
Abstract
RAS mutations are frequent in non-small cell lung cancer (NSCLC). However, targeting RAS or the downstream/upstream effectors, such as tyrosine kinase inhibitors (TKIs), has been proved to be difficult. Here, we found that the stemness of KRAS-mutant NSCLC cells but not the KRAS-wild type NSCLC cells was promoted by TKIs treatment, as evident by the increase of ALDH1 activity, stemness marker expression and spheroid formation ability. Notably, SHP2 activation was found in KRAS-mutant NSCLC cells with TKIs treatment, as judged by the increase of tyrosine 542 phosphorylation (pSHP2 Y542), which activates the RAS/MEK/ERK pathway. On the contrary, inhibition of MEK was followed by a SHP2 activation in KRAS-mutant NSCLC cells. Additionally, inhibition of SHP2 attenuates the enhanced stemness of KRAS-mutant NSCLC cells induced by TKIs, characterized by decreasing ALDH1 activity, stemness marker expression and spheroid formation capacity, while had little effects on cell viability. Finally, we revealed that SHP2 inhibitor increased the sensitivity of TKIs and chemotherapy, which was potentiated by MEK inhibition. Our results suggest a possibility of using a combination of SHP2 inhibitor and TKIs for KRAS-mutant NSCLC treatment.
Introduction
Lung cancer is one of the most common malignant tumors, about 80% of lung cancer is non-small cell lung cancer (NSCLC) and the 5-year survival rate of advanced NSCLC patients is only about 15% [Citation1]. In recent years, the small molecule inhibitors targeting tyrosine kinase (TK) have been widely used in the treatment of advanced NSCLC with mutation of epidermal growth factor receptor (EGFR) due to their high specificity and mild adverse effects [Citation2]. Accordingly, EGFR mutation has been clinically established as an effective predictor of therapeutic sensitivity of TK inhibitors (TKI). However, the response to TKIs is poor in patients with KRAS mutation, which is often an important adverse prognostic factor for progression-free survival of NSCLC patients [Citation3]. At present, there is no approved targeted drug in clinic, and lung cancer patients with KRAS mutation are more likely to be resistant to chemotherapy [Citation4].
Previous studies have shown that TKIs contributes to MAP kinase kinase (MEK) activation in KRAS-mutant (mut) tumors, however, combined with MEK inhibitors and TKIs cannot achieve better therapeutic effects in KRAS-mut lung cancer [Citation5]. Notably, a recent work shows that MEK inhibitor-resistant KRAS-mut NSCLC cells present an initial reduction of phosphorylated extracellular signal-regulated kinase (pERK) level, which is rescued within 72 h, together with an increase of Src homology region 2-containing protein tyrosine phosphatase 2 (SHP2) activation, as evident by the increase of tyrosine 542 phosphorylation (pSHP2 Y542) under EGFR-limiting condition [Citation6]. Additionally, the previous work has shown that inhibition of TKI signaling activates MEK pathway through SHP2 [Citation7]. Therefore, targeting SHP2 may overcome the resistance of TKIs in KRAS-mut NSCLC cells [Citation6], implying the importance of elucidating the mechanism by which SHP2 regulates the drug resistance of KRAS-mut NSCLC cells.
Cancer stem cells (CSCs) are regarded as the origin of tumor progression and drug resistance [Citation8]. The previous study has shown that KRAS mutations in hematopoietic stem cells cause RAS-associated autoimmune lymphoproliferative syndrome-like disease and the status of KRAS in induced pluripotent stem cells (iPSCs) impacts the self-renewal and differentiation propensity [Citation9]. The inflammatory cytokine IL-22 could promote KRAS-mut lung cancer progression via driving the cell stemness [Citation10]. Additionally, KRAS activation in Apc-driven tumours results in the expansion of the CSCs compartment by increasing ®-catenin intracellular stabilization [Citation11]. However, the mechanisms underlying the stemness of KRAS-mut NSCLC cells are still unclear.
Here, we found that that the stemness of KRAS-mut NSCLC cells but not the KRAS-wild type NSCLC cells was promoted by TKIs treatment, complied with activation of SHP2. Inhibition of SHP2 or knocking out SHP2 suppressed the stemness of KRAS-mut NSCLC cells under growth factor–limiting conditions, this effect was enhanced by MEK inhibition. Therefore, our results suggest that inhibition of SHP2 can be explored to treat KRAS-mut NSCLC cells combined with TKIs.
Material and methods
Cell culture and inhibitors
The KRAS-mutant NSCLC cell lines A549, H2122 and KRAS-wild type NSCLC cell lines NCI-H1975 and H1299 were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All of these cell lines were cultured in RPMI-1640 medium containing 10% fetal bovine serum supplemented with 1% ampicillin/streptomycin at 37 °C in 5% CO2. SHP2 inhibitor SHP099 (50 nM), MEK inhibitor PD0325901 (0.5 nM), MEK activator PAF C-16 (5 μM), Erlotinib (5 nM), Gefitinib (50 nM) and Cisplatin (100 nM) were purchased from Selleck Chemicals (Houston, TX, USA).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer's protocols. Subsequently, RNA was reverse transcribed into cDNA with M-MLV reverse transcriptase (Promega, Fitchburg, WI, USA). qRT-PCR was run with SYBR Green qPCR Master Mix (Vazyme, Nanjing, China) on the Applied Biosystems 7500 system. GAPDH was served as an internal control. Relative levels of mRNA were calculated using the 2−ΔΔCt method.
Western blotting
The cells were lysed in RIPA buffer to harvest total protein, and the protein was quantified with the BCA protein assay kit (Beyotime, Beijing, China). An equal quantity of protein was separated with the sodium dodecyl sulfate polyacrylamide gel electrophoresis. Subsequently, the proteins were transferred to polyvinylidene difluoride membranes. Then the membranes were blocked with 10% nonfat milk for 1.5 h at room temperature. After blocking, the membranes were incubated with primary antibodies at 4 °C overnight. The membranes were then washed with TBST (Tris-buffered saline, 0.1% Tween 20) buffer, hybridized with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature, and washed with TBST buffer. The membranes were visualized with ECL (Thermo, Waltham, MA, USA).
Cell viability assay
CCK8 was used to assess cell viability. Cells were seeded in 96-well plate at the density of 3 × 103 cells/well in triplicate for 48 h. Then the cells were treated with 10 μl CCK8 for 1 h. Next, the absorbance at 450 nm was measured by a microplate reader (BioTek, Winooski, VT, USA).
Spheroid formation assay
Cells were seeded in ultra-low-attachment six-well plates and cultured in serum-free medium Dulbecco's modified Eagle medium/F12 supplemented with 20 ml/L B27, 10 ng/ml EGF and 20 ng/ml bFGF for 2 weeks. Cells were replenished with fresh medium every 5 days. Sphere formation was captured two weeks after seeding and the number of spheroids was counted.
ALDH1 activity
ALDH1 Activity Assay Kit was used to detect ALDH1 activity following the manufacturer's instructions. Briefly, 1 × 106 cells were homogenized with 200 µl ice cold ALDH Assay Buffer, and then centrifuged to remove nuclei and insoluble material. 1–50 µl of the supernatant were added into 96-well plates and ALDH assay buffer was used to adjust the final volume to 50 µl. Add 50 µl of the reaction mix to each well, and the absorbance at 450 nm was measured with a microplate reader (BioTek).
Statistical analysis
All data were shown as the mean ± SD. Student's t-test or one-way ANOVA was used to evaluate statistically significant differences between two or multiple groups, respectively. All analyses were carried out with GraphPad Prism 6.0 software. p < .05 was considered statistically significant.
Results
The stemness of KRAS-mut NSCLC cells not KRAS-wild type NSCLC cells was promoted by TKIs treatment
Since CSCs contribute to drug resistance, we wondered whether TKIs treatment could regulate the stemness of NSCLC cells. KRAS-mut (KRAS-mut) NSCLC cells (A549 and H2122) and KRAS-wild type (KRAS-wt) cells (NCI-H1975 and H1299) were treated with Erlotinib (2 nM), an EGFR inhibitor. After 48 h, the expression of stemness markers (ALDH1 and Nanog) was examined. As shown in , Erlotinib significantly decreased the expression of stemness markers in KRAS-wt cells, but increased their expression in KRAS-mut cells. Additionally, the spheroid formation capacity was reduced in KRAS-wt cells, but enhanced in KRAS-mut cells (). Furthermore, a consistent result was obtained by measuring ALDH1 activity in KRAS-mut and -wt cells with Erlotinib treatment (). Notably, another EGFR inhibitor (gefitinib) was used and the consistent results were obtained (). These results suggest that TKI may exert different effects on the stemness of KRAS-mut and -wt NSCLC cells.
Figure 1. The stemness of KRAS-mut NSCLC cells not KRAS-wild type NSCLC cells was promoted by Eriotinib treatment. (A) The mRNA levels of ALDH1 and Nanog were detected in KRAS-mut and -wt NSCLC cells with or without Eriotinib treatment. (B) The protein levels of ALDH1 and Nanog were examined in the cells described in (A). (C and D) The spheroid formation capacity was evaluated in the cells depicted in (A) by measuring spheroid size (C) and number (D). (E) The ALDH activity was determined in the cells described in (A). Data were presented as mean ± SD; n ≥ 3 **p < .01 versus control.
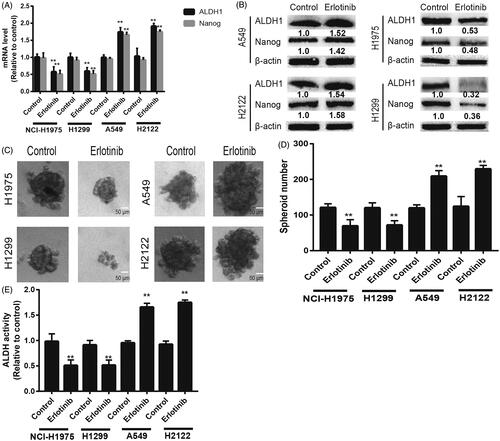
Figure 2. The stemness of KRAS-mut NSCLC cells not KRAS-wild type NSCLC cells was promoted by Gefitinib treatment. (A) The mRNA levels of ALDH1 and Nanog were detected in KRAS-mut and -wt NSCLC cells with or without Gefitinib treatment. (B) The protein levels of ALDH1 and Nanog were examined in the cells described in (A). (C and D) The spheroid formation capacity was evaluated in the cells depicted in (A) by measuring spheroid size (C) and number (D). (E) The ALDH activity was determined in the cells described in (A). Data were presented as mean ± SD; n ≥ 3 **p < .01 versus control.
SHP2 is activated in KRAS-mut NSCLC cells with TKIs treatment followed by MEK activation
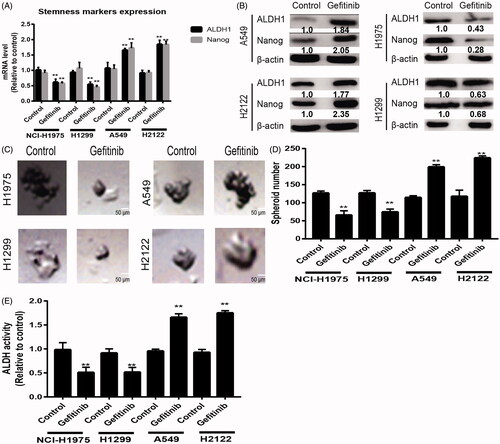
As previous work has shown that inhibition of TKI signaling activates MEK pathway through SHP2 [Citation7], we determined the status of SHP2 in KRAS-mut and -wt NSCLC cells with Erlotinib treatment and found that the expression of tyrosine 542 phosphorylation (pSHP2 Y542) was increased in KRAS-mut cells but not in KRAS-wt cells (). Additionally, MEK signaling was activated in KRAS-mut cells but not in KRAS-wt cells with Erlotinib treatment, as judged by the increase of p-ERK levels (). Since MEK inhibitor was used for KRAS-mut NSCLC treatment, we detected the effects of MEK inhibitor on the status of SHP2. Notably, SHP2 knockdown rescued the activation of MEK upon Erlotinib treatment, this effect was partially reversed by E76K SHP2 overexpression, an active mutant of SHP2 (). As shown in , PD0325901 (0.5 nM), a MEK inhibitor, exhibited an initial reduction of p-ERK levels, which was restored within 72 h in KRAS-mut cells but not in KRAS-wt cells. Importantly, SHP2 was activated by PD0325901 in a time-dependent manner (). These results indicate that SHP2 activation may be the compensatory effects of TKI or MEK inhibition.
Figure 3. SHP2 is activated in KRAS-mut NSCLC cells with TKIs treatment followed by MEK activation. (A) The expression levels of pSHP2 and pERK were detected in KRAS-mut and -wt NSCLC cells with or without Eriotinib treatment plus SHP2 knockdown and E76K SHP2 overexpression or not. (B) The expression levels of pSHP2 and pERK were detected in KRAS-mut cells with MEK inhibitor treatment at different times as indicated – (48 h), cells collected after overnight serum starvation.
Inhibition of SHP2 suppressed the stemness of KRAS-mut NSCLC cells via inactivating MEK signaling
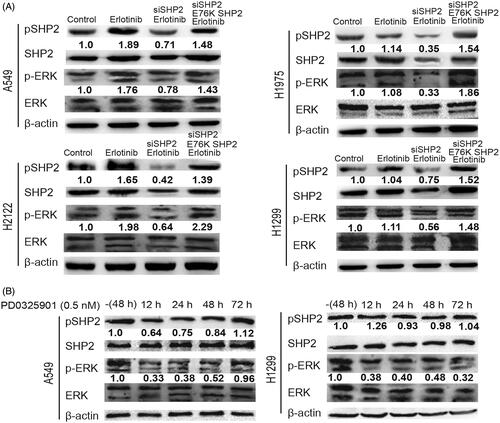
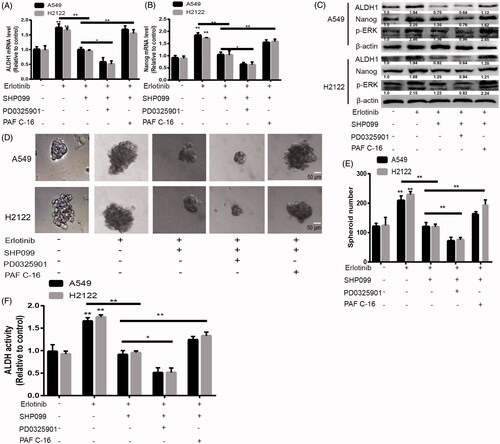
Next, we wondered whether SHP2 activation is responsible for the stemness of KRAS-mut NSCLC cells. KRAS-mut NSCLC cells were treated with SHP099 (100 nM), an inhibitor of SHP2. As shown in , the increased expression of stemness markers induced by Erlotinib was attenuated by inhibiting SHP2, this effect was enhanced by MEK inhibition and rescued by MEK activation (PAF C-16, 50 nM, an activator of MEK). Furthermore, the increased spheroid formation capacity mediated by Erlotinib was reduced by SHP2 inhibition, which was promoted by MEK inhibition and restored by MEK activation (). Additionally, ALDH1 activity detection displayed a consistent result (). These results suggest that SHP2 suppresses the stemness of KRAS-mut NSCLC cells through MEK signaling under EGFR-limiting conditions.
Figure 4. Inhibition of SHP2 suppressed the stemness of KRAS-mut NSCLC cells via inactivating MEK signaling. (A and B) The mRNA levels of ALDH1 and Nanog were examined in A549 and H2122 cells with treatment as indicated. (C) The protein levels of ALDH1 and Nanog were detected in A549 and H2122 cells with treatment as indicated. (C and D) The spheroid formation ability was evaluated in A549 and H2122 cells with treatment as indicated. (E) ALDH1 activity was determined in A549 and H2122 cells with treatment as indicated. Data were presented as mean ± SD; n ≥ 3, *p < .05, **p < .01 versus control.
Inhibition of SHP2 had little effects on the stemness of KRAS-mut NSCLC cells without Erlotinib treatment
We detected the effects of SHP2 inhibition on the stemness of KRAS-mut NSCLC cells without Erlotinib treatment. Interestingly, we found that SHP2 inhibition had little effects on the expression of stemness markers in KRAS-mut NSCLC cells (). Additionally, the spheroid formation ability was unaffected by SHP2 inhibition in KRAS-mut NSCLC cells (). Moreover, SHP2 inhibition did not change the ALDH1 activity in KRAS-mut NSCLC cells (). Thus, our results demonstrate that inhibition of SHP2 had little effects on the stemness of KRAS-mut NSCLC cells under normal cell culture condition.
Figure 5. Inhibition of SHP2 had little effects on the stemness of KRAS-mut NSCLC cells without Erlotinib treatment. (A and B) The mRNA levels of ALDH1 and Nanog were detected in A549 and H2122 cells with or without SHP099 treatment. (C) The protein levels of ALDH1 and Nanog were examined in A549 and H2122 cells with or without SHP099 treatment. (D and E) The spheroid formation capacity was determined in A549 and H2122 cells with or without SHP099 treatment. (F) ALDH1 activity was measured in A549 and H2122 cells with or without SHP099 treatment. Data were presented as mean ± SD; n ≥ 3, ns, no significance.
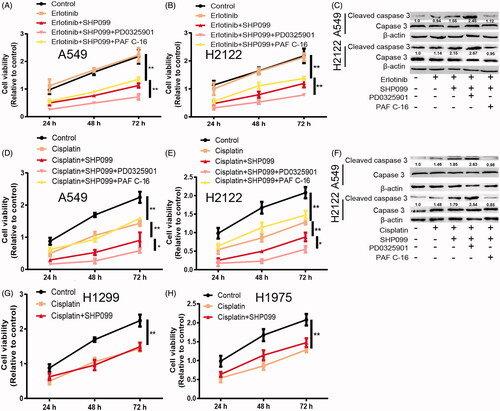
SHP2 inhibition enhances TKI and chemotherapeutic sensitivity in KRAS-mut NSCLC cells, which is potentiated by MEK inhibition
Finally, we investigated whether SHP2 inhibition could enhance TKI and chemotherapeutic sensitivity in KRAS-mut NSCLC cells. As expected, SHP2 inhibition enhanced Erlotinib sensitivity in KRAS-mut NSCLC cells, characterized as the decreased cell viability () and increased expression of apoptotic executor (Cleaved caspase3; ). This effect was enhanced by MEK inhibition and rescued by MEK activation. However, it had no effects on cell viability alone (). Furthermore, since CSCs contribute to chemoresistance, we evaluated whether SHP2 inhibition could enhance chemotherapeutic sensitivity in KRAS-mut NSCLC cells. As shown in , the cisplatin sensitivity was promoted by SHP2 inhibition in KRAS-mut NSCLC cells, as evident by the decreased cell viability and increased expression of apoptotic executor, which was potentiated by MEK inhibition and partially abrogated by MEK activation. However, inhibition SHP2 had little effects on cisplatin sensitivity in KRAS-wild type cells (). Therefore, these results demonstrate that SHP2 inhibition could specifically attenuate the stemness of KRAS-mut NSCLC cells, responsible for TKI and chemotherapeutic sensitivity.
Figure 6. SHP2 inhibition enhances TKI and chemotherapeutic sensitivity in KRAS-mut NSCLC cells, which is potentiated by MEK inhibition. (A and B) Cell viability was detected in A549 and H2122 cells with the treatment as indicated. (C) The expression of Cleaved caspase 3 was examined in A549 and H2122 cells with the treatment as indicated. (D and E) Cell viability was detected in A549 and H2122 cells with the treatment as indicated. (F) The expression of Cleaved caspase 3 was examined in A549 and H2122 cells with the treatment as indicated. (G and H) Cell viability was examined in H1299 and H1975 cells with cisplatin treatment as well as SHP2 inhibition. Data were presented as mean ± SD; n ≥ 3, *p < .05, **p < .01 versus control.
Discussion
Lung cancer is the leading cause of cancer death worldwide the highest mortality rate [Citation1]. Lung cancer can be divided into two categories: NSCLC and small cell lung cancer (SCLC), most of which are NSCLC. The efficacy of traditional chemotherapy has reached a plateau and molecular targeted therapy has become an important method of NSCLC treatment, such as TKIs, Erlotinib and Gefitinib. However, TKIs have been shown to treat to KRAS-mut NSCLC cells. Therefore, it is important to elucidate the mechanism contributing to TKIs resistance in KRAS-mut NSCLC cells.
The promoting roles of SHP2 in tumor progression have been established in NSCLC [Citation6], melanoma [Citation12], breast cancer [Citation13] and oral squamous cell carcinoma [Citation14]. However, its roles in tumor stemness have never been shown. Previous works have shown that SHP2 counteracts with Src to regulate the dynamics and organization of vimentin filaments during cell migration [Citation15]. And SHP2 knockdown in an endothelial cell monolayer increases VE-cadherin internalization and protects endothelial barrier permeability through suppressing MET and ARF1 activation [Citation16]. As the epithelial-mesenchymal transition process is closely related with the stemness of tumors [Citation17], these effects suggest that SHP2 may play critical roles in regulating the stemness of tumors. Recent studies have confirmed that SHP2 inhibition could restore the sensitivity of anaplastic lymphoma kinase inhibitor in NSCLC cells [Citation18]. Additionally, SHP2 inhibition attenuates the adaptive resistance to MEK inhibitors in multiple malignancies [Citation19]. SHP2 drives adaptive resistance to ERK signaling inhibition and combined targeting SHP2 and ERK signaling significantly prevents this resistance [Citation20]. Since CSCs have been regarded as the root of drug resistance, these result promoted us to explore SHP2 roles in regulating the stemness of KRAS-mut cells with TKI treatment. To our surprise, inhibition of SHP2 attenuated the stemness of KRAS-mut NSCLC cells with TKI treatment, while it had little effects under normal cell culture conditions, this suggests that SHP2 exerts its effects within specific microenvironments. Notably, inhibition of SHP2 had no effects on cell viability and as CSCs are a small part of tumour cells, we speculate that inhibition of SHP2 could specifically target CSCs in KRAS-mut NSCLC cells with TKI treatment. Furthermore, our results showed that inhibition of SHP2 suppressed the stemness of KRAS-mut NSCLC cells via inactivating MEK signaling, which is consistent with the previous studies proving the SHP2-ERK signaling [Citation20–23]. Importantly, we must admit that further in vivo experiments should be conducted to confirm our conclusion.
Collectively, our results demonstrate that SHP2 activation facilitates the stemness of KRAS-mut NSCLC cells with TKIs treatment, suggesting that combined use of TKIs and SHP2 inhibitor or chemotherapy may be used for the treatment of KRAS-mut NSCLC cells.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Lemjabbar-Alaoui H, Hassan OU, Yang YW, et al. Lung cancer: biology and treatment options. Biochim Biophys Acta. 2015;1856:189–210.
- Xu J, Yang H, Jin B, et al. EGFR tyrosine kinase inhibitors versus chemotherapy as first-line therapy for non-small cell lung cancer patients with the L858R point mutation. Sci Rep. 2016;6:36371.
- Jiang AG, Lu HY. k-RAS mutations in non-small cell lung cancer patients treated with TKIs among smokers and non-smokers: a meta-analysis. Contemp Oncol (Pozn). 2016;20:124–129.
- Westcott PM, Halliwill KD, To MD, et al. The mutational landscapes of genetic and chemical models of KRAS-driven lung cancer. Nature. 2015;517:489–492.
- Kim WY, Prudkin L, Feng L, et al. Epidermal growth factor receptor and K-Ras mutations and resistance of lung cancer to insulin-like growth factor 1 receptor tyrosine kinase inhibitors. Cancer. 2012;118:3993–4003.
- Mainardi S, Mulero-Sanchez A, Prahallad A, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. 2018;24:961–967.
- Grossmann KS, Rosario M, Birchmeier C, et al. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res. 2010;106:53–89.
- Zheng L, Guo Q, Xiang C, et al. Transcriptional factor six2 promotes the competitive endogenous RNA network between CYP4Z1 and pseudogene CYP4Z2P responsible for maintaining the stemness of breast cancer cells. J Hematol Oncol. 2019;12:23.
- Kubara K, Yamazaki K, Ishihara Y, et al. Status of KRAS in iPSCs impacts upon self-renewal and differentiation propensity. Stem Cell Rep. 2018;11:380–394.
- Khosravi N, Caetano MS, Cumpian AM, et al. IL22 promotes KRAS-mutant lung cancer by induction of a protumor immune response and protection of stemness properties. Cancer Immunol Res. 2018;6:788–797.
- Ghazvini M, Sonneveld P, Kremer A, et al. Cancer stemness in Apc- vs. Apc/KRAS-driven intestinal tumorigenesis. PloS One. 2013;8:e73872.
- Xiao P, Guo Y, Zhang H, et al. Myeloid-restricted ablation of Shp2 restrains melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-γ production in tumor microenvironment. Oncogene. 2018;37:5088–5100.
- Zhao H, Martin E, Matalkah F, et al. Conditional knockout of SHP2 in ErbB2 transgenic mice or inhibition in HER2-amplified breast cancer cell lines blocks oncogene expression and tumorigenesis. Oncogene. 2019;38:2275–2290.
- Cai Z, Hao XY, Liu FX. MicroRNA-186 serves as a tumor suppressor in oral squamous cell carcinoma by negatively regulating the protein tyrosine phosphatase SHP2 expression. Arch Oral Biol. 2018;89:20–25.
- Yang CY, Chang PW, Hsu WH, et al. Src and SHP2 coordinately regulate the dynamics and organization of vimentin filaments during cell migration. Oncogene. 2019;38:4075–4094.
- Zhang J, Huang J, Qi T, et al. SHP2 protects endothelial cell barrier through suppressing VE-cadherin internalization regulated by MET-ARF1. FASEB J. 2019;33:1124–1137.
- Zheng L, Xiang C, Li X, et al. STARD13-correlated ceRNA network-directed inhibition on YAP/TAZ activity suppresses stemness of breast cancer via co-regulating Hippo and Rho-GTPase/F-actin signaling. J Hematol Oncol. 2018;11:72
- Dardaei L, Wang HQ, Singh M, et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat Med. 2018;24:512–517.
- Fedele C, Ran H, Diskin B, et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018;8:1237–1249.
- Ahmed TA, Adamopoulos C, Karoulia Z, et al. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep. 2019;26:65–78 e65.
- Choi E, Kikuchi S, Gao H, et al. Mitotic regulators and the SHP2-MAPK pathway promote IR endocytosis and feedback regulation of insulin signaling. Nat Commun. 2019;10:1473
- Lu H, Liu C, Velazquez R, et al. SHP2 inhibition overcomes RTK-mediated pathway re-activation in KRAS mutant tumors treated with MEK inhibitors. Mol Cancer Ther. 2019;18:1323.
- Ryu HH, Kim T, Kim JW, et al. Excitatory neuron-specific SHP2-ERK signaling network regulates synaptic plasticity and memory. Sci Signal. 2019;12: pii: eaau5755.