammatory factors were quantified by qRT-PCR assay at mRNA level. (E) Immunoblot assay was performed to detect inflammatory factors. (F) ELISA was performed to assess the protein expression of inflammatory factors. Data was the mean of three independent experiments, each conducted in triplicate, ±SD. nsp > .05; *p < .05; **p < .01; ***p < .001. ns: not significant; LPS: lipopolysaccharide; CTRL: control; PARP: poly(ADP-ribose) polymerase; IL-6: interleukin-6; TNF-α: tumor necrosis factor alpha; CCK-8: cell counting kit-8; FITC/PI: fluorescein isothiocyanate/propidium iodide; qRT-PCR: quantitative reverse transcription PCR; ELISA: enzyme-linked immuno sorbent assay.
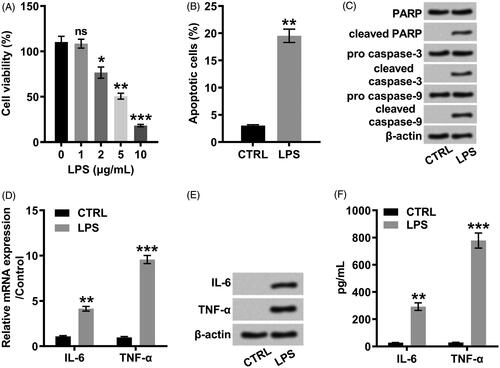
Figure 2. LPS positively modulated the expression of LINC00305 in a concentration-dependent relationship. LINC00305 was quantified by qRT-PCR. ATDC5 cells were treated by LPS at the indicated concentrations (0, 1, 2, and 5 μg/mL) for 12 h. Data was the mean of three independent experiments, each conducted in triplicate, ±SD. nsp > .05; *p < .05; **p < .01. ns: not significant; LPS: lipopolysaccharide; qRT-PCR: quantitative reverse transcription PCR.
LINC00305 participated in LPS-induced inflammatory progress
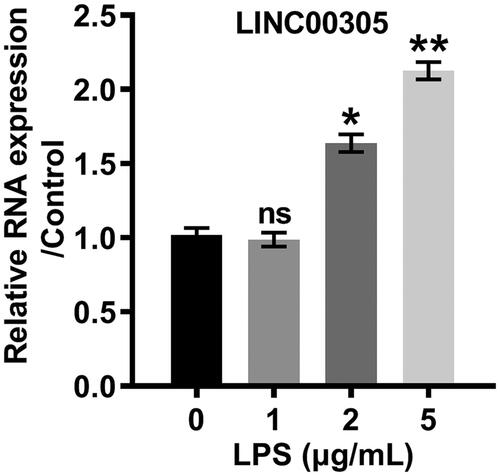
For elucidating that LINC00305 was a central mediator in the inflammation, we artificially elevated (p < .001) or silenced (p < .01) LINC00305 (). Correspondingly, we observed that LPS further repressed cell viability in LINC00305-transfected cells, while elevated cell viability in LINC00305-deficient cells (both p < .05) (). As for apoptosis, LPS facilitated apoptosis process in LINC00305-transfected cells in comparison with the un-transfected cells (p < .01) (). However, the apoptosis was mitigated by LPS in LINC00305-deficient ATDC5 cells (p < .01) (). In line with apoptosis profile, the cleavage of PARP, caspase-3 and caspase-9 was remarkably promoted by LPS in ATDC5 cells which were deficient in LINC00305, whereas retarded in LINC00305-transfected ATDC5 cells (). To investigate the function of LINC00305 in inflammation, we analyzed the expression of IL-6 and TNF-α at mRNA and protein levels. LPS-induced IL-6 (p < .05) and TNF-α (p < .01) was notably higher in ATDC5 cells than LINC00305-overexpressed ATDC5 cells (). However, LINC00305-deficient cells expressed IL-6 (p < .05) and TNF-α (p < .01) at a lower mRNA level after stimulation with LPS in comparison with sh-NC-transfected ATDC5 cells (). Consistently, when ATDC5 cells were transfected with LINC00305 the obvious up-regulation of IL-6 and TNF-α was observed at protein levels in the presence of LPS administration (both p < .05) (). By contrast, LINC00305 silence evoked the remarkable down-regulation of IL-6 and TNF-α (p < .05) (). All these results indicated that LINC00305 participated in the LPS-induced inflammatory damages.
Figure 3. LINC00305 overexpression was necessary for LPS-mediated inflammatory damages in ATDC5 cells. (A) ATDC5 cells were transfected with LINC00305 or sh-LINC00305. The expression of LINC00305 was analyzed by qRT-PCR. ATDC5 cells were transfected and then treated with 5 μg/mL LPS for 12 h. (B) After transfection and LPS administration, ATDC5 cells viability was examined by CCK-8 assay. (C) Apoptotic cells were observed by a flow cytometry after stained by Annexin V-FITC/PI. (D) Immunoblot assay was used to determine apoptosis-related proteins. (E) Inflammatory factors at mRNA level were examined by qRT-PCR. (F) Immunoblot assay was performed to detect inflammatory factors. (G) Inflammatory factors at protein level were quantified by ELISA. Data was the mean of three independent experiments, each conducted in triplicate, ±SD. nsp > .05; *p < .05; **p < .01; ***p < .001. sh: short hairpin; CTRL: control; LPS: lipopolysaccharide; NC: negative control; PARP: poly(ADP-ribose) polymerase; IL-6: interleukin-6; TNF-α: tumor necrosis factor alpha; qRT-PCR: quantitative reverse transcription PCR; CCK-8: cell counting kit-8; FITC/PI: fluorescein isothiocyanate/propidium iodide; ELISA: enzyme-linked immuno sorbent assay.
MiR-124 down-regulation was required for LINC00305 to involve in LPS-induced inflammation
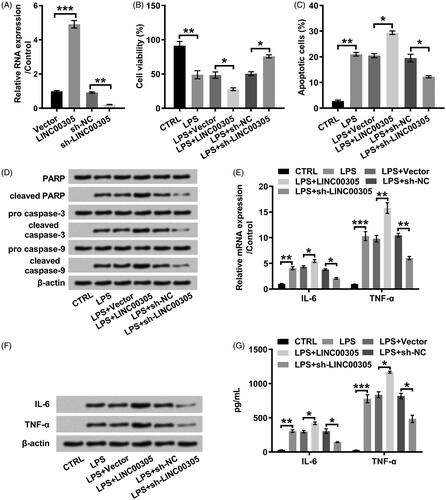
We also determined the function of LINC00305 on miR-124 level. MiR-124 expression was obviously decreased in ATDC5 cells transfected with LINC00305 (p < .05) (). Conversely, the up-regulation of miR-124 was noticed in ATDC5 cells transfected with sh-LINC00305 (p < .05) (). To investigate whether down-regulation of miR-124 was required for LINC00305 to participate in LPS-induced inflammatory injury, we artificially promoted exogenous expression of miR-124 by transfection with miR-124 mimic (p < .01) (). Obviously, the cell viability was restored in LPS-treated ATDC5 cells co-transfected with LINC00305 and miR-124 mimic in relative to singly transfected with LINC00305 (p < .05) (). We also observed that miR-124 overexpression visibly blocked apoptotic progress (p < .05) () which was evidenced by the cleavage of PARP, caspase-3 and caspase-9 (). Additionally, miR-124 overexpression notably impeded the expression of IL-6 (both p < .05) and TNF-α (both p < .01) at mRNA and protein levels (). Together, our results suggested that LINC00305 down-regulated miR-124 and then contributed to inflammatory damages of ATDC5 cells in presence of LPS administration.
Figure 4. miR-124 was positively modulated by LINC00305. ATDC5 cells were transfected with LINC00305 or sh-LINC00305. miR-124 expression was evaluated by qRT-PCR. Data was the mean of three independent experiments, each conducted in triplicate, ±SD. *p < .05. qRT-PCR: quantitative reverse transcription PCR; miR-124: microRNA-124.
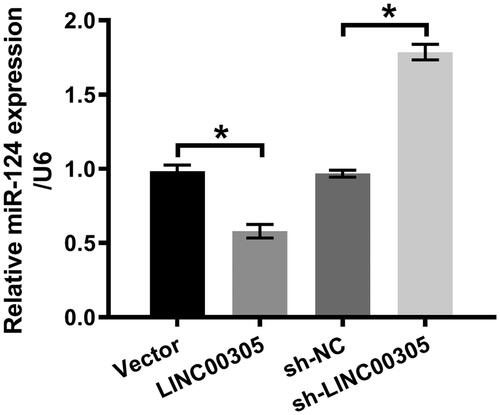
Figure 5. miR-124 down-regulation was required for LINC00305 to involve in inflammatory damages induced by LPS. (A) miR-124 mimic was transfected into ATDC5 cells and qRT-PCR was applied to examine miR-124 level. ATDC5 cells were transfected with LINC00305 and miR-124 mimic or NC and then treated with 5 μg/mL LPS for 12 h. (B) Cell viability and (C) apoptosis was assessed by CCK-8 and Annexin V-FITC/PI combined with a flow cytometry, respectively. (D) Immunoblot assay was conducted for quantification of proteins associated with apoptosis. The expression of inflammatory factors was evaluated by qRT-PCR, Western blot and ELISA at (E) mRNA and (F, G) protein levels. Data was the mean of three independent experiments, each conducted in triplicate, ±SD. *p < .05; **p < .01; ***p < .001. NC: negative control; miR-124: microRNA-124; CTRL: control; LPS: lipopolysaccharide; PARP: poly(ADP-ribose) polymerase; IL-6: interleukin-6; TNF-α: tumor necrosis factor alpha; qRT-PCR: quantitative reverse transcription PCR; CCK-8: cell counting kit-8; FITC/PI: fluorescein isothiocyanate/propidium iodide; ELISA: enzyme-linked immuno sorbent assay.
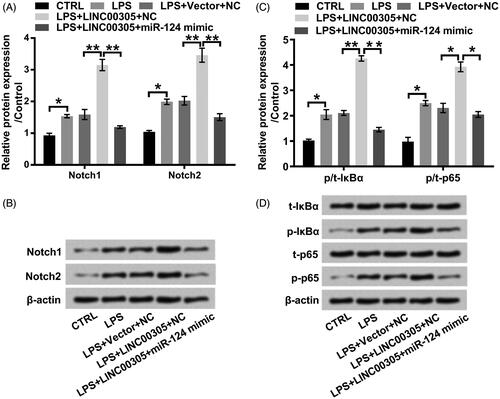
LINC00305-evoked activation of Notch/NF-κB pathway was blocked by miR-124 in the presence of LPS
Strikingly, LPS elevated Notch1 and Notch2 at protein levels (both p < .05) (), suggesting that Notch/NF-κB pathway is activated by LPS. The effect of LPS on Notch1 and Notch2 was aggrandized in LINC00305-transfected ATDC5 cells (both p < .01) (). However, LPS down-regulated Notch1 and Notch2 in LINC00305 and miR-124 co-transfected ATDC5 cells in comparison with only transfected with LINC00305 (both p < .01) (). As for the phosphorylation of IκBα and p65, we noticed that LPS further enhanced phosphorylated IκBα (p < .01) and p65 (p < .05) in LINC00305-transfected ATDC5 cells compared with untransfected cells (). However, this phosphorylation was retarded in ATDC5 cells co-transfected with LINC00305 and miR-124 mimic compared with only transfected with LINC00305 (p < .05 or p < .01) (). Conclusively, LPS resulted in the activation of Notch/NF-κB pathway through facilitating LINC00305 expression and then repressing miR-124 expression.
Figure 6. LINC00305-triggered activation of Notch/NF-κB pathway is abolished by miR-124 overexpression in the presence of LPS. ATDC5 cells were transfected with LINC00305 and miR-124 mimic or NC and then treated with 5 μg/mL LPS for 12 h. Immunoblot assay was performed to detect (A, B) Notch1, Notch2, (C, D) t-IκBα, p-IκBα, t-p65, and p-p65. Data was the mean of three independent experiments, each conducted in triplicate, ±SD. *p < .05; **p < .01. NC: negative control; miR-124: microRNA-124; CTRL: control; LPS: lipopolysaccharide; t: total; p: phospho; IκBα: inhibitor of nuclear factor kappa-B alpha.
Discussion
The role of LINC00305 in inflammation has been partially defined [Citation18]. Although LINC00305 is identified as a novel pro-inflammatory lncRNA, it is possible that LINC00305 has an indirect role in inflammation and might exert a pro-inflammatory activity dependent on the regulation of miRs. Correspondingly, we presented a regulatory mechanism associated with LINC00305 (Supplementary Figure 1).
Inflammatory mechanisms are hypothesized to conduce to the risk of OA onset and progression after injury [Citation24]. In the pathogenesis of knee OA, the expression of pro-inflammatory cytokines IL-6 and TNF-α is associated with cartilage loss, suggesting that inflammation plays a role [Citation25]. ATDC5 cell line was selected in our studies because ATDC5 is an excellent in vitro model to investigate molecular mechanisms [Citation26]. After LPS administration, we observed a significant decrement of cell viability, enhancement of apoptosis and the overexpression of IL-6 and TNF-α. Exogenous IL-6 promotes calcium-containing crystal formation and up-regulatory expression of genes involved in calcification, which finally result in experimental OA [Citation27]. As for the function of TNF-α, the activation of the apoptotic pathway is triggered by TNF-α in human chondrocytes [Citation28]. We considered that the inflammatory insult cell model was established with ATDC5 cells in the present study.
The transcriptome of lncRNAs has been studied in cartilage and its significant differential expression might play a role in cartilage homoeostasis [Citation29]. Arguably, a set of lncRNAs emerge as the potential enhancers or suppressors by transcriptionally modulating gene expression in response to inflammation [Citation30]. The accumulation of LINC00305 was detected in atherosclerotic plaques and monocytes and its up-regulation triggers the activation of cytokine expression [Citation18]. In our current study, LPS drove the overexpression of LINC00305 in ATDC5 cells in a dose-dependent manner. In addition, ATDC5 cells overexpressed LINC00305 in response to inflammation while its overexpression contributed to an excessive inflammatory insult. In fact, lncRNAs expression are generally altered following cytokine stimulus and in turn, mediate the inducible expression of cytokines [Citation31]. Although major research efforts have been centered on the function of LINC00305 on inflammation, relatively little is known about a more fundamental aspect of the potential mechanisms.
Another noncoding RNA miRs are also emphasized to play functional roles in chronic inflammation [Citation32], for instance, in OA [Citation33]. Emerging findings from studies of cell and mouse models suggested that miR-124 apparently inhibits inflammation in injury [Citation20,Citation21]. Of note, miR-124 ameliorates adjuvant-induced arthritis in the rat by post-transcriptionally suppressing cytokine expression [Citation22]. Besides, the amelioration of collagen-induced arthritis by vitamin D might be ascribed to miR-124-mediated suppression of IL-6 [Citation34]. Actually, we evaluated whether up-regulated miR-124 was able to moderate LINC00305-elicited inflammatory lesions in ATDC5 cells. Our results clearly demonstrated that LINC00305-driven inflammation was strongly attenuated in miR-124-overexpressed ATDC5 cells.
Since LINC00305 plays a pivotal role in the inflammatory progress of OA, counteracting its pro-inflammatory activities with miR-124 might be a candidate therapy. Here, we provided clear evidence that LINC00305 overexpression repressed miR-124 expression, vice versa, LINC00305 silence facilitated miR-124 expression. At the regulatory mechanism aspect, a multiplayer regulatory circuitry objectively controls this progress. LncRNA might function as a key competing endogenous RNA to link the regulatory function of miRs on the core transcription factors [Citation16]. Similar results were provided that lncRNA emerges as sponges for miRs to regulate the expression of transcription factors [Citation17]. In the hypoxia-caused apoptotic progress, an apoptosis-promoting function of LINC00305 was proposed in human umbilical vein endothelial cells and bioinformatics analysis confirmed that LINC00305 acts as a miR-136 sponge since there is a complementary base-pairing reaction between LINC00305 3’-untranslated region (3’-UTR) and miR-136 [Citation19].
The physiologic and pathological phenotypes are modulated by complex multi-functional signalling regulatory circuitry. The blockage of the Notch signalling pathway by the restoration of the miR level is conducive to the rescue of joint degeneration [Citation35]. An intra-articular administration of IκBα kinase inhibitor represses the phosphorylation and then prevents deleterious inflammation [Citation36]. Signalling pathway analysis was also conducted in our study. Our results suggested that LINC00305 activated Notch/NF-κB pathway by down-regulating miR-124 in the presence of LPS. Nonetheless, the underlying mechanisms require further research. Another study made a novel observation that miR-124 suppresses p65 expression by directly binding to its 3’-UTR [Citation37] and this study might shed new light on the pathogenesis of OA.
Conclusion
On the basis of our results and previously published results, inflammatory damages might be ascribed to ATDC5-overexpressed LINC00305 which was evoked by LPS. LINC00305 may emerge as a novel suppressor for miR-124. LINC00305-caused miR-124 silence compromises ATDC5 cell viability and ultimately results in inflammatory insults. Consequently, targeted strategies proposed to restore miR-124 level appear to be an absorbing therapy for OA.
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Deshpande BR, Katz JN, Solomon DH, et al. Number of persons with symptomatic knee osteoarthritis in the US: impact of race and ethnicity, age, sex, and obesity. Arthritis Care & Res. 2016;68:1743–1750.
- Thomas AC, Hubbard-Turner T, Wikstrom EA, et al. Epidemiology of posttraumatic osteoarthritis. J Athl Train. 2017;52:491–496.
- Niu J, Clancy M, Aliabadi P, et al. Metabolic syndrome, its components, and knee osteoarthritis: the Framingham Osteoarthritis Study. Arthritis & Rheumatol. 2017;69:1194–1203.
- Silverwood V, Blagojevic-Bucknall M, Jinks C, et al. Current evidence on risk factors for knee osteoarthritis in older adults: a systematic review and meta-analysis. Osteoarthr Cartil. 2015;23:507–515.
- Robinson WH, Lepus CM, Wang Q, et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12:580–592.
- Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthr Res Ther. 2017;19:18.
- Wang T, He C. Pro-inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine & Growth Fact Rev. 2018;44:38–50.
- Marques-Rocha JL, Samblas M, Milagro FI, et al. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J: Offic Publ Feder Am Soc Exper Biol. 2015;29:3595–3611.
- Chinzei N, Rai MF, Hashimoto S, et al. Evidence for genetic contribution to variation in posttraumatic osteoarthritis in mice. Arthr Rheumatol. 2019;71:370–381.
- Barter MJ, Young DA. Epigenetic mechanisms and non-coding RNAs in osteoarthritis. Curr Rheumatol Rep. 2013;15:353.
- Li Y, Li S, Luo Y, et al. LncRNA PVT1 regulates chondrocyte apoptosis in osteoarthritis by acting as a sponge for miR-488-3p. DNA and Cell Biol. 2017;36:571–580.
- Zhang G, Wu Y, Xu D, et al. Long noncoding RNA UFC1 promotes proliferation of chondrocyte in osteoarthritis by acting as a sponge for miR-34a. DNA and Cell Biol. 2016;35:691–695.
- Hu G, Zhao X, Wang C, et al. MicroRNA-145 attenuates TNF-alpha-driven cartilage matrix degradation in osteoarthritis via direct suppression of MKK4. Cell Death Dis. 2017;8:e3140.
- Li ZC, Han N, Li X, et al. Decreased expression of microRNA-130a correlates with TNF-alpha in the development of osteoarthritis. Int J Clin Exper Pathol. 2015;8:2555–2564.
- Yoon JH, Abdelmohsen K, Gorospe M. Functional interactions among microRNAs and long noncoding RNAs. Sem Cell & Dev Biol. 2014;34:9–14.
- Wang Y, Xu Z, Jiang J, et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25:69–80.
- Cesana M, Cacchiarelli D, Legnini I, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011;147:358–369.
- Zhang DD, Wang WT, Xiong J, et al. Long noncoding RNA LINC00305 promotes inflammation by activating the AHRR-NF-kappaB pathway in human monocytes. Sci Rep. 2017;7:46204.
- Zhang BY, Jin Z, Zhao Z. Long intergenic noncoding RNA 00305 sponges miR-136 to regulate the hypoxia induced apoptosis of vascular endothelial cells. Biomed Pharmacother. 2017;94:238–243.
- Huang S, Ge X, Yu J, et al. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J: Offic Publ Feder Am Soc Exper Biol. 2018;32:512–528.
- Sun Y, Li Q, Gui H, et al. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. 2013;23:1270–1283.
- Nakamachi Y, Ohnuma K, Uto K, et al. MicroRNA-124 inhibits the progression of adjuvant-induced arthritis in rats. Ann Rheum Dis. 2016;75:601–608.
- Wang XN, Zhang LH, Cui XD, et al. lncRNA HOXA11-AS is involved in fracture healing through regulating mir-124-3p. Euro Rev Med Pharmacol Sci. 2017;21:4771–4776.
- Lieberthal J, Sambamurthy N, Scanzello CR. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthr Cartil. 2015;23:1825–1834.
- Stannus O, Jones G, Cicuttini F, et al. Circulating levels of IL-6 and TNF-alpha are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr Cartil. 2010;18:1441–1447.
- Yao Y, Wang Y. ATDC5: an excellent in vitro model cell line for skeletal development. J Cell Biochem. 2013;114:1223–1229.
- Nasi S, So A, Combes C, et al. Interleukin-6 and chondrocyte mineralisation act in tandem to promote experimental osteoarthritis. Ann Rheum Dis. 2016;75:1372–1379.
- Lopez-Armada MJ, Carames B, Lires-Dean M, et al. Cytokines, tumor necrosis factor-alpha and interleukin-1beta, differentially regulate apoptosis in osteoarthritis cultured human chondrocytes. Osteoarthr Cartil. 2006;14:660–669.
- Ajekigbe B, Cheung K, Xu Y, et al. Identification of long non-coding RNAs expressed in knee and hip osteoarthritic cartilage. Osteoarthr Cartil. 2019;27:694–702.
- Mathy NW, Chen XM. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J Biol Chem. 2017;292:12375–12382.
- Carpenter S, Fitzgerald KA. Cytokines and long noncoding RNAs. Cold Spring Harbor Persp Biol. 2018;10:a028589.
- Alexander M, O'Connell RM. Noncoding RNAs and chronic inflammation: micro-managing the fire within. BioEssays: News and Rev Mol, Cell Dev Biol. 2015;37:1005–1015.
- Nugent M. MicroRNAs: exploring new horizons in osteoarthritis. Osteoarthr Cartil. 2016;24:573–580.
- Zhou L, Wang J, Li J, et al. 1,25-Dihydroxyvitamin D3 ameliorates collagen-induced arthritis via suppression of Th17 cells through miR-124 mediated inhibition of IL-6 signaling. Front Immunol. 2019;10:178.
- Guan YJ, Li J, Yang X, et al. Evidence that miR-146a attenuates aging- and trauma-induced osteoarthritis by inhibiting Notch1, IL-6, and IL-1 mediated catabolism. Aging Cell. 2018;17:e12752.
- Murahashi Y, Yano F, Kobayashi H, et al. Intra-articular administration of IkappaBalpha kinase inhibitor suppresses mouse knee osteoarthritis via downregulation of the NF-kappaB/HIF-2alpha axis. Sci Rep. 2018;8:16475.
- Qiu S, Feng Y, LeSage G, et al. Chronic morphine-induced microRNA-124 promotes microglial immunosuppression by modulating P65 and TRAF6. J Immunol (Baltimore, Md: 1950). 2015;194:1021–1030.