
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Aim
2-HP-β-cyclodextrin-PLGA nanoparticle complexes were prepared to enhance the aqueous humour delivery of Triamcinolone acetonide.
Materials & methods
Drug-loaded 2-HP-β-CD/PLGA nanoparticle complexes prepared by adapting a quasi-emulsion solvent evaporation technique. In vitro drug release, in vitro transcorneal permeation study, histopathological study and in vivo transcorneal penetration of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes were evaluated.
Results
Particle size distributions of 2-HP-β-CD/PLGA nanoparticle complexes were 149.4 ± 3.7 nm and presented stable system. Corneal penetration studies revealed steady sustained drug release (First-order); 2-HP-β-CD/PLGA nanoparticle complexes increased ocular bioavailability by increasing dispersion in the tear film and improving drug release.
Conclusion
2-HP-β-CD/PLGA nanoparticle complex formulation is a promising alternative to conventional eye drops.
Graphical Abstract
Introduction
Normal ocular structure comprises of special physiological structures, such as the corneal barrier, blood-aqueous barrier, and blood-retinal barrier, that may obstruct the delivery of ocular drugs, and thus, hinder them from achieving satisfactory bioavailability [Citation1,Citation2]. The application of drug delivery systems such as nano eye drops, contact lenses, conjunctival implants, and non-implantable ocular surface drug delivery devices increases the amount of penetration into the aqueous humour by topical eye drops [Citation3–5]. Polymeric carrier materials are widely used because of their suitable biomechanical, physical, and chemical properties, strong plasticity and low irritation potential, all of which make them biocompatible and safe for application [Citation6,Citation7]. Polylactic-co-glycolic acid (PLGA) polymer can overcome the limitations caused by these physiological barriers. It can improve the permeability, mucosal adhesion, sustained release, and controlled release properties of different drug molecules that, in turn, improves the bioavailability of the drug. Silva-Abreu et al. [Citation8] investigated the in vitro scleral permeability of pioglitazone-loaded PLGA nanoparticles to demonstrate that the nanoparticles had better permeability and could be more effective against inflammation. Kalam et al. [Citation9] examined the pharmacokinetics of aqueous extracts of tacrolimus PLGA nanoparticles after aqueous eye drops. The results showed that the nanoparticles had longer t1/2 in aqueous humour and the bioavailability was significantly higher than that of aqueous solutions as control group. The PLGA nanoparticles loaded with sparfloxacin can be retained in the anterior cornea for a long time and are not easily eliminated by systemic circulation [Citation10]. Since PLGA is a copolymer of lactic acid (LA) and glycolic acid (GA) monomers, its rate of degradation in the body, drug encapsulation, and in vitro release can be adjusted by changing the ratio of the two monomers and the molecular weight that each contributes to the polymer. Nanoparticles loaded with bevacizumab were prepared using PLGA (molecular weight of 7000–17,000 and a 50/50 copolymerisation ratio). The average retention time and bioavailability of bevacizumab-loaded PLGA nanoparticles in the vitreous increased by 3.28 and 1.39 times, respectively, compared to that in the control group [Citation11]. Numerous studies have shown that the cells have a significant size dependence on the uptake efficiency and targeting of the vector. The PLGA nanoparticles with particle sizes between 1 and 1000 nm can better dissolve, couple, or entrap drugs in the eye for sustained long-term release. Hamed et al. [Citation12] injected a brinzolamide-loaded PLGA nanoparticle with an average particle size distribution of 100 nm under the conjunctiva of normal rabbits and compared their results with the same-site injection of a brinzolamide suspension. The results of intraocular pressure monitoring showed that nanoparticles can reduce intraocular pressure for 10 days; the reduction and duration of intraocular pressure were better for nanoparticles than for the brinzolamide suspension. The effect of improving drug release and penetration can be achieved by modifying a specific polymer on the surface of the PLGA carrier. Cyclodextrins (CDs) are cyclic oligosaccharides with a hydrophilic outer surface and a hydrophobic inner cavity that can interact with small molecules to form a total inclusion complex or form a partial inclusion complex with a macromolecular drug through a hydrophobic side chain [Citation13]. Estefanía Vega et al. [Citation14] added hydroxypropyl-β-cyclodextrin to PLGA and flurbiprofen to PLGA-PEG di-block copolymers. In vitro excised corneal permeability results indicated that the presence of CDs reduces the burst effect and allows a more sustained release of the drug. The strong hydrophilicity of CDs not only provides the drug with the ability to penetrate the tear film but also increases drug concentration on the surface of the eye, thereby enhancing the ability of the drug to enter the eye. Zikhona et al. [Citation15] compared the effect of CD-modified nano-drug delivery system for the eyes on corneal permeability in vitro; the results showed that in the presence of CDs, drug penetration was >50% within 15 min. The control group had a penetration of less than 30% in 30 min.
The work presented here focussed on the preparation of a hydrophilic polymer and PLGA nanoparticles loaded with triamcinolone acetonide (TA-NPs) for ocular drug delivery. Triamcinolone acetonide was selected as a model drug because of its highly lipophilic nature. Polylactic-co-glycolic acid, with its excellent encapsulation efficiency in combination with 2-hydroxypropyl-beta-cyclodextrin (2-HP-β-CD), was used as a hydrophilic polymer so that it could increase the hydrophilicity of the nanoparticles and aid in making suitable formulations for ocular application.
Materials and methods
Materials
Triamcinolone acetonide (TA) was purchased from Jin Hui Pharmaceutical Group Co., Ltd (Tianjin, China). Poly (lactic-co-glycolic acid) (PLGA, 50:50, MW 10,000 Da) copolymer was obtained from Dai Gang Bioengineering Co., Ltd (Jinan, China). Polyvinyl alcohol (PVA) was supplied by Cola International Trade Co., Ltd (Shanghai, China), while 2-HP-β-CD was gifted by Qian Hui Biotechnology Co., Ltd (Zibo, China). All other reagents and solvents were analytical grade reagents.
Preparation of PLGA NPs and 2-HP-β-CD/PLGA nanoparticle complexes
PLGA NPs were prepared by the emulsion solvent evaporation method based on the previous description with slight modification [Citation16,Citation17]. Briefly, 10 mg of TA and 100 mg of PLGA (LA/GA = 50:50) in 5 ml of acetone/ethanol (4:1) was slowly injected into 40 ml of 2% (w/v) PVA aqueous solution in ice bath using a probe sonicator set at 150 W of energy output (JY88-IIN, Scientz Biotechnology Co, Ltd, People’s Republic of China) for 600 s with pulses of 3 s on and 3 s off to form an oil-in-water (O/W) emulsion. Then this emulsion was diluted with DI water, followed by the evaporation of organic solvents under magnetic stirring for overnight to obtain a PLGA nanoparticle nanosuspension.
To prepare 2-HP-β-CD/PLGA-nanoparticle complexes, 2-HP-β-CD (1.5% w/v) was dissolved in the PVA aqueous phase. All other steps were the same as in the original method, and the 2-HP-β-CD/PLGA-nanoparticle complexes were obtained. 2-HP-β-CD/PLGA-nanoparticle complexe is a composite system comprising 2-HP-β-CD/PLGA nanoparticles and a small amount of 2-HP-β-CD inclusion compound. PLGA NPs and 2-HP-β-CD/PLGA-nanoparticle complexes were dried in a lyophilizer using mannitol (1% w/v) as cryoprotectant to prepare for subsequent experiments.
Characterisation of the two types of nanoparticle complexes
Particle size and zeta-potential
The particle size (diameter, nm), polydispersity index (PDI), and zeta-potential of PLGA NPs and the 2-HP-β-CD/PLGA nanoparticle complexes were determined using a laser diffraction instrument (Delsa Nano C® particle size and ζ-potential analyser, Beckman Coulter Inc., CA, USA). All measurements were performed in triplicates, and the results are reported as means ± SD of the three replicates (n = 3) for each formulation.
Morphological observations
The surface morphology of PLGA NPs and 2-HP-β-CD/PLGA nanoparticle complexes were determined using transmission electron microscopy (TEM) (JEM-1400, JEOL, JAPAN). Few microliters of samples of the NPs suspension were dropped on a copper grid and dried at room temperature. After complete drying, the samples were stained using 2% w/v phosphotungstic acid solution and observed using TEM.
Loading capacity and entrapment efficiency
Since triamcinolone acetonide is a fat-soluble drug, the experiment first uses low-speed centrifugation (1000 rpm, 10 min) to separate the nanoparticles from the undissolved drug, and the undissolved drug sinks to the bottom layer; the upper layer of liquid is removed to an ultrafiltration tube and centrifuged (9000 rpm, 30 min, 4 °C) to separate a small amount of dissolved drug from the nanoparticles. The loading capacity (LC) and entrapment efficiency (EE) were calculated by the following equation.
(1)
(1)
(2)
(2)
where W0 = total amount of TA; W1 = amount of TA in the upper liquid at low speed centrifugation; W2= amount of TA in ultrafiltration filtrate; WN = weight of NPs.
Evaluation of the interactions of nanoparticle complexes
Fourier transform infra-red spectroscopy (FTIR)
Many studies have employed FTIR spectra to study various miscible polymer blend systems [Citation18,Citation19]. Furthermore, the existence of a certain group can be inferred, as well as the combination method of the nanoparticle system can be deduced. FTIR spectra were obtained using FTIR-spectrometer (Perkin–Elmer, spectrum 100, Waltham, USA). Dried samples were mixed with KBr powder and compressed into discs. The each samples pellet was scanned at 4 mm/s at a resolution of 2 cm over a wave number region of 400–4000 cm−1.
Differential scanning calorimetry (DSC)
Differential scanning calorimetry can be used to evaluate the changes in the crystal forms of the drug in the preparations and the interaction between them [Citation20]. The change in enthalpy was the main parameter that was used to judge whether the excipients had altered. DSC thermograms were obtained using an automatic thermal analyser DSC system (DSC400, Perkin–Elmer, Norwalk, CT, USA). The dried samples were loaded and sealed into an aluminium pan and scanned between 30 and 300 °C at heating rate of 10 °C/min, under constant purging nitrogen atmosphere.
X-ray diffraction (XRD)
XRD was used to investigate the physical form (crystalline or amorphous) of drug dispersion within the PLGA matrix of the nanoparticles. XRD analysis was carried out using a D8-Advance X-ray diffractometer (Bruker, Germany) equipped with a position-sensitive detector allowing all angles between 8°Cand 80°Cto be read simultaneously at a scan rate of 1°/min. The system was operated at 40 kV and 40 mA, using the Cu Kα as the radiation source (λ = 1.54 Å).
In vitro drug-release studies
In vitro release studies were evaluated in phosphate buffer saline (PBS, pH 7.4), simulated tear fluids (STF, pH 7.4) [Citation21], and simulated aqueous fluids (SAF, pH 7.4) [Citation22] using a dialysis tube with a molecular weight cut-off of 8000–14,000. Two millilitres of the formulation was placed into a dialysis tube and immersed in 25 ml of release media maintained at 34.5 °C in a shaking water bath (100 rpm). At predetermined intervals, 2.0 ml of the release medium was withdrawn and the same volume of fresh release medium was added to maintain sink conditions. The amount of TA released was quantified by HPLC. The release study was carried out in triplicate. The equation (EquationEquation (3)(3)
(3) ) used to calculate the cumulative percentage of drug release was as follows:
(3)
(3)
where Cn is the drug concentration (μg mL−1) at time t, Ci is the drug concentration at the i-th sampling point, C0 is the total amount of drug in the solution, and V is the sampling volume.
In vitro transcorneal permeation study
The transcorneal experiment was performed with a modified Franz vertical diffusion cell [Citation23]. New Zealand white rabbit s were humanely killed by an intravenous injection of excess urethane, and the whole eyes were enucleated. The method of dissection of the cornea was the same as those reported previously [Citation24]. The receptor compartment was filled with 5 ml of SAF (pH 7.4), while 0.5 of sample (equivalent to 0.1 mg of TA) were applied to the epithelial side. The opening of the donor compartment was sealed with a cover slip and the receptor compartment was maintained at 34 °C with constant stirring, using a magnetic stir bead. At predetermined intervals, 2.0 ml of permeation medium was withdrawn and the same volume of fresh permeation medium was added. The amount of TA permeated was quantified by HPLC. All the experiments were conducted in three repeats. The cumulative permeation amount, (EquationEquation (4)
(4)
(4) ), the apparent permeability coefficient,
(EquationEquation (5)
(5)
(5) ), and the steady state flow rate, Jss (Equation (6)), were calculated for each sample.
(4)
(4)
where Cn is the drug concentration at time t, Ci is the drug concentration at the time point before the last sampling time point t, V0 is the total volume of the medium in the receiving pool (5 ml), and V is the sampling volume (2 ml).
(5)
(5)
where C0 is the initial drug concentration in the supply pool, and △Q/△t is the steady-state slope of the linear portion of the cumulative permeated amount of drug in the receiving chamber (Q) plotted versus time (t).
(6)
(6)
Histopathological study
The in vivo evaluation of the tested formulations for possible ocular irritation potential was conducted following the low-volume eye test procedure, which was actually a modification of the Draize Test [Citation25–27]. The tested eyes were observed at 1, 2, 4, 12, 24, 48, and 72 h to compare changes in the cornea, iris, and the conjunctival secretion while controlling for the bulbar conjunctival oedema. Long-term irritation tests were the same as those of single-dose eye irritation but lasted for 7 days. After irritation tests, the rabbits were sacrificed by air injection. The eyeball was fixed in 4% formaldehyde and embedded in paraffin for histopathological assessment.
In vivo transcorneal penetration
The study was approved by the Institutional Animal Care and Use Committee of Guangdong Pharmaceutical University (Guangzhou, PR China), which ensured that the care and use of animals conformed to the National Institutes of Health guidelines for the care and use of laboratory animals. Studies were performed on nine healthy male New Zealand albino rabbits (weighing 2–2.5 kg) that were randomly classified into 3 groups, receiving aqueous suspension of TA, PLGA NPs, and 2-HP-β-CD/PLGA nanoparticle complexes, respectively.
Rabbits were anaesthetised with the injection of urethane. During the experiment, the rabbit under study was kept anaesthetised throughout the procedure. A CMA-30 microdialysis probe was implanted into the anterior chamber of each rabbit eye as described [Citation28]. microdialysis probe inlet and outlet lines were tunnelled beneath the conjunctiva, under the upper eyelid. The outlets of probe were fixed to prevent any disturbances during the sample collection. After the probe had been implanted, the microdialysis physiological saline solution was used as the perfusate at a perfusion rate of 0.5 After perfusion for 1 h to alleviate the local minimally invasive state, 180 µL (equivalent to 36 μg of TA) of the preparation was dropped into the conjunctival sac of the eye. After administration, samples were collected continuously every 30 min for 6 h. Each sample was processed without treatment, and thus, could be directly tested using HPLC.
Results
Characterisation of the two types of nanoparticle complexes
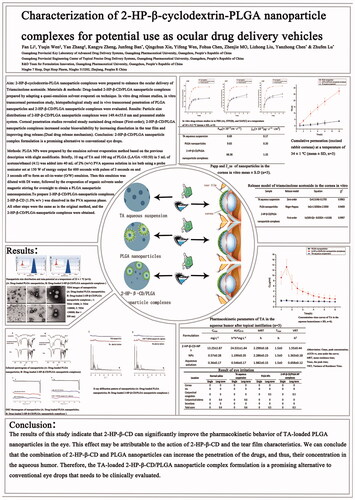
Particle size distributions of the PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes are shown in , respectively. The average particle size of the PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes were 163.2 ± 2.8 and 149.4 ± 3.7 nm, respectively. The PDI values were all within 0.2, i.e., the particle size distribution range was narrow, and the system was relatively uniform. As shown in , the PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes (, respectively) showed negative values of zeta-potential (−16.14 ± 1.23 and −14.05 ± 0.39 mV, respectively).
Figure 1. Size distribution and zeta-potential of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes at a temperature of 25 ± 1 °C (n = 3). Particle size and zeta-potential of PLGA nanoparticles (A) and 2-HP-β-CD/PLGA nanoparticle complexes(B), respectively. PLGA, Polylactic-co-glycolic acid; 2-HP-β-CD, 2-HP-β-cyclodextrin.
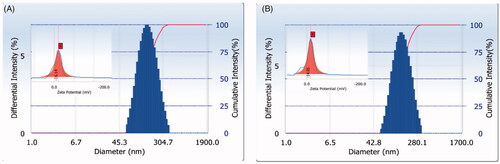
The TEM image of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes are shown in , respectively. The particle size observed under TEM was larger than that recorded using the laser particle size analyser, which may be due to sample drying that causes nanoparticle aggregation during pre-treatment of sample for TEM. Under magnification of 20,000× and 50,000×, PLGA nanoparticles () and 2-HP-β-CD/PLGA nanoparticle complexes () appeared to be spherical and uniformly dispersed. Their particle size was uniform, which further indicated that the prepared nanoparticle system was similar as well.
Figure 2. TEM images of PLGA nanoparticles (A) and 2-HP-β-CD/PLGA nanoparticle complexes (B). (1: TEM ×5000; 2: TEM ×20000; 3: TEM ×50000; Bar = 200 nm). PLGA, Polylactic-co-glycolic acid; 2-HP-β-CD, 2-HP-β-cyclodextrin.
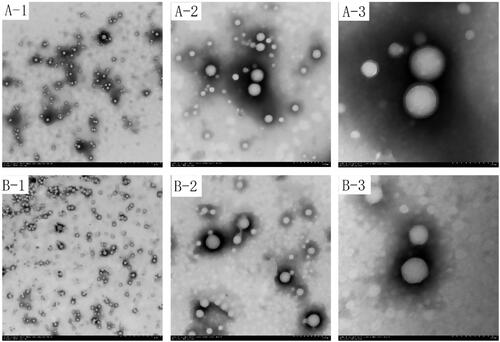
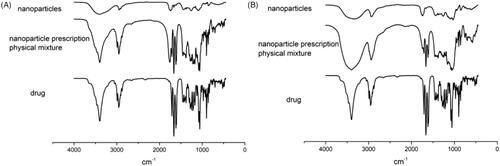
Figure 3. Infrared spectrogram of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes (A: PLGA nanoparticles; B: 2-HP-β-CD/PLGA nanoparticle complexes).
The loading capacity and entrapment efficiency of PLGA NPs and 2-HP-β-cyclodextrin-PLGA nanoparticle complexes was 0.19 ± 0.014% and 18.4 ± 0.7%, 0.52 ± 0.035% and 76.0 ± 4.8% respectively. The addition of cyclodextrin effectively increases the entrapment efficiency, which should benefit from the increased solubility of the drug and the stability of the system due to the presence of cyclodextrin.
Evaluation of interactions in the nanoparticle complexes
The nature of interactions between the drugs and PLGA or 2-HP-β-CD was established with FTIR spectroscopy since any kind of physicochemical interaction that may take place, such as the formation of hydrogen bonds between the drugs and PLGA or 2-HP-β-CD, will automatically lead to frequency shifts or splitting of the absorption peaks. The FTIR spectra of pure TA, the physical mixtures of each component, PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes were interpreted. The results are shown in . From the infra-red spectrum, it can be seen that due to the vibrations of the carbon-carbon double bond (C=C) and ketonic carbonyl (C=O), the TA showed four strong absorption peaks between 1708 cm−1 and 1600 cm−1. The vibration of the aliphatic C-H bond was observed at 2960 cm−1 and 2871 cm−1. The physical mixture also had the same characteristic peaks in these wavelength ranges. In the infra-red spectrum of PLGA nanoparticles () and 2-HP-β-CD/PLGA nanoparticle complexes (), all the characteristic peaks of TA disappeared, while the ketonic carbonyl absorption peak of 1708 cm−1 exhibited a blue-shift to 1740 cm−1. The characteristic peak of TA at 2960 cm−1 exhibited a red-shift to the amorphous state of 2931 cm−1. It is speculated that the interaction between TA and the nanoparticles may be due to the self-assembly of the carrier via hydrogen bonding and the like. The modification of PLGA nanoparticles by 2-HP-β-CD did not affect the presence of TA in the nanoparticles.
Differential scanning calorimetry (DSC) of the PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes were carried out in order to examine the possible changes in the specific heat of PLGA. The result is shown in . It can be seen from the DSC spectrum that TA exhibited a melting endothermic peak at about 290 °C, where the physical mixture also had a characteristic peak, but its absorption peak signal was weak, which may be due to the low content of TA in the sample. In PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes, the characteristic absorption peaks of TA disappeared. This amorphization was in accordance with the XRD patterns (), which indicated that TA was not in a crystalline state [Citation29] within the nanoparticles, as well as with the FTIR spectra (), which indicated the conversion of drug to amorphous and distribution at nano/uniform level in the NPs.
Figure 4. DSC thermogram of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes (A: Drug-loaded PLGA nanoparticles; B: Drug-loaded 2-HP-β-CD/PLGA nanoparticle complexes).
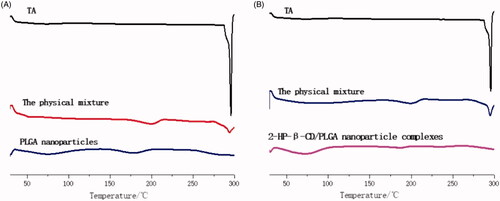
Figure 5. X-ray diffraction pattern of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes (A: Drug-loaded PLGA nanoparticles; B: Drug-loaded 2-HP-β-CD/PLGA nanoparticle complexes).
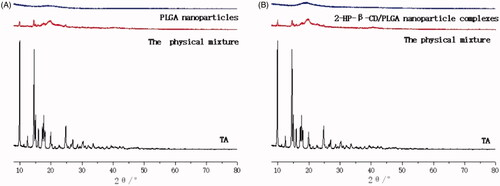
For further identification of the physical state of the drugs incorporated in PLGA nanoparticles, XRD analysis was used and the patterns of pure drugs, as well as TA-loaded PLGA nanoparticles and TA-loaded 2-HP-β-CD/PLGA nanoparticle complexes were obtained. The results from this analysis are shown in . It can be seen from the XRD pattern that the characteristic map of TA showed a plurality of sharp high-intensity peaks at 8–30°, indicating that TA existed in the form of crystals. The physical mixture also exhibited a weak characteristic peak at the corresponding position, which may be due to the small amount of raw material in the physical mixture. Furthermore, the amorphous substance containing PVA, PLGA, or the like was more in amount, resulting in poor crystallinity. However, it was still apparent that the drug was present in its crystalline form in the physical mixture, and no change in its crystallinity occurred. In PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes, the crystal diffraction peak of TA disappeared, which may be due to TA forming a molecular dispersion or an amorphous nanodispersion within the PLGA matrix of the nanoparticles [Citation30,Citation31].
In vitro drug-release studies
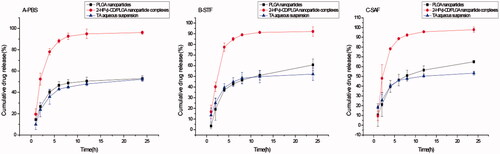
In vitro drug release studies can preliminarily predict the release behaviour of nanoparticles in vivo. Since the bioavailability of the drug is usually low in the form of local eye drops, frequent administration is required, and it is difficult to achieve a therapeutic effect. Therefore, it is important to prepare a nanoparticle capable of releasing and maintaining sufficient drug concentrations for a certain period of time. It can be seen from that the cumulative drug release from 2-HP-β-CD/PLGA nanoparticle complexes in PBS, artificial tears, and artificial aqueous humour is more than 90%. The release amount and release rate of 2-HP-β-CD/PLGA nanoparticle complexes were significantly higher than that of the PLGA nanoparticles and TA aqueous suspension.
Figure 6. The results of in vitro release in PBS (A), STF(B), and SAF(C) at a temperature of 34 ± 0.5 °C (mean ± SD, n = 3).
In vitro transcorneal permeation study
Cyclodextrins have been shown to reduce drug irritation after being topically administered to the eye and to enhance chemical stability of drugs in aqueous eye drop formulations. However, increasing the amount of dissolved drug in the aqueous tear fluid by addition of CDs, while keeping the tear fluid saturated with drug, does not lower the drug activity [Citation32–34]. By modifying the PLGA nanoparticles with 2-HP-β-CD, the overall hydrophilicity of the nanoparticles increased; therefore, nanoparticles were able to better disperse in the tear film.
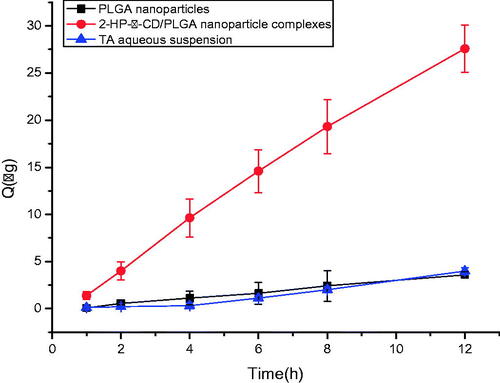
The results of in vitro drug transcorneal permeation test are shown in . The cumulative permeation amount of 2-HP-β-CD/PLGA nanoparticle complexes (27.591 ± 10.05 ) was significantly higher than that of the PLGA nanoparticles (3.973 ± 2.10
) and the TA aqueous suspension (3.567
) after 12 h.
Figure 7. Cumulative permeation (excised rabbit corneas) at a temperature of 34 ± 1 °C (mean ± SD, n = 3) Polylactic-co-glycolic acid; 2-HP-β-CD, 2-HP-β-cyclodextrin.
The cumulative permeability amount () was fitted with time (t) to calculate the apparent permeability coefficient (
). The
of the TA aqueous suspension was
As shown in , the
of TA aqueous suspension was low, which is attributable to the hydrophobic nature of the drug itself that limits its diffusion through the stromal layer. The
of 2-HP-β-CD/PLGA nanoparticle complexes were 4.93 times of PLGA nanoparticles and 7.19 times of TA aqueous suspension, respectively. Furthermore, there was a significant difference (p < .05). It can be further illustrated that the modification of 2-HP-β-CD can produce a significant penetration-enhancing effect on the PLGA nanoparticles.
Table 1. Papp and of nanoparticles in the cornea in vitro mean ± S.D (n = 3).
The steady-state flow rate (Jss) of TA aqueous suspension was The results of the steady-state flow of nanoparticles are shown in . As compared to the PLGA nanoparticles and TA aqueous suspension, the steady-state flow of 2-HP-β-CD/PLGA nanoparticle complexes was significantly improved. Maintaining a drug concentration at saturation in the tear water sample layer helped maintain the driving force for drug diffusion into the aqueous humour and increased the amount of drug that passed through the cornea [Citation35].
From the cumulative drug permeation amount-time curve (), it was deduced that TA in the nanoparticles penetrated the excised cornea at a continuously increasing rate over a period of 8 h. In order to further elucidate the release characteristics of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes, the model was used to fit the cumulative drug permeation amount and time in 8 h, and the release characteristics of different nanoparticles and TA aqueous suspension were investigated. The first-order release model equation, the Higuchi model equation, the Ritger-Peppas model equation, and the zero-order model equation were fitted; the correlation was determined using the model correlation coefficient. The results are given in .
Table 2. Release model of triamcinolone acetonide in the cornea in vitro.
Histopathological study
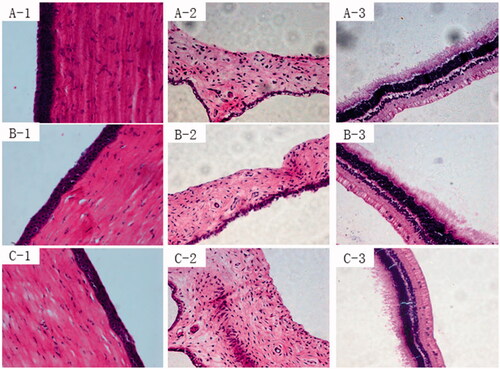
The Draize method was used to evaluate the eye irritation potential of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes, with saline solution and TA aqueous suspension as control materials. For all formulations, the corneal and iris scores were zero (). Although conjunctival hyperaemia was observed in the group administered the 2-HP-β-CD/PLGA NP complexes, there was no significant difference between the groups administered normal saline solution and TA aqueous suspension. Conjunctival hyperaemia led to conjunctival sensitivity to exogenous compounds. The total scores of all formulations were valued between 0 and 3 in a single-dose or long-term eye irritation test. These results showed that PLGA nanoparticles did not stimulate any action in the rabbit eye tissues and were less irritating than the aqueous suspension.
Table 3. Result of eye irritation.
Results from histological analysis of the corneal sections of different formulations after long-term irritation are shown in . As can be seen from , satisfactory epithelium and stroma structure with a little oedema were maintained after the administration of normal saline solution (, respectively). After long-term irritation tests, the corneal epithelial cells of eyes were treated with PLGA nanoparticles that exhibited some slight oedema (, respectively). However, there was no significant difference between the two groups (p > .05).
Figure 8. Histopathology images of the cornea (1), iris (2), and sclera (3) after treatment with: Normal saline(A), PLGA nanoparticles (B), and 2-HP-β-CD/PLGA nanoparticle complexes (C). PLGA, Polylactic-co-glycolic acid; 2-HP-β-CD, 2-HP-β-cyclodextrin.
In vivo transcorneal penetration
In this study, microdialysis was used for pharmacokinetic experiments. As compared to traditional keratotomy, microdialysis is a local minimally invasive sampling technique that can help reduce the sample size of animals and also the differences caused by having many animals as subjects in the experiment. The dialysis window of the microdialysis probe has a certain molecular weight cut-off value; if the collected sample does not contain biological macromolecules, such as proteins and enzymes, it can be directly injected and analysed without further processing [Citation36]. Therefore, sample loss that occurs during sample preparation can be avoided, and the measured results are also closer to the true drug concentration in the body. In the previous study, the positive and negative recovery rates of microdialysis probes during in vitro and in vivo reverse recovery of the anterior chamber were examined. The experimental results showed that these rates of recovery were similar, and there was no significant difference. It was indicated that the in vivo reverse recovery rate can be used to infer the true concentration of the tested drug. Both in vitro and in vivo recovery rates decreased with increasing perfusion rate, regardless of the concentration of perfusate. In the pharmacokinetic experiments, the concentration of the drug in the animal changes with time, and the probe recovery rate is only applicable if it is not affected by the concentration. Taking into account the sensitivity and practical application of the detection instrument, this study used a perfusion rate of 0.5 μL·min−1 for pharmacokinetic experiments.
It can be seen from the drug concentration-time curve in the aqueous humour () that the drug permeation of 2-HP-β-CD/PLGA nanoparticle complexes after eyedrop administration is significantly better than that of PLGA nanoparticles and aqueous drug solution. The pharmacokinetic properties of the drug in aqueous humour after administration of eye drops were investigated. The data are shown in . The peak concentration (Cmax) of 2-HP-β-CD/PLGA nanoparticle complexes group was significantly higher than that of the PLGA nanoparticles group and the TA aqueous solution group, which were 23.2 times and 36.8 times higher, respectively. It was observed that the PLGA nanoparticles that were modified by 2-HP-β-CD were beneficial for the absorption of drugs and improved their bioavailability.
Figure 9. Concentration–time curves of TA in the aqueous humor (mean ± SD, n = 6). PLGA, Polylactic-co-glycolic acid; 2-HP-β-CD, 2-HP-β-cyclodextrin.
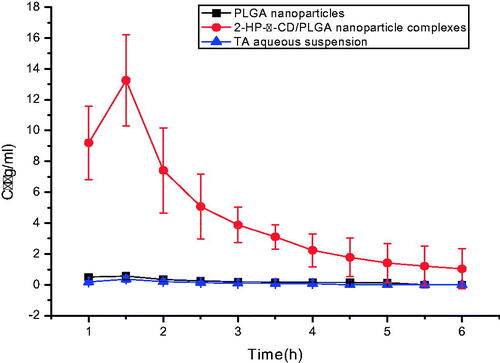
Table 4. Pharmacokinetic parameters of TA in the aqueous humour after topical instillation (n = 3).
The area under the curve (AUC) for the 2-HP-β-CD/PLGA nanoparticle complexes in aqueous humour increased by 20.5 times and 44.9 times as compared to the AUC before modification and that of the aqueous solution, respectively; the mean residence time(MRT) increased by 1.16 times and 1.15 times, respectively, as well. These data indicate that the drug can maintain an effective concentration in the aqueous humour for a longer period of time.
Discussion
PLGA has been widely reported for ocular delivery. However, due to the special environment of the ocular surface and special structure of the cornea, the overall performance of the TA-loaded PLGA nanoparticles has yet to be improved. In our current work, we have developed and evaluated a new colloidal system, 2-HP-β-CD-modified PLGA nanoparticle complexes, for TA delivery that gives better hydrophilicity to the nanoparticles and improves their corneal penetration.
The particle size and zeta-potential of nanoparticles play an important role in the uptake of nanoparticles by cells [Citation37] by affecting their transport and penetration. It has been reported in literature that nanoparticles with a particle size of less than 200 nm can carry drugs around the cornea and deliver them to the posterior segment of the eye [Citation38]. The average particle size of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticle complexes prepared in this study were all less than 200 nm, and the PDI values were all within 0.2, which was consistent with the particle size requirements for delivery to the posterior segment of the eye. The size and zeta-potential of the 2-HP-β-CD/PLGA nanoparticle complexes were both smaller than that of the PLGA nanoparticles, probably because the 2-HP-β-CD in the aqueous phase provided a lipophilic cavity for the system. It was seen in the TEM image that the outer layer of the PLGA nanoparticle was light in colour; the outer layer of 2-HP-β-CD/PLGA nanoparticle complexes was not present, probably due to the modification of 2-HP-β-CD, increasing the hydrophilicity of the outer layer of 2-HP-β-CD/PLGA nanoparticle complexes.
Moving on, DSC, XRD, and FTIR studies were performed to determine if the drug incorporated into the nanoparticle system was crystalline, amorphous, or in a combined form. The PLGA nanoparticles and the 2-HP-β-CD/PLGA nanoparticle complexes did not exhibit peaks indicating the presence of the drug, suggesting that the drug was encapsulated in the polymer matrix or might have taken an amorphous form within the polymer matrix. Combining the results of the three spectra, it can be further inferred that TA may exist in the nanoparticles in an amorphous state. It can be seen from the comparative results of the map that modification of the 2-HP-β-CD does not affect the form that the drug takes when it is present within the nanoparticles.
In vitro release profile showed an initial rapid release of 2-HP-β-CD/PLGA nanoparticle complexes. This initial rapid release of the drug may be due to the trance drug adsorbed on the surface of the 2-HP-β-CD/PLGA nanoparticle complexes [Citation39,Citation40], and also due to some of the drug being released from the 2-HP-β-CD. In this initial release, the effective drug concentration can be achieved in a short time, which is helpful in improving the bioavailability of the drug. In the medium term, TA continues to be released from the polymer due to hydration and swelling of the polymer [Citation41–43]. The 24 h cumulative release of 2-HP-β-CD/PLGA nanoparticle complexes was higher than 90%, and the release amount and release rate were significantly higher than PLGA nanoparticles and TA suspension. The reason may be that the addition of cyclodextrin to the polymer system can enhance drug release by acting as a channelling or wicking agent or by promoting erosion of the matrix [Citation44,Citation45]. It is also possible that cyclodextrin promotes hydration of polymer systems [Citation46]. It was seen in the TEM image that the surface of the 2-HP-β-CD/PLGA nanoparticles is rougher than the surface of the PLGA nanoparticles, and there are more black spots, which may be grooves or voids on the surface of the nanoparticle. These all promote the erosion of the matrix and the release of the drug.
The results of in vivo transcorneal penetration experiments show that 2-HP-β-CD-modified PLGA nanoparticles can significantly increase the cumulative permeation of nanoparticles so that it becomes much higher than that of the PLGA nanoparticles and TA aqueous suspension. Pharmacokinetic experiments indicated that 2-HP-β-CD-modified PLGA significantly increased the area under the drug concentration-time curve of the nanoparticles in the aqueous humour, the average residence time in the body, and the peak drug concentration in the aqueous humour. 2-HP-β-CD/PLGA nanoparticle complexes significantly increased the bioavailability of TA in the eye. The result is consistent with the in vitro transcorneal permeation study. This indicates that a hydrophilic modification is added to facilitate corneal permeation of the nanoparticles. This may be due to the fact that hydrophobic PLGA is easily lost by tears flushing into the nasolacrimal duct, while hydrophilic 2-HP-β-CD can penetrate the tear film by carrying a lipophilic molecule that is poorly soluble in water. Therefore, the drug can be passively transported into the anterior chamber of the eye to improve the bioavailability of the drug.
In summary, our findings suggest that 2-HP-β-CD/PLGA nanoparticle complexes may achieve desired therapeutic effects by improving dispersion of TA in the tear film and enhancing corneal permeation, supporting improved bioavailability and effective delivery of drugs to eye.
Conclusion
The results of this study indicate that 2-HP-β-CD can significantly improve the pharmacokinetic behaviour of TA-loaded PLGA nanoparticles in the eye. This effect may be attributable to the action of 2-HP-β-CD and tear film characteristics. We can conclude that the combination of 2-HP-β-CD and PLGA nanoparticles can increase the penetration of the drugs, and thus, their concentration in the aqueous humour. Therefore, the TA-loaded 2-HP-β-CD/PLGA nanoparticle complex formulation is a promising alternative to conventional eye drops that needs to be clinically evaluated.
| Abbreviations | ||
| PLGA | = | Polylactic-co-glycolic acid |
| NPs | = | nanoparticles |
| TA | = | Triamcinolone acetonide |
| PVA | = | Polyvinyl alcohol |
| PBS | = | phosphate buffer saline |
| STF | = | simulated tear fluids |
| SAF | = | simulated aqueous fluids |
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Mo Z, Ban J, Zhang Y. Nanostructured lipid carriers-based thermosensitive eye drops for enhanced, sustained delivery of dexamethasone. Nanomedicine-UK. 2018; 13(11):1239–1253.
- Ban J, Zhang Y, et al. Corneal permeation properties of a charged lipid nanoparticle carrier containing dexamethasone. Int J Nanomedicine. 2017;12:1329–1339.
- Tavazzi S, Rossi A, Picarazzi S, et al. Polymer-interaction driven diffusion of eye shadow in soft contact lenses. Cont Lens Anterior Eye. 2017;40(5):335–339.
- Upendra N. Recent advances in ocular drug delivery system. J Adv Pharm Technol Res. 2014;5(4):151.
- Tg DSM, Alf DAC, Centelhas AC, et al. Use of biodegradable collagen-glycosaminoglycan copolymer matrix implant to reduce postoperative fibrosis in strabismus surgery. Taiwan J Ophthalmol. 2017;7(4):227–229.
- Almeida H, Lobã OP, et al. Preparation, characterization and biocompatibility studies of thermoresponsive eyedrops based on the combination of nanostructured lipid carriers (NLC) and the polymer Pluronic F-127 for controlled delivery of ibuprofen. Pharm Dev Technol. 2017;22(3):336–349.
- Imperiale JC, Acosta GB, Sosnik A. Polymer-based carriers for ophthalmic drug delivery. J Control Release. 2018;285:106.
- Silva-Abreu M, Calpena AC, et al. Optimization, biopharmaceutical profile and therapeutic efficacy of pioglitazone-loaded PLGA-PEG nanospheres as a novel strategy for ocular inflammatory disorders. Pharm Res. 2018;35(1):11.
- Kalam MA, Alshamsan A. Poly (d, l-lactide-co-glycolide) nanoparticles for sustained release of tacrolimus in rabbit eyes. Biomed Pharmacother. 2017;94:402–411.
- Gupta H, Aqil M, Khar RK, et al. Sparfloxacin-loaded PLGA nanoparticles for sustained ocular drug delivery. Nanomedicine-UK. 2010;6(2):324–333.
- Varshochian R, Riazi-Esfahani M, et al. Albuminated PLGA nanoparticles containing bevacizumab intended for ocular neovascularization treatment. J Biomed Mater Res.. 2015;103(10):3148–3156.
- Salama HA, Ghorab M, Mahmoud AA, et al. PLGA nanoparticles as subconjunctival injection for management of glaucoma. AAPS Pharmscitech. 2017;18(7):2517–2512.
- Aachmann FL, Otzen DE, Larsen KL, et al. Structural background of cyclodextrin-protein interactions. Protein Eng Design Select. 2003;16(12):905–912.
- Vega E, Egea MA, Calpena AC, et al. Role of hydroxypropyl-β-cyclodextrin on freeze-dried and gamma-irradiated PLGA and PLGA–PEG diblock copolymer nanospheres for ophthalmic flurbiprofen delivery. 2012;7:1357–1371.
- Hayiyana Z, Choonara YE, Makgotloe A, et al. Ester-based hydrophilic cyclodextrin nanosponges for topical ocular drug delivery. Curr Pharm Design. 2016;22(46):6988–6997.
- Lutfi G, Muzeyyen D. Preparation and characterization of polymeric and lipid nanoparticles of pilocarpine HCl for ocular application. Pharm Dev Technol. 2013;18(3):701–709.
- Huang X. Preparation and preliminary study of targeting of cyclodextrin coated PLGA nanoparticles [master’s thesis]. Guangzhou: Guangdong Pharmaceutical University; 2018.
- Varnell DF, Coleman MM. FT i.r. studies of polymer blends: V. Further observations on polyester-poly(vinyl chloride) blends. Polymer. 1981;22(10):1324–1328.
- Varnell DF, Moskala EJ, Painter PC, et al. On the application of fourier transform infrared spectroscopy to the elucidation of specific interactions in miscible polyester‐poly(vinyl chloride) blends. Polym Eng Sci. 2010;23(12):658–662.
- Pani NR, Nath LK, Acharya S, et al. Application of DSC, IST, and FTIR study in the compatibility testing of nateglinide with different pharmaceutical excipients. J Therm Anal Calorim. 2012;108(1):219–226.
- Deepthi S, Jose J. Novel hydrogel-based ocular drug delivery system for the treatment of conjunctivitis. Int Ophthalmol. 2018;39(6):1355–1366.
- Macri A, Marini V, Sangalli G, et al. An artificial aqueous humor as a standard matrix to assess drug concentration in the anterior chamber by high performance liquid chromatography methods. Clin Lab. 2015;61(1–2):47–52.
- Paolo M, Simone E, et al. Trans-scleral diffusion of triamcinolone acetonide. Curr Eye Res. 2005;30(5):355–361.
- Malhotra M, Majumdar DK. In vitro transcorneal permeation of ketorolac tromethamine from buffered and unbuffered aqueous ocular drops. Indian J Exp Biol. 1997;35(9):941–947.
- Wilhelmus KR. The Draize eye test. Surv ophthalmol. 2001;45(6):493–515.
- Abdelkader H, Pierscionek B, Carew M, et al. Critical appraisal of alternative irritation models: three decades of testing ophthalmic pharmaceuticals. Brit Med Bull. 2015;113(1):59–71.
- Wolfe GW. Preclinical safety study design templates and estimated costs. Hoboken, NJ: John Wiley & Sons, Inc., 2010.
- L Nnroth P, Jansson PA, Smith U. A microdialysis method allowing characterization of intercellular water space in humans. Am J Phys. 1987;253(1):228–231.
- Bikiaris D, Papageorgiou GZ, et al. Physicochemical studies on solid dispersions of poorly water-soluble drugs. Thermochim Acta. 2005;439(1-2):58–67.
- Kaushal AM, Gupta P, Bansal AK. Amorphous drug delivery systems: molecular aspects, design, and performance. Crit Rev Ther Drug. 2004;21(3):133.
- Gupta P, Bansal AK. Devitrification of amorphous celecoxib. AAPS Pharmscitech. 2005;6(2):E223–E230.
- Ahuja M, Dhake AS, Sharma SK, et al. Topical ocular delivery of NSAIDs. AAPS J. 2008;10(2):229–241.
- Rodriguez-Aller M, Guinchard S, et al. New prostaglandin analog formulation for glaucoma treatment containing cyclodextrins for improved stability, solubility and ocular tolerance. Eur J Pharm Biopharm. 2015;95:203–214.
- Bozkir A, Denli ZF, Basaran B. Effect of hydroxypropyl-beta-cyclodextrin on the solubility, stability and in-vitro release of ciprofloxacin for ocular drug delivery. Acta Pol Pharm. 2012;69(4):719–724.
- Loftsson T, Stefánsson E. Cyclodextrins and topical drug delivery to the anterior and posterior segments of the eye. Int J Pharmaceut. 2017;531(2):413–423.
- Rittenhouse KD, Pollack GM. Microdialysis and drug delivery to the eye. Adv Drug Deliver Rev. 2000;45(2–3):229–241.
- Ha H, Kim JW, Lee M, et al. Cellular uptake and cytotoxicity of β-lactoglobulin nanoparticles: the effects of particle size and surface charge. Asian-Australas J Anim Sci. 2015;28(3):420–427.
- Sah AK, Suresh PK. Recent advances in ocular drug delivery, with special emphasis on lipid based nanocarriers. Recent Pat Nanotechnol. 2015;9(2):94–105.
- Mogi T, Ohtake N, et al. Sustained release of 17β-estradiol from poly (lactide- co -glycolide) microspheres in vitro and in vivo. Coll Surf B Biointerf. 2000;17(3):153–165.
- Meng J, Sturgis TF, Youan B-BC. Engineering tenofovir loaded chitosan nanoparticles to maximize microbicide mucoadhesion. Eur J Pharm Sci. 2011;44(1–2):57–67.
- Fazil M, Md S, Haque S, et al. Development and evaluation of rivastigmine loaded chitosan nanoparticles for brain targeting. Eur J Pharm Sci. 2012;47(1):6–15.
- Amidi M, Romeijn SG, Borchard G, et al. Preparation and characterization of protein-loaded N-trimethyl chitosan nanoparticles as nasal delivery system. J Control Release. 2006;111(1–2):107–116.
- Makino K, Mogi T, et al. Pulsatile drug release from poly (lactide-co-glycolide) microspheres: how does the composition of the polymer matrices affect the time interval between the initial burst and the pulsatile release of drugs?. Coll Surf B Biointerf. 2000;19(2):173–179.
- Villar-López ME, Nieto-Reyes L, Anguiano-Igea S, et al. Formulation of triamcinolone acetonide pellets suitable for coating and colon targeting. Int J Pharm. 1999;179(2):229–235.
- Bibby DC, Davies NM, Tucker IG. Mechanisms by which cyclodextrins modify drug release from polymeric drug delivery systems. The Netherlands: Elsevier B.V; 2000. p. 1–11.
- Gã Rsoy A, Tã Rkoä Lu M, Senyã Cel B, et al. Evaluation of tableted microspheres of dipyridamole. Drug Dev Commun. 1995;21(4):503–507.