
ABSTRACT
Avian influenza viruses (AIV) have been classified on the basis of 16 subtypes of hemagglutinin (HA) and 9 subtypes of neuraminidase. Here we describe genomic evidence for a new candidate HA subtype, nominally H19, with a large genetic distance to all previously described AIV subtypes, derived from a cloacal swab sample of a Common Pochard (Aythya ferina) in Kazakhstan, in 2008. Avian influenza monitoring in wild birds especially in migratory hotspots such as central Asia is an important approach to gain information about the circulation of known and novel influenza viruses. Genetically, the novel HA coding sequence exhibits only 68.2% nucleotide and 68.5% amino acid identity with its nearest relation in the H9 (N2) subtype. The new HA sequence should be considered in current genomic diagnostic AI assays to facilitate its detection and eventual isolation enabling further study and antigenic classification.
Introduction
Metapopulations of wild aquatic birds (waterfowl and shorebirds) represent the primary natural reservoir of avian influenza A viruses (AIVs) of which 16 hemagglutinin (HA) and nine neuraminidase (NA) subtypes are currently known. Two further subtypes, H17 and H18, were recently discovered in Little yellow-shouldered (Sturnia lilium) and a flat-faced fruit bats (Artibeus planirostris) respectively, but not in any avian species or human to date [Citation1,Citation2]. The most recent IAV subtype identified in an avian species was the H16 subtype which was isolated from a Black-headed gull in 2005 [Citation3]. An important part of our knowledge regarding low pathogenicity avian influenza viruses (LPAIVs) has been acquired via wildlife monitoring studies. Such wild bird studies have been carried out over the past few decades, especially after emergence and geographical expansion of high pathogenicity AIVs (HPAIVs) of the H5Nx subtype since 2005. Yet, no evidence for another new AIV subtype has been reported since, until now [Citation4–12].
We have examined approximately 8000 oropharyngeal and cloacal samples in the framework of an AIV monitoring programme in wild birds in Kazakhstan from 2006 to 2011. Molecular screening of samples and subsequent sequencing of AIV positive samples revealed a number of known AIV subtypes but a single influenza A virus HA sequence could not be classified within the existing subtypes. Here we describe sequence analyses of this sample providing genomic evidence for a novel HA subtype of AIV from a Common Pochard (Aythya ferina). With more than 30% genetic distance [Citation3,Citation13], based on the HA coding region nucleotide sequence, to its closest relatives in the H9 subtype, the novel sequence represents a potential candidate new subtype, nominally H19.
Materials and methods
Study design
In the framework of an AIV wild bird monitoring programme in Kazakhstan during 2002 to 2009, approximately 4800 cloacal and oropharyngeal swab samples from 155 different species of wild birds were collected and analysed [Citation11]. The study was previously described comprehensively [Citation11], in summary, samples were tested using virus culture in embryonated chicken eggs, conventional RT–PCR, and a generic quantitative real-time RT–PCR (RT-qPCR) targeting the AIV nucleoprotein (NP) gene [Citation14]. In total, 95 AIV from different subtypes, especially H3, H4, and H13 subtypes, were isolated. In addition, 99 positive samples were identified using conventional RT–PCR and quantitative RT–PCR (RT-qPCR). A cloacal swab sample with sample ID of Kz52, that was collected on 23.11.2008 from a Common Pochard (Aythya ferina) was among the RT-qPCR positives. Positive RT-qPCR samples, including Kz52 were used for re-inoculation in embryonated chicken SPF (specific pathogen free) eggs, MDCK II (Madin-Darby canine kidney) cell culture cultivation, genome amplification, and sequencing.
Virus isolation and characterization
Virus isolation was attempted in 11-day-old embryonated SPF chicken eggs and MDCK II cell cultures based on standard procedures [Citation15]. The samples were passaged two or three times before being considered negative based on lack of hemagglutination activity and negative generic RT-qPCR tests.
RNA extraction
RNA was extracted from the original swab sample using the QIAamp Viral RNA kit (Qiagen) for the conventional RT–PCR or sequencing, according to the manufacturer’s instructions. RNA for RT-qPCR analysis was extracted directly from the samples by an automated RNA extraction robot (Freedom Evo 3000, Tecan) using the NucleoSpin 96 Virus Core kit (Macherey & Nagel).
Quantitative real-time RT–PCR (RT-qPCR)
A previously validated generic quantitative real-time RT–PCR (RT-qPCR), using forward and reverse primers (1448-F and 1543-R) and probe (1473-FAM), targeting the AIV NP gene was used for screening of the samples or evaluation of cultivated cell cultures, and viral load in original sample was estimated [Citation14,Citation16].
RT–PCR and sequencing
Reverse transcription-PCR (RT–PCR) assays were performed via one-step protocols using the Qiagen RT–PCR Kit according to the manufacturers’ instructions. Hoffmann primers were used for full-length amplification of genomic RNA and further sequencing of different viral segments, including polymerase basic 2 (PB2), polymerase basic 1 (PB1), polymerase acidic (PA), HA, NP, NA, matrix (M), and non-structural protein (NS) genes [Citation17]. PCR products of the anticipated size range were purified using the QIAquick Gel Extraction Kit (Qiagen, Germany). Purified DNA fragments were cycle sequenced in both directions using the same primers as for RT–PCR. The Prism Big Dye Terminator v1.1 cycle sequencing kit (Applied Biosystems) was utilized and amplicons were analysed on an automatic sequencer (ABI-377, Applied Biosystems). Assembled nucleotide sequences were then used in BlastN2 database searches for subtype specification.
A second round of sequencing focused specifically on the HA gene of sample Kz52 and was conducted in collaboration with Virology Research Services (Sandwich, UK) and was achieved via two approaches. Firstly, the sequence of the N-terminal two-thirds (∼1150 bp) was obtained by one strategy, followed by the C-terminal one third (∼600 bp) by an alternative approach, both described below. For the former, total extracted RNA was retro-transcribed using random hexamer and influenza HA-specific primers (details below) to initiate first round cDNA synthesis via Superscript Reverse Transcriptase (ThermoFisher) priming at 65°C for 5 min (min), reverse transcribed at 55°C for 10 min, then deactivated at 80°C for 10 min. HA sequences from this cDNA were then amplified by polymerase chain reaction (PCR) using HA-specific primers (details below) and Phusion Hi-Fidelity DNA Polymerase (ThermoFisher) according to manufacturer’s instructions, using the thermal cycling; initially 98°C for 30 s, then 25 cycles of 98°C for 10 s + 52°C for 30 s + 72°C for 30 secs, with final extension at 72°C for 5 min. PCR products (∼300–400 bp) were purified by gel electrophoresis and extraction. These products were Sanger sequenced (Source Bioscience, Cambridge, UK) and used to design novel primers to produce further nested PCR products for sequencing. These sequences were assembled into the ∼1150 bp consensus. Despite several attempts, it was not possible to extend this sequence further by the same methodology, possibly due to target degradation. Consequently, a second “gene walking” approach was employed. This involved using a PCR primer based on a known sequence adjacent to unknown region (5′-AAAGAGACACCGCTCAAG-3′ using above cycle) to produce ssDNAs of varying lengths from the cDNA above, followed by non-specific binding of reverse primers to generate dsDNAs (single cycle of 98°C for 30 s + 45°C for 30 s + 72°C for 3 min). A second round of nested PCR was conducted (30 cycles, as above) to produce more amplicons that were gel purified and sequenced. Lastly, the N and C-terminal sequences were assembled as a single 1686 nucleotide contiguous open reading frame.
Sequences of primers utilized in above methodologies:
Primers used for RNA retro-transcription reaction I:
Rev2: 5′-AAAGGTACTCTTGTTCTGATT-3′
Rev1: 5′-TCAGATACAAATGGTGCATTTG-3′
Primers used for RNA retro-transcription reaction II:
Rev1: 5′-TCAGATACAAATGGTGCATTTG-3′
Primers used for RNA retro-transcription reaction III:
Rev3: 5′-CGAAGTTATCTTATCAATGGCC-3′
Primers used for PCR reactions:
HAFor: 5′-ATGTGGAAACTAGCATTAGTAACG-3′
HARev: 5′-AAAGGTACTCTTGTTCTGATT-3′
HA1For: 5′-TTAGTAACGACTTTTTTGATGC-3′
HA1Rev: 5′-ACTTGAGGAAGAGAATAAGAC-3′
HA2For: 5′-AAAGAGACACCGCTCAAG-3′
HA2Rev: 5′-ATTTTTAAAGTCTGATTGGGC-3′
HA3For: 5′-GAGTTTCAAGTCAACATTCGG-3′
HA3Rev: 5′-CGAAGTTATCTTATCAATGGCC-3′
HA4For: 5′-AATTCAGAGGGAACAGGAATGG-3′
HA4Rev: 5′-TCAGATACAAATGGTGCATTTG-3′
Estimates of evolutionary divergence between sequences
The number of base substitutions per site between sequences were calculated using the Tajima-Nei model implemented within MEGA 11 software. The rate variation among sites was modelled with a gamma distribution. Codon positions included were 1st + 2nd + 3rd + Noncoding. All positions with less than 95% site coverage were eliminated and ambiguous bases were allowed at any position (partial deletion option). There were a total of 1680 positions in the final dataset. Evolutionary analyses were conducted in MEGA 11.
Prediction of potential N-glycosylation sites
Potential N-glycosylation sites (PGS) of the HA were predicted using the NetNGlyc 1.0 Server (https://services.healthtech.dtu.dk/services/NetNGlyc-1.0).
Phylogenetic analyses
Phylogenetic analyses were carried out for the complete open reading frame (ORF) of the HA gene of A/Common Pochard/Kazakhstan/Kz52/2008 (Kz52 in brief) AIV and representatives of other influenza A virus subtypes. Ninety-one representative sequences of all AIV and bat-origin HA subtypes for phylogenetic analysis were retrieved from the NCBI Influenza Database, aligned, and trimmed to equal lengths using BioEdit 7.2.5.
The trees were generated using a maximum-likelihood (ML) approach based on a General Time Reversible (GTR) model with rate heterogeneity and a proportion of invariant sites with 1000 bootstrap replicates implemented within MEGA 11 software. In addition, all trees were reconstructed and confirmed using the neighbor-joining algorithm based on the Tamura 3-parameter model, rate heterogeneity and 1000 bootstrap replicates.
Three-dimensional (3D) structural modelling of HA
A 3D model of the Kz52 HA polypeptide was predicted using the free software Phyre2 [Citation18; www.sbg.bio.ic.ac.uk/phyre2]. Modelling compared the novel HA sequence with that of other HAs for which structures have been determined. The best structural alignment was with H16 (A/black-headed gull/Sweden/2/99; NCBI accession number AY684888.1). Phyre 2 “normal mode” default settings were used to predict the 3D models of HA. The Phyre 2 PDB file was then imported into the PyMOL program (www.pymol.org) in order to highlight specific features on the HA structure.
Results
One cloacal swab sample, Kz52, that was collected from a Common Pochard was considered positive in a generic AIV-specific RT-qPCR with low viral load of about 102–103 viral genome copies per 100 µl of cloacal swab suspension (Ct value equal to 34.12 in original swab material). No virus could be propagated either in embryonated chicken SPF eggs, or MDCK cell cultures, possibly because the sample had already experienced several cycles of freezing and thawing following initial analysis, storage, shipment, and RNA extraction phases, before any attempts at virus isolation were made and the sample included low virus load. RNA extracted from the residual sample was only adequate for complete sequencing of the hemagglutinin and short fragments of three other segments.
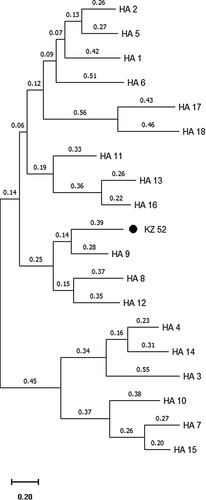
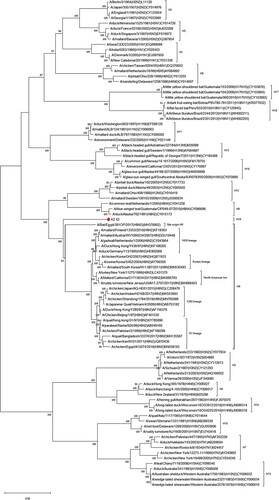
The sequencing of the HA gene via different complimentary approaches and protocols finally revealed a complete sequence of the HA open reading frame (ORF) comprising 1686 nucleotides (including start and stop codons) and 561 amino acids (). Extensive database searches and estimation of evolutionary divergence revealed that the HA gene shared only 68.2% nucleotide and 68.5% amino acid sequence identity with its closest related sequences, all found among strains of the H9 subtype isolated from wild and domestic birds and bats (https://blast.ncbi.nlm.nih.gov/Blast.cgi), such as A/duck/NZL/76/1984(H9N1), A/chicken/Korea/GH2/2007(H9N2) and A/Bat/Egypt/381OP/2017(H9N2), respectively. Kz52 is placed as a sister clade to the H9 subtype with subtypes H8 and H12 being closely related to the sister clade ((a and b)).
Figure 1. The complete open reading frame (ORF) of the HA gene of A/ Common Pochard/Kazakhstan/Kz52/2008 influenza virus (new subtype), compared to closely related H9 subtype isolates. Representative isolates for each lineage included: H9-bat: A/Bat/Egypt/381OP/2017(H9N2), H9-Y280: A/Chicken/Beijing/1/97(H9N2), H9-G1: A/quail/Hong Kong/G1/97(H9N2), H9-Y439: A/Duck/Hong Kong/Y439/97(H9N2), H9-N. America: A/Mallard/California/D1713634/2017(H9N2), H9-Korea: A/chicken/Korea/GH2/2007(H9N2).

Figure 2. Phylogenetic analysis of Kz52 HA coding region nucleotide sequences and the representative sequences of other influenza subtypes using obtained from publicly available sequence datasets (GenBank). (a) The distances and (b) the corresponding bootstrap values. Analysis was based on full-length coding sequence of the HA gene. Only bootstrap values >80 are shown. Bar represents 0.01 nt substitutions.
Due to the low viral load of the specimen, the gene-specific RT–PCR assays did not reveal any positive PCR product for PB2, PA, NA, and M genes of Kz52; and partial sequences were generated only for PB1 (620 of 2370 nt), NP (310 of 1594 nt), and NS (330 of 919 nt) genes.
Analysis of partial sequences of the NP gene shows 98.4% sequence identity with A/mallard/Sweden/124418/2010 (H4N6), NS with 93% identity with A/mallard/Sweden/104690/2009 (H4N6), and PB1 with 93.7% identity with A/mallard/Netherlands/10/1999 (H1N8) revealing much higher sequence identity to known naturally occurring wild bird influenza viruses of the Eurasian lineage compared to the HA. However, the phylogenetic analyses indicated that Kz52 NS and PB1 sequences form a separate branch even compared to closely related correspondent sequences (Figures S1–S3). The HA sequence has been deposited in Genbank database with accession number ON637239.
The Kz52 HA cleavage site was defined as PIKETR↓GLF (with G at position 340). Despite the fact that Kz52 HA showing the highest sequence identity and 3D predicted HA model similarity () with H9 subtypes, its cleavage site pattern was identical to that of the bat influenza strain A (H18N11) isolated in South America [Citation2]. The cleavage motif of Kz52 has two basic amino acids that are seen often in low pathogenic H5 AIV strains. Historically, amino acid substitutions at positions E190D and G225D for H1N1 [Citation19] and Q226L with G228S in the case of H2N2 and H3N2 (H3 numbering) [Citation20] within the HA receptor-binding site (RBS) of all influenza viruses have been shown to play a key role in transition from avian-like to human-like terminal sialic acid receptor recognition. The Kz52 HA RBS has typical avian residues at the corresponding positions: E190, G225, and G228 but has a substitution Q226I that differed from the closest H9 subtype sequences that have leucine or glutamine at this position. In addition, the HA protein sequence of Kz52 has tyrosine 98 (Y98), serine 136 (S136), tryptophan 153 (W153), and histidine 183 (H183) forming the base of the RBS which is conserved among all HA subtypes [Citation21].
Figure 3. Schematics showing predicted 3D models of the Kz52 HA Ribbon (right) and surface (left) formats generated using Phyre2 software. The images show the HA1 and HA2 subunits (green and yellow respectively), the cleavage site (red), and receptor-binding sites (blue).
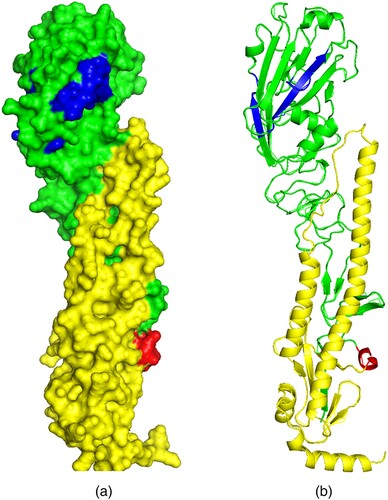
Six potential N-glycosylation sites (PGSs) at positions 30, 142, 193, 249, 299, and 306 in HA1, and one 493 in HA2 were identified. Two of those PGSs, at positions 193 and 249, have not been observed in avian and bat-origin H9 viruses [Citation22]. None of these PGS affected the RBSs.
Discussion
Our knowledge regarding the circulation of AIVs in wild bird populations as potential donors of previous pandemic causing influenza viruses, the relative prevalence of various subtypes in different species, susceptibility or resistance of different bird species to different genetic lineages of AIV subtypes, mainly stems from monitoring and surveillance studies in an extensive range of countries [Citation4–11,Citation23]. Thus, continued influenza monitoring of wild bird populations is essential to assess potential disease impact in these species, domestic birds, and mammals, including man.
As a result of such activities, the genomic sequence of a potentially novel HA subtype of AIVs was identified from a cloacal swab sample of a Common Pochard (Aythya ferina), A/ Common Pochard/Kazakhstan/Kz52/2008. Unfortunately, due to the low virus load in the bird, the virus itself could not be isolated. Despite successful sequencing of the entire HA ORF, it was not possible to obtain the complete sequence of the Kz52 genome or further complete ORFs with the remaining viral genomic RNA. Nevertheless, partial sequences of other genes (NP, NS, and PB1) were obtained, providing some further genetic information.
The HA of this virus is genetically significantly divergent from all known AIVs, and appears to represent a separate monophyletic group established as a sister clade to the H9 phylogenetic branch. The H9 HA sequence obtained from bats in Egypt is likewise close to the root of H9, although it is reported to represent a true H9 subtype with an identity of 73% to avian H9 viruses [Citation22]. The full HA coding region of Kz52 showed only 68.2% sequence homology with closest related H9 subtypes viruses. In addition, as the genetic distance on the basis of amino acid sequences from other known subtypes was more than 30%, in accordance with current AIV subtype classification [Citation3,Citation13], thus we suggest the Kz52 as a potential candidate for a new subtype, although antigenic classification is essential to confirm this, as described in WHO guidelines [Citation24]. Nevertheless, this represents the first new avian HA sequence as substantially distinct from all other subtypes, as of the most recent additions, H16 AIV in 2005 [Citation3], and two bat-origin influenza viruses identified a decade ago [H17, H18: 1-2].
Since this new HA subtype remained undetected despite extensive AIV monitoring in wild birds, it is possible that the corresponding AIV may only propagate in very limited numbers of avian species, or there may be a strict geographic restriction. The intensified monitoring of wild birds at the location where this sample has been obtained would be a logical strategy to obtain further material for analysis of this new subtype candidate, including isolating the virus itself. A specifically designed RT-qPCR would aid in high throughput screening of such new samples and existing sample banks [Citation25]. The sample was collected in the Delta of Ural River and the North Caspian region, one of the important areas for stopover, accumulation, and nesting of migratory birds, where the Black Sea-Mediterranean and East African-West Asian migratory flyways intersect. The retrospective confirmation of circulation of H16 subtype AIVs among gulls in the Northern Caspian since 1976 [Citation26] and first isolation of the H14 subtype from this region [Citation13] indicates that the Northern Caspian is one of the potential epicenters for emerging novel and rare influenza A virus subtypes.
The close phylogenetic association of Kz52 with bat-origin H9 sequences [Citation22] is strikingly paralleled by its HA cleavage site which was similar to bat H18N11 influenza viruses [Citation2] but has a typical avian RBS (at positions of E190, G225, and G228). However, the unique substitution Q226I was observed at the RSB key position of Kz52, while H9 subtype viruses mainly contain leucine (L) or glutamine (Q) at the position 226 [Citation27]. Although the novel subtype hemagglutinin possesses avian-like RBS, experimental studies are necessary to determine the role of its RBS in host specificity.
We were not able to amplify the Neuraminidase (NA) gene using a range of sequencing primers previously used for all other known NAs. This may indicate that NA was also genetically different from previously identified NAs. Among three partial sequences of other viral segments, only NP showed very high genetic identity (98%) with NP of other circulating Eurasian AIVs in wild birds. NS and PB1 fragments showed approximately 93% genetic relatedness with other NS and PB1 of AIVs. Although short fragment sequencing of other segments such as NP, NS, and PB1 revealed different levels of genetic relatedness with commonly circulating genes in wild birds, without access to the virus or complete genome of the virus, a definitive conclusion about the genetic background of the virus is not possible. A small amount of original genomic RNA was preserved for future investigations, when genome sequencing techniques might permit further refined analysis, prior to reconstructing a replication-competent virus by reverse genetics. There are several Next Generation Sequencing approaches, which might be applied, such as the multisegment RT–PCR method [Citation28], or more recent Oxford Nanopore minION direct RNA sequencing platform [Citation29], however, the current scarcity of sample material is a barrier to progress. This highlights the need for further wildlife surveillance to provide new material, combined with improved detection with novel PCR primers. Nevertheless, we have cloned the HA gene into expression vectors and are currently devising serological tests for antigenic subtyping and potential subtype delineation [unpublished data].
With the current intensified AIV surveillance in wild and domestic animals, it is probable that further viruses closely related to Kz52 will be detected, and indeed further AIV subtypes. The development of diagnostic kits and updating the primers for AIV screening using the results of the current study may facilitate further identification of related influenza viruses and aid in specific monitoring in a range of avian species [Citation30]. Increasing usage of whole genome sequencing (WGS)/metagenomics as a diagnostic tool could be considered more with wild bird samples, which will help identify rare and divergent subtypes. Identification of a such divergent HA sequence of AIV in a Common Pochard indicates the continuing emergence of influenza viruses and emphasizes the intriguing variability of these viruses. It seems that the full spectrum of variability of AIV remains to be unravelled yet.
Supplemental Material
Download PDF (80.6 KB)Supplemental Material
Download PDF (72.8 KB)Supplemental Material
Download PDF (79.5 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Tong S, Li Y, Rivailler P, et al. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci USA. 2012;109(11):4269–4274. doi:10.1073/pnas.1116200109
- Tong S, Zhu X, Li Y, et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013;9(10):e1003657. doi:10.1371/journal.ppat.1003657
- Fouchier RA, Munster V, Wallensten A, et al. Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J Virol. 2005;79(5):2814–2822.
- Baek YH, Pascua PN, Song MS, et al. Surveillance and characterization of low pathogenic H5 avian influenza viruses isolated from wild migratory birds in Korea. Virus Res. 2010;150(1-2):119–128. doi:10.1016/j.virusres.2010.03.002
- Brown IH. Summary of avian influenza activity in Europe, Asia, and Africa, 2006-2009. Avian Dis. 2010;54(1 Suppl):187–193. doi:10.1637/8949-053109-Reg.1
- Busquets N, Alba A, Napp S, et al. Influenza A virus subtypes in wild birds in north-eastern Spain (Catalonia). Virus Res. 2010;149(1):10–18. doi:10.1016/j.virusres.2009.12.005
- Fereidouni SR, Werner O, Starick E, et al. Avian influenza virus monitoring in wintering waterbirds in Iran, 2003-2007. Virol J. 2010;7:43. doi:10.1186/1743-422X-7-43
- Fouchier RA, Olsen B, Bestebroer TM, et al. Influenza A virus surveillance in wild birds in Northern Europe in 1999 and 2000. Avian Dis. 2003;47(3 Suppl):857–860. doi:10.1637/0005-2086-47.s3.857
- Henriques AM, Fagulha T, Barros SC, et al. Multiyear surveillance of influenza A virus in wild birds in Portugal. Avian Pathol. 2011;40(6):597–602. doi:10.1080/03079457.2011.618943
- Hoye BJ, Munster VJ, Nishiura H, et al. Surveillance of wild birds for avian influenza virus. Emerg Infect Dis. 2010;16:1827–1834.
- Kydyrmanov A, Sayatov M, Karamendin K, et al. Monitoring of influenza A viruses in wild bird populations in Kazakhstan in 2002-2009. Arch Virol. 2017;162(1):147–155. doi:10.1007/s00705-016-3076-4
- Miller BJ. Why unprecedented bird flu outbreaks sweeping the world are concerning scientists. Nature. 2022;606(7912):18–19. doi:10.1038/d41586-022-01338-2
- Kawaoka Y, Yamnikova S, Chambers TM, et al. Molecular characterization of a new hemagglutinin, subtype H14, of influenza A virus. Virology. 1990;179(2):759–767. doi:10.1016/0042-6822(90)90143-f
- Fereidouni SR, Harder TC, Gaidet N, et al. Saving resources: avian influenza surveillance using pooled swab samples and reduced reaction volumes in real-time RT-PCR. J Virol Meth. 2012;186:119–125.
- EC (European Commission). Commission Decision 2020/689/EC of 4 August 2006 approving a Diagnostic Manual for avian influenza as provided for in Council Directive 2005/94/EC; 2019. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri = CELEX:32020R0689&from = en.
- Fereidouni SR, Starick E, Beer M, et al. Highly pathogenic avian influenza virus infection of mallards with homo- and heterosubtypic immunity induced by low pathogenic avian influenza viruses. PLoS One. 2009;4(8):e6706. doi:10.1371/journal.pone.0006706
- Hoffmann E, Stech J, Guan Y, et al. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146:2275–2289.
- Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi:10.1038/nprot.2015.053
- Gamblin SJ, Haire LF, Russell RJ, et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science. 2004;303(5665):1838–1842. doi:10.1126/science.1093155
- Liu J, Stevens DJ, Haire LF, et al. Structures of receptor complexes formed by hemagglutinins from the Asian influenza pandemic of 1957. Proc Natl Acad Sci USA. 2009;106(40):17175–17180. doi:10.1073/pnas.0906849106
- Martín J, Wharton SA, Lin YP, et al. Studies of the binding properties of influenza hemagglutinin receptor-site mutants. Virology. 1998;241(1):101–111. doi:10.1006/viro.1997.8958
- Kandeil A, Gomaa MR, Shehata MM, et al. Isolation and characterization of a distinct influenza A virus from Egyptian bats. J Virol. 2019;93(2):e01059-18. doi:10.1128/JVI.01059-18
- Lewis NS, Javakhishvili Z, Russell CA, et al. Avian influenza virus surveillance in wild birds in Georgia: 2009-2011. PLoS One. 2013;8(3):e58534. doi:10.1371/journal.pone.0058534
- World Health Organization (WHO). A revision of the system of nomenclature for influenza viruses: a WHO memorandum. Bull World Health Organ. 1980;58(4):585–591.
- Shriner SA, Root JJ, Lutman MW, et al. Surveillance for highly pathogenic H5 avian influenza virus in synanthropic wildlife associated with poultry farms during an acute outbreak. Sci Rep. 2016;6:36237. doi:10.1038/srep36237
- Yamnikova SS, Gambarian AS, Aristova VA, et al. [A/H13 and A/H16 influenza viruses: different lines of one precursors]. Vopr Virusol. 2009;54(4):10–17. Russian. Abstract in English.
- Wan H, Perez DR. Amino acid 226 in the hemagglutinin of H9N2 influenza viruses determines cell tropism and replication in human airway epithelial cells. J Virol. 2007;81(10):5181–5191. doi:10.1128/JVI.02827-06
- Zhou B, Donnelly ME, Scholes DT, et al. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza a viruses. J Virol. 2009;83(19):10309–10313. doi:10.1128/JVI.01109-09
- Keller MW, Rambo-Martin BL, Wilson MM, et al. Direct RNA sequencing of the coding complete influenza A virus genome. Sci Rep. 2018;8(1):14408. doi:10.1038/s41598-018-32615-8
- Panzarin V, Marciano S, Fortin A, et al. Redesign and validation of a real-time RT-PCR to improve surveillance for avian influenza viruses of the H9 subtype. Viruses. 2022;14(6):1263. doi:10.3390/v14061263