
Abstract
Circular antisense non-coding RNA in the INK4 locus (cANRIL) participated in inflammation of endothelial cells. However, whether cANRIL is associated with inflammatory injury of HK-2 cells, thereby affecting chronic kidney disease has not been investigated. We tested the hypothesis that cANRIL participated in inflammatory response in vitro. HK-2 cells were stimulated by lipopolysaccharides (LPS). RT-qPCR was executed for cANRIL expression assessment. After transfection, cell viability, apoptosis, inflammatory cytokines and ROS generation were appraised to evaluate the impact of silencing cANRIL on LPS-induced inflammatory injury. The regulatory relationship between cANRIL and microRNA-9 (miR-9) was verified. In addition, whether miR-9 affected LPS-induced inflammatory injury was measured after miR-9 inhibitor transfection. Western blot was utilized to detect NF-κB and JNK/p38 pathway-related proteins. The results showed that LPS promoted cANRIL expression and cell injuries in HK-2 cells. Furthermore, silencing cANRIL alleviated inflammatory injuries by promoting viability, suppressing apoptosis, inflammatory cytokines and ROS generation in HK-2 cells. In addition, miR-9 expression was accelerated by silencing cANRIL. Meanwhile, miR-9 down-regulation invalidated the effect of silencing cANRIL on inflammation and NF-κB and JNK/p38 pathways. The study clarified that silencing cANRIL hindered NF-κB and JNK/p38 pathways by positively regulating miR-9, thereby protecting HK-2 cells from LPS-induced injury.
Introduction
Chronic kidney disease (CKD) is characterized by the loss of glomerular filtration rate and an inevitable slump in kidney function [Citation1]. Accompanied by 8–16% pervasiveness around the world, CKD has been appraised as an exceedingly communal healthy matter [Citation2]. It should be noted that folks who are diagnosed with CKD are exceptionally exposed to two major complications: acute kidney injury [Citation3] and cardiovascular disease, all of which are the principal causes of high mortality in CKD [Citation4,Citation5]. The most common concurrent cardiovascular disorder is characterized by accelerated systemic inflammation in confirmed CKD process [Citation6,Citation7]. Renal tubular inflammatory disease, one of the pathological manifestations of the chronic inflammatory response, is prevailing in patients with coexisting CKD and atherosclerosis by stimulating inflammatory factors and activating oxidative stress [Citation8]. Therefore, it is worthy to find molecular biomarkers that can fight against inflammation or prevent atherosclerosis, thus contributing to the clinical therapeutic method of CKD.
CircRNAs are non-coding RNA molecules with covalently closed-ring structures, which are reported to play a vital role in modulating multiple miRNAs, regulating transcription and splicing and bridging protein–protein cooperation in neuronal, cardiovascular and even cancer progression [Citation9,Citation10]. The antisense non-coding RNA in the INK4 locus (ANRIL) is located at the human CDKN2A/B locus [Citation11,Citation12]. Shaping circular RNA (circRNA) is an increasingly recognized aspect in ANRIL transcripts [Citation13]. Recently, a report published that circular ANRIL (cANRIL) elicited p53 activation, accelerated cell apoptosis and alleviated cell proliferation to modulate atherosclerosis [Citation14]. Suppressed cANRIL could prevent coronary atherosclerosis by reducing vascular endothelial cells apoptosis and inflammatory response [Citation15]. Although a multitude of mechanisms between cANRIL and atherosclerosis were proposed, there were no studies evaluating the impact of cANRIL on inflammatory response in patients with CKD.
MicroRNAs (miRNAs), a class of small non-coding RNA molecules, regulate protein expression at the post-transcriptional level by binding to target mRNA to induce degradation or translational suppression. Some studies reported that miRNAs, such as miR-1825 [Citation16], miR-21 [Citation17] and miR-223-3p [Citation18], were abnormally expressed in CKD samples, thereby participating in further deterioration of the kidney diseases. In the study by Wu et al., miR-9 was identified as an injury-associated molecule in renal tubular cell [Citation19]. Meanwhile, miR-9 was showed to play a critical role in uric acid-induced renal injury of rat [Citation20]. However, the direct involvement of miR-9 in the cell injury of CKD has not been studied.
In this study, lipopolysaccharides (LPS) solution was utilized to treat HK-2 cells, establishing nephritic injury cell model. We explored the underlying mechanisms how cANRIL regulated the inflammatory response of LPS-treated HK-2 cells with an aim of exploring new molecular target for preventing and treating CKD. Overall, our findings demonstrated that silencing cANRIL alleviated inflammatory response and blocked NF-κB and JNK/p38 pathways by positively regulating miR-9 in the LPS-injured HK-2 cells. These findings might provide a potential molecular therapeutic target for CKD.
Materials and methods
Cells and culture
HK-2 cells were attained from American Type Culture Collection (Manassas, VA, USA) and cultured in K-SFM (Gibco, Rockville, MD, USA), which were enriched with EGF (5 ng/mL) and bovine pituitary extract (40 pg/mL). The condition for cell culture was 5% CO2 and 37 °C. Cell cultures were stimulated by 2 μg/mL LPS (Solarbio, Beijing, China) for 6 h.
Cell transfection
Small interfering RNA of cANRIL (si-cANRIL) and si-NC was colligated by Sangon (Shanghai, China). MiR-9 inhibitor or NC inhibitor were purchased from Life Technology (Carlsbad, CA, USA). According to the sectionalization, si-cANRIL, si-NC, miR-9 inhibitor or NC inhibitor were specifically transfected into cells with lipofectamine 2000 reagent (Life Technology). After two days, cells were collected and utilized in the next related experiments.
Reverse transcription-quantitative real-time PCR (RT-qPCR)
TRIzol (Beyotime, Shanghai, China) was utilized to separate total RNA from transfected cell cultures. Reverse transcription of RNA was implemented by utilizing SuperRT cDNA Synthesis Kit (Cwbio, Jiangsu, China). The reverse transcription conditions were 10 min at 25 °C, 30 min at 48 °C, and a final step of 5 min at 95 °C. SYBR® Green PCR Kit (Qiagen, Hilden, Germany) was employed for qPCR analysis to detect cANRIL and miR-9 expression. qPCR was executed on iQ5 Real-Time PCR apparatus (Bio-Rad, Hercules, CA, USA). The expression of cANRIL was normalized by GAPDH. The miR-9 expression were normalized with U6. The 2–ΔΔCt values were calculated for the quantification of the relative cANRIL and miR-9 expression [Citation21].
Cell viability experiment
Cells (5 × 103/well) were cultured in 96-well plates for 48 h. Cell viability was assessed by a Cell Counting Kit-8 (CCK-8, Solarbio). In brief, after LPS treatment and cell transfection, 10 μL CCK-8 reagents were infused to the cultures that were then cultivated for 1 h. Microplate Reader (Bio-Rad) was utilized to test OD450 value.
Cell apoptosis assay
Annexin V-FITC/PI staining kit (Invitrogen) was utilized to examine cell apoptosis ratio. Cells (1 × 106/well) were transferred in 6-well plates. Next, PI (100 μg/mL) and Annexin V-FITC (20 mg/mL) solutions were utilized to dispose the cells that were transfected with si-cANRIL, si-NC, miR-9 inhibitor or NC inhibitor and then stimulated by LPS. Next, flow cytometry was performed to sort apoptotic cells by utilizing FACS can (Becton Dickinson, San Jose, CA, USA).
Enzyme-linked immunosorbent assay (ELISA)
Cell suspension was collected and the levels of inflammatory cytokines were analyzed by ELISA. The levels of IL-1β and IL-6 were detected by utilizing ELISA kit (Solarbio) according to the protocols. The OD value at 450 nm was collected by Microplate Reader (Bio-Rad).
Reactive oxygen species (ROS) assay
The 2,7-dichlorofluorescein diacetate (DCFH-DA, Solarbio) was applied to perform ROS assay. Cells were cultured in a 6-well plate. After treatment and transfection, the cell medium was moved and cultures were co-incubated with the medium including DCFH-DA (final concentration for 10 μM) at 37 °C for 20 min. Then, cells were rinsed and re-suspended to 500 μL PBS buffer. The fluorescence was evaluated utilizing a flow cytometer (488 nm excitation and 521 nm emission, Millipore Guava, Billerica, MA, USA).
Western blot
Total proteins were isolated utilizing RIPA lysis buffer (Cwbio) added with phenylmethylsulfonyl fluoride (PMSF, Solarbio). The proteins were quantified by the Super-Bardford Pritein Assay Kit (Cwbio) and loaded into 12% polyacrylamide gel on the Bis-Tris Gel system (Bio-Rad). Then, the extractions were moved to polyvinylidene fluoride (Solarbio) membranes from gel. Next, the membranes were cultivated with primary antibodies for 12 h that were diluted in 5% blocking buffer. Primary antibodies were purchased from Abcam (Cambridge, MA, USA), including anti-Bcl-2 (#ab182858), anti-Bax (#ab182733), anti-cleaved-caspase-3 (#ab2302), anti-IL-1β (#ab82558), anti-IL-6 (#ab9324), anti-t-IκBα (#ab7217), anti-p-IκBα (#ab133462), anti-t-p65 (#ab16502), anti-p-p65 (#ab86299), anti-t-JNK (#ab208035), anti-p-JNK (#ab219584), anti-t-p38 (#ab170099), anti-p-p38 (#ab4822), and anti-β-actin (#ab179467). Then, the PVDF membranes were rinsed and hatched with goat anti-rabbit IgG (#ab6721) and goat anti-mouse IgG (#ab205719) for 1 h at 25 °C. After rinsing, the membranes carried HRP-blots that were stimulated by ChemiDoc™ XRS system (Bio-Rad). Then, chemiluminescence signal was captured and evaluated using Image Lab™ (Bio-Rad).
Statistical analysis
Each group of experimentation is repeated three times. Graphpad 6.0 (San Diego, CA, USA) was utilized to do the statistical analysis, which was shown as the mean + SD. Analysis of variance (ANOVA) and the Student t-test were utilized to appraise the p values. The p < .05 was regarded as a statistical difference.
Results
LPS elicited the cANRIL expression in HK-2 cells
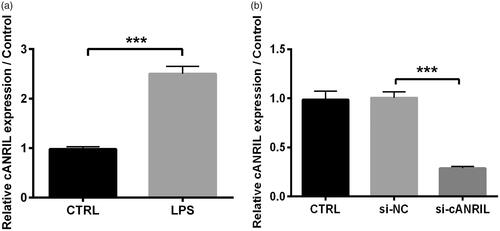
According to the preceding study reported by Shi et al. [Citation22], the kidney injury cell model was established by the treatment of 2 μg/mL LPS for 6 h towards HK-2 cells. Initially, we affirmed that LPS elicited cANRIL expression in HK-2 cells when compared to the without LPS group (p < .001, . To further investigate the significance of cANRIL in LPS-induced inflammatory cells, we employed si-cANRIL to restrain the expression of cANRIL. As expected, we observed that the relative cANRIL expression was apparently declined in si-cANRIL group compared with that in the si-NC group (p < .001, ), hinting efficient silencing in HK-2 cells.
Figure 1. LPS elicited the cANRIL expression in HK-2 cells. (A) HK-2 cells were exposed in LPS (2 μg/mL) environment for 6 hours. After treating with LPS or not, the expression of cANRIL in HK-2 cells was detected by RT-qPCR. (B) After si-cANRIL transfection, the transfection efficiency was detected by using RT-qPCR. GAPDH was utilized as an internal reference. N = 3. Results were expressed as mean ± SD. ***p < .001.
Silencing cANRIL weakened LPS-induced inflammatory injury in HK-2 cells
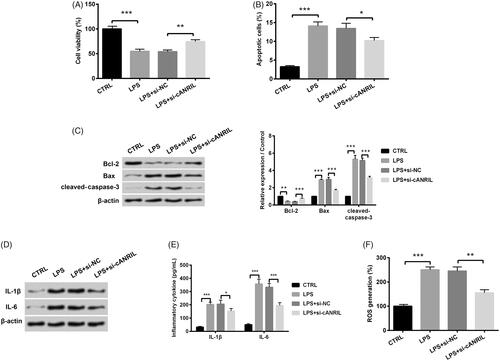
The CCK-8 assay displayed that LPS treatment significantly attenuated HK-2 cell viability (p < .001, . The depressed viability triggered by LPS was ameliorated when cANRIL was silenced (p < .01, . On the contrary, LPS elicited cell apoptotic potential (p < .001), while silencing cANRIL reversed the cell apoptotic ability (p < .05, . Meanwhile, LPS significantly increased the expression levels of pro-apoptotic proteins (Bax and cleaved-caspase-3) and reduced the anti-apoptotic factor (Bcl-2) expression (p < .01 or p < .001, . On the contrary, silencing cANRIL reversed the increased levels of Bax, cleaved-caspase-3 and the suppressed Bcl-2 expression (p < .001, . Moreover, western blot and ELISA assays presented that LPS expedited the expression and secretion of inflammatory cytokines (IL-1β and IL-6), whereas cANRIL down-expression impeded the expression and secretion (p < .05 or p < .001, ). An increased ROS generation was observed in LPS-injured cells than that in the control cells (p < .001, . But, the ROS generation was suppressed due to the reduction of cANRIL (p < .01, . Taken together, the aforementioned discoveries indicated that silencing cANRIL degraded LPS-induced inflammatory response in HK-2 cells.
Figure 2. Silencing cANRIL attenuated LPS-induced inflammatory response in HK-2 cells. Cells were transfected with si-cANRIL or the corresponding control (si-NC) and then treated with LPS for 6 hours. (A) Cell viability was examined by CCK-8 assay. (B) Apoptotic cells were counted by flow cytometry. (C) Expression of apoptosis-related proteins (Bcl-2, Bax and cleaved-caspase-3) was measured by western blot. (D) Expression of inflammatory cytokines (IL-1β and IL-6) was measured by western blot. The relative expression of proteins was normalized by β-actin. (E) The content of inflammatory factors (IL-1β and IL-6) in cell cultures were examined by ELISA. (F) ROS generation was assessed by ROS assay. N = 3. Results were expressed as mean ± SD. *p < .05, **p < .01, ***p < .001.
The expression of miR-9 was down-regulated by LPS treatment but was up-regulated by silencing cANRIL
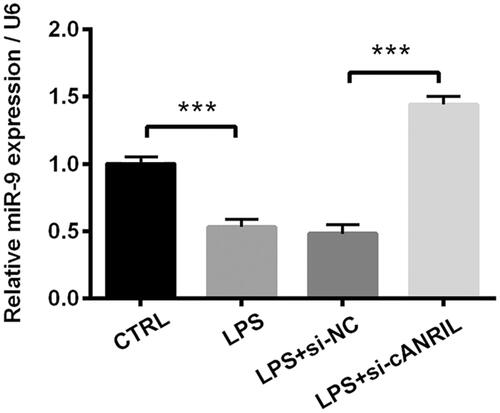
To get insights into the modulation of silencing cANRIL on miR-9, the miR-9 expression was detected. RT-qPCR identified the significant down-regulation of miR-9 in response to LPS (p < .001, . By contrast, the decreased miR-9 expression was reversed by silencing cANRIL (p < .001). The aforementioned discoveries demonstrated that miR-9 expression was positively regulated by silencing cANRIL in LPS-injured HK-2 cells.
Figure 3. The expression of miR-9 was positively increased by silencing cANRIL in LPS-stimulated HK-2 cells. Cells were transfected with si-cANRIL or the corresponding controls (si-NC) and then were treated with LPS for 6 hours. Next, the relative miR-9 expression was examined by RT-qPCR. U6 served as an internal control. N = 3. Results were expressed as mean ± SD. ***p < .001.
Silencing cANRIL attenuated LPS-induced inflammatory response via regulating miR-9
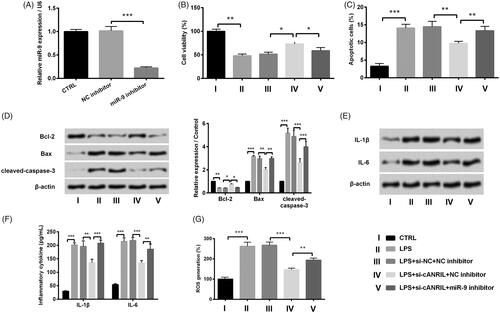
Considering that miR-9 has been identified as an inflammatory-associated miRNA [Citation23], we clarified whether miR-9 was involved in LPS-led inflammatory response in HK-2 cells. Inhibition efficiency was examined by RT-qPCR. The expression of cells group transfected with miR-9 inhibitor was obviously down-modulated compared to that transfected with NC inhibitor (p < .001, . The results of disclosed that miR-9 inhibitor obviously overturned the increase in cell viability and cell apoptosis inhibition in LPS-treated and si-ANRIL-transfected cells (p < .05 or p < .01). By comparison with LPS + si-cANRIL + NC inhibitor group, the protein expression levels of Bax and cleaved-caspase-3 were increased and the Bcl-2 expression was reduced in the LPS + si-cANRIL + miR-9 inhibitor group (p < .05, p < .01 or p < .001, . In addition, displayed that miR-9 inhibitor also reversed the silencing of cANRIL-induced down-regulated expression and secretion of inflammatory cytokine in LPS-treated cells (p < .01 or p < .001). Furthermore, the inhibition of ROS generation in LPS + si-cANRIL + NC inhibitor group was also reversed in LPS + si-cANRIL + miR-9 inhibitor group (p < .01, . The above-mentioned data manifested that cANRIL knockdown significantly ameliorated LPS-induced inflammatory response via up-regulating miR-9 in HK-2 cells.
Figure 4. Silencing cANRIL attenuated the LPS-induced inflammatory response via regulating miR-9. (A) After miR-9 inhibitor or NC inhibitor transfection, the miR-9expression was evaluated by RT-qPCR. After si-cANRIL (or miR-9 inhibitor) or the corresponding controls transfection, cells were exposed in LPS solutions for 6 hours. (B) Cell viability was examined by CCK-8 assay. (C) Apoptotic cells were calculated by flow cytometry. (D) The protein expression of Bcl-2, Bax, cleaved-caspase-3 was measured by western blot. (E) The protein expression of IL-1β and IL-6 was measured by western blot. The relative expression of proteins was normalized by β-actin. (F) The inflammatory factors (IL-1β and IL-6) in the cell cultures were examined by ELISA. (G) ROS generation of HK-2 cells was appraised by ROS assay. N = 3. Results were expressed as mean ± SD. *p < .05, **p < .01, ***p < .001.
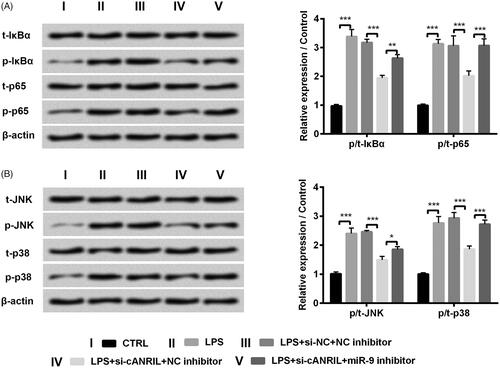
Silencing cANRIL blocked NF-κB and JNK/p38 pathways by up-regulating miR-9 expression
To further clarify whether the downstream signal pathways affected the functions of cANRIL in LPS-induced inflammatory response, we then detected the expression of NF-κB and JNK/p38-associated proteins by western blot. The results presented that the protein levels of p-IκBα, p-p65, p-JNK and p-p38 were obviously elevated in LPS-exposed HK-2 cells (p < .001, ). Silencing cANRIL impeded the expression of the above-mentioned proteins in comparison with the group transfected with si-NC (p < .001, ). Further, miR-9 inhibitor reversed the down-regulated protein levels of p-IκBα (p < .01), p-p65 (p < .001), p-JNK (p < .05) and p-p38 (p < .001) triggered by silencing cANRIL in LPS-exposed cells (). The above-mentioned consequences indicated that silencing cANRIL blocked NF-κB and JNK/p38 pathways by up-regulating miR-9 expression in LPS-treated HK-2 cells.
Figure 5. Silencing cANRIL blocked NF-κB and JNK/p38 pathways by positively regulating miR-9 in LPS-treated HK-2 cells. After si-cANRIL (or miR-9 inhibitor) or the corresponding controls transfection, cells were exposed in LPS solutions for 6 hours. (A,B) The protein levels of t-IκBα, p-IκBα, t-p65, p-p65, t-JNK, p-JNK t-p38 and p-p38 were examined by western blot. The relative expression of proteins was normalized by β-actin. N = 3. Results were expressed as mean ± SD. *p < .05, **p < .01, ***p < .001.
Discussion
Chronic systemic inflammation, which appears as low-grade inflammatory cytokines, is likely to cause kidney injury through affecting inflammation-associated diseases [Citation24,Citation25]. Hence, it is imperative to discover impactful therapeutic targets reducing inflammatory response in CKD. In the present report, our data showed that cANRIL was over-expressed in LPS-treated cells. After plasmid transfection, LPS-induced injury was relieved by silencing cANRIL. Further, we observed that miR-9 expression was up-regulated by cANRIL knockdown, and the effect of silencing cANRIL on LPS-induced inflammatory injury was reversed the miR-9 inhibition. Finally, we indicated that down-expression of cANRIL blocked NF-κB and JNK/p38 pathways by positively modulating miR-9 in LPS-injured HK-2 cells.
Recent researches revealed that the tubular epithelial cells were dysfunctioned due to CKD, which appeared as inflammatory responses [Citation26,Citation27]. LPS, a fundamental component of cell wall in gram-negative bacteria, can be defined as endotoxin and utilized to establish renal inflammatory models [Citation28]. It is almost certain that LPS is considered a contributory factor to the obvious increasing of cell apoptotic capacity and inflammation in HK-2 cells [Citation29]. Our results were in accord with previous studies indicating that LPS evoked inflammatory response of HK-2 cells through inhibiting cell viability, aggravating cell apoptotic capacity, promoting IL-1β and IL-6 secretion and accelerating ROS generation.
To date, there have been a number of longitudinal studies involving circRNAs and inflammation response. Zhang et al. held the view that circRNA ARF3 acted as an endogenous miR-103 sponge to impede mitophagy-interfered adipose inflammatory response [Citation30]. Additionally, Zhou et al. suggested that circRNA_0033186 was obviously up-regulated and promoted apoptosis while inhibited proliferation in osteoarthritis cells and mouse model [Citation31]. Similar findings were obtained in Nd2O3-induced human bronchial epithelial cells, in which circRNA_0039411 enhanced inflammation and dysfunction through acting as a sponge of miR-93-5p [Citation32]. However, what is not yet clear is whether cANRIL is linked to inflammation in LPS-exposed cells. The data of this paper suggested that the cANRIL expression was increased in LPS-exposed HK-2 cells. However, silencing cANRIL promoted cell viability, suppressed cell apoptotic capacity, restrained IL-1β, IL-6 expression and ROS generation in LPS-exposed cells. The present results indicated that cANRIL played a crucial role in the inflammatory response of HK-2 cells.
Regarding the potential mechanisms of cANRIL in LPS-treated HK-2 cells, we get insights into the possibility that cANRIL may act as a ceRNA [Citation33]. We discovered that LPS significantly down-regulated miR-9 expression while LPS-induced down-expression of miR-9 was reversed by silencing cANRIL. Recent study showed that miR-9 participated in the regulation of inflammation in several diseases. For instance, miR-9 declined inflammasome activation in oxLDL-stimulated human atherosclerosis cells [Citation23]. On the contrary, emerging study showed that miR-9 overexpression alleviated inflammation and promoted tube formation via inhibiting NF-κB expression in lymphatic endothelial cells [Citation34]. Consistent with the previous results, our present study also showed that miR-9 inhibitor reversed the increase of cell viability and the declination of cell apoptosis, inflammatory cytokines and ROS generation that were caused by silencing cANRIL in LPS-stimulated HK-2 cells. It was suggested that silencing cANRIL might be a pivotal step in the progress of LPS-induced inflammatory response through strengthening miR-9 expression in HK-2 cells.
NF-κB is known to be linked to many physiological behaviors such as proliferation, motility, cell death and immune responses, with increasing participation in recruiting inflammatory cytokines [Citation35–37]. It has been reported that circRNA_0068087 down-regulation could suppress inflammation by restraining TLR4/NF-κB/NLRP3 complexes in human umbilical vein endothelial cells [Citation38]. Another study implied that circRNA_0075932 activated AuroraA/NF-κB pathway and then induced inflammatory response in human dermal keratinocytes [Citation39]. Moreover, evidence also revealed that JNK/p38 signaling pathways were blocked by β-carotene in LPS-induced inflammation of macrophages [Citation40]. In this study, our results indicated that silencing cANRIL significantly attenuated NF-κB and JNK/p38 pathways via positively regulating miR-9. These finding indicated that NF-κB and JNK/p38 pathways may be involved in regulating the role of cANRIL in LPS-induced inflammatory injury.
In general, the data of this research supported the idea that silencing cANRIL hindered NF-κB and JNK/p38 pathways by positively regulating miR-9, thereby protecting HK-2 cells from LPS-induced inflammatory injury. This was the first study that evaluated the effectiveness of silencing cANRIL in LPS-induced inflammatory response and provided neoteric insights into the rational design of the therapeutic target of the CKD. Nevertheless, the main weakness of this study is the paucity of in vivo experiments, which should be operated in the future.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Related Research Data
References
- Vassalotti JA, Centor R, Turner BJ, et al. Practical approach to detection and management of chronic kidney disease for the primary care clinician. Am J Med. 2016;129:153–162.e7.
- Jha V, Garcia-Garcia G, Iseki K, et al. Chronic kidney disease: global dimension and perspectives. Lancet (London, England). 2013;382:260–272.
- Hsu RK, Hsu CY. The role of acute kidney injury in chronic kidney disease. Semin Nephrol. 2016;36:283–292.
- Tanaka K, Watanabe T, Takeuchi A, et al. Cardiovascular events and death in Japanese patients with chronic kidney disease. Kidney Int. 2017;91:227–234.
- Mathew RO, Bangalore S, Lavelle MP, et al. Diagnosis and management of atherosclerotic cardiovascular disease in chronic kidney disease: a review. Kidney Int. 2017;91:797–807.
- Stubbs JR, House JA, Ocque AJ, et al. Serum trimethylamine-N-oxide is elevated in CKD and correlates with coronary atherosclerosis burden. JASN. 2016;27:305–313.
- Pant S, Deshmukh A, Gurumurthy GS, et al. Inflammation and atherosclerosis – revisited. J Cardiovasc Pharmacol Ther. 2014;19:170–178.
- Hassan MO, Duarte R, Dix-Peek T, et al. Transforming growth factor-beta protects against inflammation-related atherosclerosis in South African CKD patients. Int J Nephrol. 2018;2018:Article ID 8702372.
- Hsiao KY, Sun HS, Tsai SJ. Circular RNA – new member of noncoding RNA with novel functions. Exp Biol Med (Maywood). 2017;242:1136–1141.
- Ebbesen KK, Hansen TB, Kjems J. Insights into circular RNA biology. RNA Biol. 2017;14:1035–1045.
- Kong Y, Hsieh CH, Alonso LC. ANRIL: a lncRNA at the CDKN2A/B locus with roles in cancer and metabolic disease. Front Endocrinol (Lausanne). 2018;9:405.
- Chi JS, Li JZ, Jia JJ, et al. Long non-coding RNA ANRIL in gene regulation and its duality in atherosclerosis. J Huazhong Univ Sci Technol Med Sci. 2017;37(6):816–822.
- Burd CE, Jeck WR, Liu Y, et al. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233
- Holdt LM, Stahringer A, Sass K, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7:12429.
- Song C-L, Wang J-P, Xue X, et al. Effect of circular ANRIL on the inflammatory response of vascular endothelial cells in a rat model of coronary atherosclerosis. Cell Physiol Biochem. 2017;42:1202–1212.
- Muralidharan J, Ramezani A, Hubal M, et al. Extracellular microRNA signature in chronic kidney disease. Am J Physiol Renal Physiol. 2017;1;312:F982–f991.
- Gomez IG, Nakagawa N, Duffield JS. MicroRNAs as novel therapeutic targets to treat kidney injury and fibrosis. Am J Physiol Renal Physiol. 2016;310:F931–44.
- Ulbing M, Kirsch AH, Leber B, et al. MicroRNAs 223-3p and 93-5p in patients with chronic kidney disease before and after renal transplantation. Bone. 2017;95:115–123.
- Wu J, Li DD, Li JY, et al. Identification of microRNA-mRNA networks involved in cisplatin-induced renal tubular epithelial cells injury. Eur J Pharmacol. 2019;851:1–12.
- Chen LL, Xu Y. Epigallocatechin gallate attenuates uric acid-induced injury in rat renal interstitial fibroblasts NRK-49F by up-regulation of miR-9. Eur Rev Med Pharmacol Sci. 2018;22:7458–7469.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408.
- Shi M, Zeng X, Guo F, et al. Anti-inflammatory pyranochalcone derivative attenuates LPS-induced acute kidney injury via inhibiting TLR4/NF-kappaB pathway. Molecules (Basel, Switzerland). 2017;22:1683.
- Wang Y, Han Z, Fan Y, et al. MicroRNA-9 inhibits NLRP3 inflammasome activation in human atherosclerosis inflammation cell models through the JAK1/STAT signaling pathway. Cell Physiol Biochem. 2017;41:1555–1571.
- Kimura T, Isaka Y, Yoshimori T. Autophagy and kidney inflammation. Autophagy. 2017;13:997–1003.
- Komada T, Muruve DA. The role of inflammasomes in kidney disease. Nat Rev Nephrol. 2019;15:501–520.
- Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97.
- Ferenbach DA, Bonventre JV. Acute kidney injury and chronic kidney disease: from the laboratory to the clinic. Nephrol Therap. 2016;12:S41–S8.
- Wang Y, Zhang W, Yu G, et al. Cytoprotective effect of aquaporin 1 against lipopolysaccharide-induced apoptosis and inflammation of renal epithelial HK-2 cells. Exp Ther Med. 2018;15:4243–4252.
- Bascands JL, Bachvarova M, Neau E, et al. Molecular determinants of LPS-induced acute renal inflammation: implication of the kinin B1 receptor. Biochem Biophys Res Commun. 2009;386:407–412.
- Zhang Z, Zhang T, Feng R, et al. circARF3 alleviates mitophagy-mediated inflammation by targeting miR-103/TRAF3 in mouse adipose tissue. Mol Therap Nucleic Acids. 2019;14:192–203.
- Zhou ZB, Huang GX, Fu Q, et al. circRNA.33186 contributes to the pathogenesis of osteoarthritis by sponging miR-127-5p. Mol Ther. 2019;27:531–541.
- Hua Q, Chen Y, Liu Y, et al. Circular RNA 0039411 is involved in neodymium oxide-induced inflammation and anti-proliferation in a human bronchial epithelial cell line via sponging miR-93-5p. Toxicol Sci. 2019;170:69.
- Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell. 2011;146:353–358.
- Chakraborty S, Zawieja DC, Davis MJ, et al. MicroRNA signature of inflamed lymphatic endothelium and role of miR-9 in lymphangiogenesis and inflammation. Am J Physiol Cell Physiol. 2015;309:C680–92.
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62.
- Sun T, Dong W, Jiang G, et al. Cordyceps militaris improves chronic kidney disease by affecting TLR4/NF-κB redox signaling pathway. Oxid Med Cell Longev. 2019;2019:7850863.
- Feng YL, Chen H, Chen DQ, et al. Activated NF-kappaB/Nrf2 and Wnt/beta-catenin pathways are associated with lipid metabolism in CKD patients with microalbuminuria and macroalbuminuria. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2317–2332.
- Cheng J, Liu Q, Hu N, et al. Downregulation of hsa_circ_0068087 ameliorates TLR4/NF-kappaB/NLRP3 inflammasome-mediated inflammation and endothelial cell dysfunction in high glucose conditioned by sponging miR-197. Gene. 2019;709:1–7.
- Zhang X, Chen L, Xiao B, et al. Circ_0075932 in adipocyte-derived exosomes induces inflammation and apoptosis in human dermal keratinocytes by directly binding with PUM2 and promoting PUM2-mediated activation of AuroraA/NF-kappaB pathway. Biochem Biophys Res Commun. 2019;511:551–558.
- Li R, Hong P, Zheng X. β-carotene attenuates lipopolysaccharide-induced inflammation via inhibition of the NF-κB, JAK2/STAT3 and JNK/p38 MAPK signaling pathways in macrophages. Anim Sci J. 2019;90:140–148.