
ABSTRACT
Antivirals with broad coronavirus activity are important for treating high-risk individuals exposed to the constantly evolving SARS-CoV-2 variants of concern (VOCs) as well as emerging drug-resistant variants. We developed and characterized a novel class of active-site-directed 3-chymotrypsin-like protease (3CLpro) inhibitors (C2–C5a). Our lead direct-acting antiviral (DAA), C5a, is a non-covalent, non-peptide with a dissociation constant of 170 nM against recombinant SARS-CoV-2 3CLpro. The compounds C2–C5a exhibit broad-spectrum activity against Omicron subvariants (BA.5, BQ.1.1, and XBB.1.5) and seasonal human coronavirus-229E infection in human cells. Notably, C5a has median effective concentrations of 30–50 nM against BQ.1.1 and XBB.1.5 in two different human cell lines. X-ray crystallography has confirmed the unique binding modes of C2–C5a to the 3CLpro, which can limit virus cross-resistance to emerging Paxlovid-resistant variants. We tested the effect of C5a with two of our newly discovered host-directed antivirals (HDAs): N-0385, a TMPRSS2 inhibitor, and bafilomycin D (BafD), a human vacuolar H+-ATPase [V-ATPase] inhibitor. We demonstrated a synergistic action of C5a in combination with N-0385 and BafD against Omicron BA.5 infection in human Calu-3 lung cells. Our findings underscore that a SARS-CoV-2 multi-targeted treatment for circulating Omicron subvariants based on DAAs (C5a) and HDAs (N-0385 or BafD) can lead to therapeutic benefits by enhancing treatment efficacy. Furthermore, the high-resolution structures of SARS-CoV-2 3CLpro in complex with C2–C5a will facilitate future rational optimization of our novel broad-spectrum active-site-directed 3C-like protease inhibitors.
Introduction
Coronavirus disease 2019 (COVID-19) continues to present a significant challenge to healthcare systems worldwide, with more than 768 million SARS-CoV-2 infections and more than 6.9 million deaths reported by July 26, 2023. Although 72.3% of the world's population received at least one dose of a COVID-19 vaccine (https://covid19.who.int/), new variants of SARS-CoV-2 with the potential for enhanced transmission, replication, and immune evasion continue to emerge, causing reduced vaccine effectiveness. Reports on SARS-CoV-2 Omicron BQ.1.1 and XBB.1.5 subvariants suggest that current antibody-mediated protection offered by vaccines and monoclonal antibodies is severely reduced compared to the protection provided against Omicron BA.5 [Citation1–4].
Direct-acting antivirals (DAAs) that target virus-encoded enzymes are an essential component in the fight against viral infections, and they have revolutionized treatment against viruses such as hepatitis C virus (HCV) and human immunodeficiency virus (HIV) [Citation5,Citation6]. Three DAAs (remdesivir, nirmatrelvir, and molnupiravir) targeting SARS-CoV-2-encoded viral enzymes, which are essential for viral replication, have been approved in North America. If used appropriately, they can greatly limit the morbidity and mortality in patients infected with SARS-CoV-2 VOCs [Citation7–9]. Remdesivir and molnupiravir [Citation10], which target SARS-CoV-2-encoded RNA-dependent RNA polymerase (RdRP), were the first approved DAAs [Citation10,Citation11]. Nirmatrelvir [Citation12] is a clinically approved oral covalent peptidomimetic targeting the SARS-CoV-2 3-chymotrypsin-like protease (3CLpro), also called the main protease (Mpro). Nirmatrelvir has shown effectiveness against COVID-19 when used early following infection; however, it needs to be used in combination with ritonavir (PaxlovidTM) to increase the compound half-life [Citation13]. The current COVID-19 treatments and therapeutics are all used as monotherapies for which SARS-CoV-2 may develop resistance, as has been observed for other viruses such as HCV, HIV, and influenza A, stressing the need for additional treatment options [Citation14–16]. Most notably, mutations in the SARS-CoV-2 3CLpro have been identified that may confer resistance to nirmatrelvir [Citation17–19].
SARS-CoV-2 3CLpro, which is responsible for mediating 11 out of 14 cleavages of the virus-encoded polyproteins [Citation20], has been demonstrated to be enzymatically active as a dimer [Citation21,Citation22]. Dimerization is critical for efficient enzymatic activity with the interface interactions required for correct active site structure [Citation23,Citation24]. The 3CLpro active site contains a Cys-His catalytic dyad (His41, Cys145) and can be adapted to accommodate the different cleavage sequences [Citation25]. 3CLpro inhibitors have been shown to bind different active site conformations, and this malleability is an important consideration for drug design [Citation26,Citation27]. To date, 3CLpro remains highly conserved across SARS-CoV-2 VOCs, and the active site is conserved across members of the alpha- and beta-coronaviruses. As such, DAAs targeting 3CLpro may display broadly acting antiviral activity that can be used against multiple human coronaviruses (HCoVs).
Here, we report the application of computer-aided drug design in developing a novel series of non-covalent, non-peptide SARS-CoV-2 3CLpro inhibitors (C2–C5 and C5a–C5e) that show submicromolar to single-digit micromolar IC50 values (0.6–8.7 µM) against 3CLpro in both in vitro assays and cell-based bioassays. Importantly, the most potent protease inhibitor (C5a) inhibits infection of SARS-CoV-2 Omicron subvariants with immune evasion properties (e.g. BQ.1.1 and XBB.1.5) in both human lung and colon cell lines with nanomolar half-maximal effective concentration (EC50) values. As hypothesized, C2–C5 show pan-coronavirus activity as demonstrated by the robust inhibition of seasonal human coronavirus HCoV-229E infection in human cells. Finally, we report the high-resolution structures of SARS-CoV-2 3CLpro in complex with C2–C5 and C5a, using X-ray crystallography, and we provide important structural information regarding future rationale for optimization of our novel broad-spectrum active-site-directed 3C-like protease inhibitors.
Overall, this study can inform important future investigations that are targeting SARS-CoV-2 3CLpro for developing novel DAAs to address the urgent need for improved therapeutics against current and future SARS-CoV-2 variants [Citation28,Citation29].
Materials and methods
Free energy perturbation (FEP) simulations
We first identified a potential scaffold for developing our anti-3CLpro drugs by filtering the database results from the COVID Moonshot collaboration between Postera AI and the Diamond Lightsource in the UK [Citation30]. Out of the 394 3CLpro structures with co-crystallized fragments, we focused specifically on fragments that interacted non-covalently with 3CLpro. This subset consisted of 54 structures with reported IC50 values below 50 µM. These fragments predominantly occupied the S1 and S2 subpockets.
To generate a combinatorial library for discovering new 3CLpro inhibitors, we selected the core of fragment C1 as a starting point (, https://fragalysis.diamond.ac.uk/viewer/react/preview/direct/target/Mpro/mols/x11790/L/P/C). The selection of C1 was based on unique molecular interactions observed between C1 and 3CLpro (the isoquinoline group in S1, a thiophene ring in S2, and dimethylaniline in S3′ []) and synthetic ease. The combinatorial library aimed to optimize the groups in each pocket by combining existing and in-house groups of potent fragments observed in the Diamond and Postera crystal structures. Using this methodology, we generated 360 unique compounds, which were then docked to SARS-CoV-2 3CLpro to assess their potential binding affinities and binding modes. We reduced this list to 178 by inspecting their docking scores, docking poses, and structural similarity. Specifically, we selected molecules that interacted with 3CLpro in the S1, S2, and S1’ subpockets, and which possessed synthons with high synthetic accessibility according to our synthetic expertise. Additionally, we prioritized compounds that had a core orientation (with respect to the pocket) like the core of the original starting fragment (C1). Compounds with predicted improved binding affinities were selected for further experimental evaluation. In this case, the FEP-calculated binding affinity represents the predicted improvement of binding affinities compared to the initial starting molecule, thereby providing valuable information on which de novo compounds could be prioritized for synthesis. Due to the alchemical nature of the FEP process, it was important to select compounds that have only minor changes relative to the start C1 structure. The final list of compounds selected for FEP+ evaluation is shown in .
Figure 1. Combinatorial library building pipeline and free-energy perturbations (FEP) analysis. The pipeline used for FEP de novo design of non-peptide compounds targeting the catalytic site of 3CLpro. The core of fragment C1 was used as a starting point for the design of new compounds by substituting and combining various groups in the different subpockets. Docking of the active fragment, C1 (teal sticks) in SARS-CoV-2 3CLpro (7KHP) and active derivatives was performed and the Glide docking scores of the derivatives were determined. The Δ predicted affinity (ΔPred Aff) represents the predicted changes in binding affinities (in kcal/mol) over the parental compound C1. The more negative the value, the more improved the derivative is predicted to be.
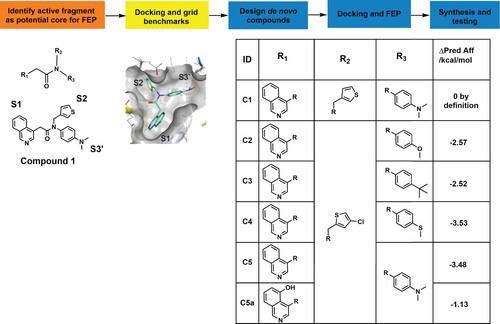
The FEP+ module from Schrödinger LLC was used to estimate the relative binding free energies of the custom fragment derivatives. Prior to FEP evaluation, the compounds of interest were grouped into subsets based on their similarity and their manageability for FEP+ calculations. The setup of the FEP calculations was automated following the FEP+ GUI methodology. First, the protein and ligand complexes were prepared with the OPLS3e force field. The force-field builder module was then used to correct the force field by generating the missing torsional angle profiles and parameters for all molecules. The REST (replica exchange with solute tempering) algorithm was used for locally enhanced sampling, and cycle closure correction was applied to calculate free energy estimates. Finally, an FEP mapper was used to analyze free energy calculations based on similarity scores between compounds. The molecular dynamics (MD) part of the FEP calculations are implemented in Desmond GPU to ensure speed and accuracy. All calculations were performed on NVidia Tesla V100 GPUs from the Vancouver Prostate Centre’s Juggernaut cluster.
Non-covalent, non-peptide SARS-CoV-2 3CLpro inhibitors (C2–C5, C5a–C5e, and C7/nirmatrelvir)
All compounds used in this study were synthesized according to the procedures described in Supplementary Materials and Methods. Each compound’s identity and purity were verified by LC-HRMS and 1H-, and by 13C-NMR (Supplementary Materials and Methods).
Cell lines and antibodies
Human Calu-3 cells (ATCC® HTB-55™) and human Caco-2 cells (ATCC® HTB-37™) were cultivated according to ATCC recommendations. All experiments were performed in these cells below passage 15. Vero E6 cells (ATCC® CRL-I1586™; used for generating SARS-CoV-2 stocks) were cultivated in MEM that was supplemented with 10% FBS, 1 mM sodium pyruvate, and 0.1 nM non-essential amino acids and used at passage <40. VeroE6/TMPRSS2 cells (JCRB 1819) were maintained in MEM supplemented with 5% FBS and penicillin and streptomycin (100 IU and 100 µg/ml, respectively). Normal human bronchial epithelial cells (NHBE), isolated from the epithelial lining of airways above the bifurcation of the lungs (NHBE-Bronchial Epi Cells; Lonza CC-2540S) were cultivated according to Lonza recommendations. The SARS-CoV-2 nucleocapsid antibody [HL344] (GTX635679) was kindly provided by GeneTex; mouse anti-dsRNA antibody (J2-1904) was purchased from Scicons English and Scientific Consulting (Long 2020); Hoechst 33,342 and secondary antibodies goat anti-mouse IgG Alexa Fluor 488 (A11001) and goat anti-rabbit IgG Alexa Fluor 555 (A21428) were obtained from Invitrogen.
3CLglow Assay – SARS-CoV-2 3CLpro endoproteolytic activity in cellulo
Caco-2 cells were seeded at a concentration of 10,000 cells/well in 96-well plates the day before transduction. Cells were transduced with two BacMam baculovirus vectors (Molecular Montana), one vector expressing the recombinant SARS-CoV-2 3CLpro (enzyme BacMam) and the other expressing an intracellular biosensor–red fluorescent protein (RFP) and mNeonGreen (mNG) fluorescent protein with an inhibitor tag. Three h after transduction, the cells were treated with a serial dilution of compounds and then incubated for 28 h, followed by fixation of the cells with 3.7% formalin for 30 min. The fixative was removed, the cells were washed with PBS, and the cells were nuclear stained with Hoechst 33,342 at 1.5 µg/mL for 30 min at room temperature in the dark. After washing with PBS, the plates were kept in the dark at 4°C until imaging was performed on a high-content screening (HCS) platform (CellInsight CX7 HCS, Thermo Fisher Scientific) with a 10X objective.
SARS-CoV-2 infection and treatment in Calu-3, Caco-2, and NHBE cells
All infections were carried out in Biosafety Level 3 (BSL3) facilities (either UBC FINDER or SFU BIO3) in accordance with Public Health Agency of Canada and UBC FINDER or SFU BIO3 regulations (UBC BSL3 Permit #B20-0105 and SFU Permit #361-2021). SARS-CoV-2 VOCs (B.1.617.2 Delta and BA.2) were kindly provided by Dr. Mel Krajden (BC Centre for Disease Control, BC, Canada). SARS-CoV-2 Delta was first isolated in Vero E6/TMPRSS2 cells (passage 1) and then passaged in Vero E6 cells (passage 2). The Delta and Omicron BA.2 virus stocks used in the experiments (passage 3) were propagated in Vero E6 cells. SARS-CoV-2 Omicron BA.1 (BC-SFU-OM6), BA.5, BQ.1.1, and XBB.1.5 were isolated by Dr. Niikura from a clinical specimen in Vero E6/TMPRSS2 cells and confirmed as BA.1, BA.5, BQ.1.1, and XBB.1.5 subvariants by complete genome sequencing (sequence available in GISAID).
Omicron BA.1, BA.2, BA.5, BQ.1.1, and XBB.1.5 were amplified in Vero-E6 TMPRSS2 cells and used in the experiments at passage 2. Cells were seeded in 96-well plates the day before infection [Calu-3 cells (10,000 cells/well); Caco-2 cells (8,000 cells/well); and NHBE cells (8,000 cells/well)]. SARS-CoV-2 stocks were diluted in cell-specific media to a multiplicity of infection (MOI) of 2.
Cells were pretreated with compounds for 3 h and then incubated with the virus for 48 h, followed by fixation of the cells with 3.7% formalin for 30 min to inactivate the virus. The fixative was removed, and the cells were washed with PBS, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 1% bovine serum albumin (BSA) for 1 h. This was followed by immunostaining with the mouse primary antibody J2 (dsRNA) and rabbit primary antibody HL344 (SARS-CoV-2 nucleocapsid) at working dilutions of 1:1000 overnight at 4°C. Secondary antibodies were used at a 1:2000 dilution and included the goat anti-mouse IgG Alexa Fluor 488 and goat anti-rabbit IgG Alexa Fluor 555 with the nuclear stain Hoechst 33,342 at 1.5 µg/mL for 1 h at room temperature in the dark. After washing with PBS, the plates were kept in the dark at 4°C until imaging was performed on a high-content screening (HCS) platform (CellInsight CX7 HCS, Thermo Fisher Scientific) with a 10X objective (Supplementary Materials and Methods).
HCoV-229E infection and treatment in Caco-2 cells
All infections with HCoV-229E (ATCC® VR-740) were carried out in a Biosafety Level 2 (BSL2) facility in accordance with Public Health Agency of Canada regulations (UBC BSL2 Permit # B17-0024). Viral stocks used in the experiments were propagated in Caco-2 cells, and all HCoV-229E infections were performed at 33°C. Caco-2 cells were seeded at a concentration of 8,000 cells/well in 96-well plates the day before infection. Cells were pretreated with compounds for 3 h, then incubated with the virus (diluted to an MOI of 1 in EMEM w/o FBS) for 48 h, followed by fixation of the cells with 3.7% formalin for 30 min to inactivate the virus. The fixative was removed, and the cells were washed with PBS, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 1% BSA for 1 h, followed by immunostaining with the mouse primary antibody J2 (dsRNA) at working dilutions of 1:1000 for 1 h at room temperature. A secondary antibody goat anti-mouse IgG Alexa Fluor 488 was used at a 1:2000 dilution with the nuclear stain Hoechst 33,342 at 1.5 µg/mL for 1 h at room temperature in the dark. After washing with PBS, the plates were kept in the dark at 4°C until imaging was performed on an HCS platform (CellInsight CX7 HCS, Thermo Fisher Scientific) with a 10X objective (Supplementary Materials and Methods).
Median effective dose (EC50) curves and cytotoxicity assays
Intracellular dose response (EC50 values) for selected compounds against SARS-CoV-2 and HCoV-229E were determined by pretreating human cells (Calu-3, Caco-2 or NHBE cells) for 3 h with serially diluted compounds, followed by virus infection for 48 h. Viral infection was detected by staining for dsRNA or nucleocapsid signal and quantified as described above. EC50 experiments were repeated at least three times for each compound with two technical replicates in each experiment. Intracellular nucleocapsid and dsRNA levels were interpolated to negative control (1% DMSO, no infection) = 0, and positive control (1% DMSO, with infection) = 100. The GraphPad Prism 9™ (GraphPad Software, Inc.) nonlinear regression fit modelling variable slope was used to generate a dose–response curve [Y = Bottom + (Top–Bottom)/(1 + 10^((LogIC50-X)*HillSlope))], constrained to top = 100, bottom = 0. Cellular viability assays were performed as previously described with PrestoBlue Cell Viability Assay (Thermo Fisher Scientific) (Supplementary Materials and Methods) [Citation31,Citation32].
Two-drug combination
The two-drug combination was tested using five-fold and ten-fold serial dilutions of C5a and N-0385 [Citation31] or bafilomycin D [Citation32] ≤ EC50, respectively. Different dilutions of the two compounds were mixed together and added to Calu-3 cells for 3 h before being infected with SARS-CoV-2 Omicron BA.5 for 48 h. All tests were conducted in duplicate in two independent experiments. To test whether the drug combinations act synergistically, the observed responses were compared with expected combination responses. The expected responses were calculated based on the highest single agent (HSA), Bliss, Loewe, and Zero interaction potency reference models using the open-source web application SynergyFinder. Synergy scores were assigned for each condition, and values above 10 were interpreted as a synergistic effect [Citation33–35].
Results
Computer-aided drug design of small molecules targeting SARS-CoV-2 3CLpro
We used a computer-aided drug design (CADD) pipeline, combining large fragment datasets [Citation30], computational docking, de novo fragment design, and free energy perturbation analysis (FEP) to design four new non-covalent, non-peptide SARS-CoV-2 3CLpro-directed compounds ( and Figure S1). To summarize our CADD pipeline, the core of fragment C1 was selected as a starting point for combinatorial library design because its scaffold interacted with distinct subpockets of SARS-CoV-2 3CLpro (see Methods for details). Fragment C1 is characterized by an isoquinoline group in S1, a β-thiophenyl ring in S2, and dimethylaniline in S3′ (). We designed and synthesized four novel non-peptide compounds (see Supplementary Methods for the synthetic details) – C2, C3, C4, and C5 – that substituted the S2 thiophenyl ring for a 4-chloro-2-thiophenyl ring to increase hydrophobic contacts with 3CLpro. We observed that the isoquinoline in the S1 group was superior to that in the other groups because it could both maintain the hydrogen bond with the His163 imidazole side chain and facilitate additional contacts within the S1 pocket through derivatization. To explore potential interaction preferences in the S3′ pocket, the dimethylaniline group was replaced by an anisole group, a tert-butylbenzene, or a thioanisole to yield C2, C3, and C4, respectively ( and Figure S2) (docking pose analysis and interaction networks in Methods and Supplementary Materials and Methods).
Based on the observed activity of C5 and the high synthetic tractability of potential analogues, we explored derivatization as a means of increasing potency. C5 primarily occupies subpockets S1, S2, and S1′/S3′, so subsequent designs explored the S3/S4 subpockets. Although these subpockets are not as critical as the S1 and S2, it has been proposed that they contribute to the potency and specificity of 3CLpro inhibitor binding [Citation36]. For example, the importance of P4 (occupying the S4 position) to effective binding is reflected by the conservation in the 11 SARS-CoV-2 cleavage sites to small, mostly hydrophobic, amino acids [Citation27,Citation37]. The improved binding to the S3 and S4 subsites likely contributes to the increased potency of C7/nirmatrelvir. After docking analysis, we elected to synthesize and test compounds C5a–C5e ( and Figure S2).
C2–C5a are potent inhibitors of SARS-CoV-2 3CLpro endoproteolytic activity in vitro
To evaluate the inhibitory activity of the novel 3CLpro inhibitors described above, we employed an in vitro assay that uses purified recombinant SARS-CoV-2 3CLpro and an internally quenched fluorogenic substrate (IQFS) as a reporter of endoproteolytic activity [Citation27,Citation37]. In this case, a 9-residue peptide (AVLQ↓SGFRY) was designed to mimic the nsp4/nsp5 cleavage junction in the polyprotein pp1a that is cleaved by the SARS-CoV-2 3CLpro during infection [Citation37,Citation38]. The peptide has a fluorescent O-aminobenzoyl (Abz) donor group at the N-terminus and a 3-nitro-tyrosine [Y(3-NO2)] acceptor – or quencher – group at the C-terminus [Citation37,Citation39,Citation40]. For the uncleaved IQFS, resonance energy transfer between the fluorophore donor and acceptor moieties results in the quenching of the fluorescent signal [Citation40]. Following the endoproteolytic cleavage of the IQFS by SARS-CoV-2 3CLpro, the quenching effect is lost and an increase in fluorescence signal can be detected and measured [Citation40].
The baseline enzymatic activity of recombinant 3CLpro (Figure S3) was measured by incubating the enzyme with a range of concentrations of the IQFS and measuring fluorescence over time. The purity of the 3CLpro substrate and cleavage into expected products were confirmed (Figures S4–S6). Initial velocity values were fitted to the Michaelis–Menten rate equation to determine Km (26.0 ± 1.3 μM) and Vmax (1.01 ± 0.12 µM/min). The kcat determined for 3CLpro was 1.7 ± 0.2 s−1 (Figure S7), in line with the previously reported value of 0.5 ± 0.04 s−1 [Citation27].
We then measured the endoproteolytic activity of recombinant 3CLpro incubated with serially diluted 3CLpro inhibitors for 15 min before the addition of the IQFS. The IC50 values (drug concentration causing a 50% reduction in enzymatic activity) were determined to be 8.5 ± 1.7 μM for C2; 5.0 ± 0.3 μM for C3; 2.3 ± 0.2 μM for C4; 0.9 ± 0.6 μM for C5; 0.7 ± 0.2 μM for C5a; 4.5 ± 1.0 μM for C5b; 1.4 ± 0.8 μM for C5c; 2.2 ± 0.7 μM for C5d; 1.4 ± 0.8 μM for C5e; and 0.03 ± 0.01 μM for C7/nirmatrelvir (, Table S1, and Figure S8). We also determined the dissociation constant (Kd) and inhibition constants (Ki) for these compounds, which presented potencies relatively consistent with IC50 values (, Table S1, and Figure S9). Based on the measured IC50 values, we found that FEP (ΔPred Aff, ) correctly ranked the compounds within an acceptable margin of error. C2 and C3 had an average improvement of ∼2.5 kcal/mol, and their IC50 values were 8.5 and 5.0 μM, respectively. FEP (ΔPred Aff, ) predicted that C4 and C5 would improve by ∼3.5 kcal/mol, and their IC50 values were 2.3 and 0.9 μM (), respectively. Additionally, the inhibition is selective because C5 and C5a do not exert significant inhibitory effects against the purified recombinant SARS-CoV-2 papain-like protease (PLpro) in vitro (Figure S19).
Figure 2. Novel 3CLpro molecules (C2–C5a) inhibit the endoproteolytic activity of SARS-CoV-2 3CLpro in vitro and in cellulo. (A-E) The proteolytic activity of purified recombinant 3CLpro was measured using the IQFS in the presence of increasing concentrations of selected candidate compounds (C2–C5a). (F-J) The inhibitory Kd measures were performed by microscale thermophoresis (MST). (K-O) Caco-2 cells were transduced with vectors to express the recombinant SARS-CoV-2 3CLpro and 3CLglow biosensor following treatment with serial dilution concentrations of C2–C5a (black circles) and the toxicity is reported by biosensor RFP (open circles). Fluorescence intensity was measured with a CellInsight CX7 High Content Screening System. The GraphPad Prism 9™ (GraphPad Software, Inc.) nonlinear regression was used for IC50 curve-fitting (n = 3).
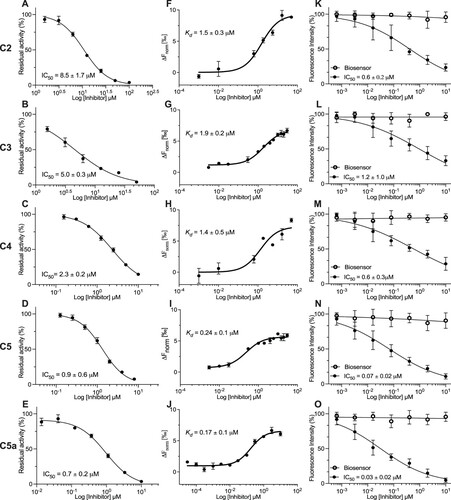
C2–C5a are potent inhibitors of SARS-CoV-2 3CLpro endoproteolytic activity in cellulo
We used a live-cell assay, which can measure SARS-CoV-2 3CLpro activity with a reporter based on a green fluorescence protein that emits only after cleavage by 3CLpro. The assay consists of two modified baculovirus vectors (BacMam) that can be used to transiently express proteins in mammalian cells. One vector expresses the recombinant SARS-CoV-2 3CLpro while the other expresses an intracellular biosensor–red fluorescent protein (RFP) and mNeonGreen (mNG) fluorescent protein with an inhibitor tag. The RFP biosensor is constitutively expressed and is a marker for successful transfection and cell viability. The mNG biosensor is unable to properly fold and fluoresce because of the inhibitor tag. The transduction with both vectors results in red and green fluorescence within cells as intracellularly expressed SARS-CoV-2 3CLpro cleaves off the mNG biosensor inhibitor tag [Citation41,Citation42]. Transduced Caco-2 cells were treated 3 h post-transduction with a serially diluted compound, and after 28 h of additional transduction, the cells were fixed and the red and green fluorescence intensity was measured. The IC50 values for our lead 3CLpro inhibitors in cellulo were determined to be 0.6 ± 0.2 M for C2; 1.2 ± 1.0 μM for C3; 0.6 ± 0.3 μM for C4; 0.07 ± 0.02 μM for C5; 0.03 ± 0.02 μM for C5a; and 0.05 ± 0.02 μM for C7/nirmatrelvir ( and Table S1). These results demonstrate the efficacy of our novel non-covalent, non-peptide compounds (C2–C4) in inhibiting the endoproteolytic activity of recombinant SARS-CoV-2 3CLpro expressed in human cells. The results also demonstrate that C5 and C5a are the most potent SARS-CoV-2 3CLpro inhibitors discovered in our study, with nanomolar IC50 values (70 and 30 nM, respectively), that are equivalent to the value determined for C7/nirmatrelvir (IC50, 50 nM; Table S1).
Crystal structures of SARS-CoV-2 3CLpro bound with C2–C5a
To gain further insight into the mechanism of binding of our novel 3CLpro inhibitors (C2–C5a), we determined the X-ray crystal structures in complex with ancestral SARS-CoV-2 3CLpro ((C) and Table S5), for which the co-crystal structure has been previously reported with C7/nirmatrelvir [Citation12].
Figure 3. The binding mechanism for the non-covalent, non-peptide inhibitor series C2–C5 of SARS-CoV-2 3CLpro. (A) Interaction schematic for inhibitor series C2–C5 showing interactions for a common scaffold. Hydrogen bonds are shown as blue dashed lines, hydrophobic interactions as green dashed lines, and pi-stacking interactions as black dashed lines. The varied groups (R) are shown on right. (B) Dimeric 3CLpro (7KHP) with one monomer shown as cartoon coloured from the N- (blue) to C-terminus (red); the other monomer is shown as a grey surface with the active site boxed. (C) Zoomed-in active site from B showing bound inhibitor series C2–C5, colours indicated in (A). A model of the 3CLpro C-terminal auto-cleavage sequence is superimposed and shown as lines with carbon black, nitrogen blue, and oxygen red. Subsites S1, S2, and S3’ are labelled.
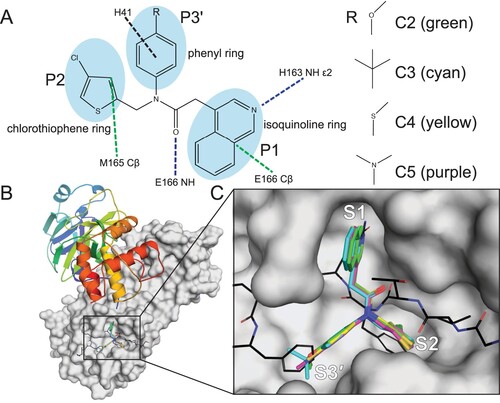
The structures of compounds C2–C5 show binding similar to the parent compound C1 ((A–C)), and the isoquinoline is bound in the S1 pocket with the nitrogen forming a hydrogen bond with His163 Nϵ2. The core carbonyl forms a hydrogen bond with the backbone amide nitrogen of Glu166, and the chlorothiophene binds in the S2 subsite sandwiched between Met49 and Met165 with the Cl atom pointing into the pocket. The varied substituent (R3 in (A)) forms a strong edge-on pi-stacking interaction with the imidazole ring of His41 mediated by the common benzene group while the anisole (C2), tert-butylbenzene (C3), thioanisole (C4), and dimethylaniline (C5, C5a) extend into the S3′ subsite with primarily hydrophobic interactions with Thr25, His41, Thr45, Ser46, and Met49. The 3CLpro active site conformation, specifically the S2 subsite, is similar to the unliganded active site or to the active site when bound to the C-terminal auto-cleavage substrate (nsp5/6 junction, P5-P1: SGVTFQ [Citation27]) with the chlorothiophene acting as the Phe(P2) surrogate ((C)). Inhibitors targeting this active site conformation with a Phe(P2) surrogate and a functional group occupying the S3′ site effectively mimic the binding of the enzyme–substrate (ES) complex [Citation27] ((C)) and represent promising leads for further development.
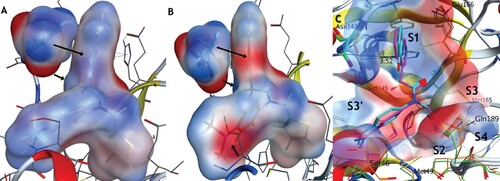
Comparing C5 and C5a, which differ only by a phenolic group, the active site remains largely unchanged with the notable exception of Ser46 and Asn142 (). In C5a, the loop containing Ser46 takes a more closed conformation, while the dimethyl aniline ring has rotated to expose its electron-rich face. This allows for the chiral proton of Ser46 to form a new weak hydrogen bond with the nitrogen, with the C–N distance decreasing by 0.7 Å. Further, Asn142’s sidechain moves closer to the isoquinoline by ∼1 Å, to better benefit from the much more polar pi-pi stacking opportunity now available. To further visualize these compounds in a theoretically more “in vivo” setting and to avoid potential crystal packing artifacts in Asn142, we performed low-mode molecular dynamics (LMMD) simulations in implicit solvation (see Supplementary Information for details). In both cases, we saw that Asn142 prefers to pi-stack with the ring, however, C5a has a much more electronegative surface near the NH2 due to the phenolic oxygen, while the phenolic hydrogen near the Asn142 sidechain carbonyl is much more electropositive compared with the CH2 group. Overall, we postulate that the increased affinity between C5 and C5a stems from the increased opportunity for H-bonding interactions and the complimentary dipoles now present in the respective pi orbitals of Asn142 and the isoquinoline system. Such electrostatic complementation has been previously described during the optimization from micromolar to picomolar compounds [Citation43]. We have similarly compared the binding between C5a, C7/nirmatrelvir, and the newly reported Wu-04 compound [Citation44]. Unlike us, that research team found that the installation of the OH group at position 7 reduced binding affinity, likely owing to differences in the interaction pattern between Wu-04 and SARS-CoV-2 3CLpro (Figure S18).
Figure 4. Comparison of the electrostatic potential interaction surface for C5 (A) and C5a (B) and Asn142 after low-mode molecular dynamics (C). Both compounds are modelled in the AMBER14:EHT forcefield, using their respective crystal structures as starting points (C5: 8CYU, C5a: 8SXR). For both compounds, Asn142 forms a pi-pi stacking interaction in the same orientation. The surfaces have been coloured blue for positive, red for negative, and white for neutral. Arrows highlight the differences in interaction potential as seen by Asn142. The increased polarity of the C5a isoquinoline system and its alignment with the electronegative surface of Asn142, combined with a new hydrogen donation from the chiral proton of Ser46 to the electron-rich dimethyl amino system, likely account for the increased affinity observed. (C) Superposition of crystal structures 8CYU and 8SXR with residue and subsite labelling. The Asn142 moves ∼1 Å to permit better interaction with C5a.
C2–C5 are pan-SARS-CoV-2 inhibitors of SARS-CoV-2 Delta and Omicron (BA.1 and BA.2) in human lung (Calu-3) cells
Very few mutations have been reported in the 3CLpro of SARS-CoV-2 VOCs, suggesting that 3CLpro inhibitors should retain high potency against variants [Citation45]. First, we pre-treated Calu-3 cells with the serially diluted compound for 3 h prior to infection with SARS-CoV-2 Delta, Omicron BA.1, or Omicron BA.2. Relative quantification of virus-infected cells was used to determine EC50 values (drug concentration required to reduce infection by 50%). We then performed an antiviral dose–response analysis as described above of our novel compounds (C2–C5) compared to C7/nirmatrelvir against SARS-CoV-2 Delta (Figure S10), Omicron BA.1 (Figure S11), and Omicron BA.2 (Figure S12). Similar EC50 values in the low micromolar range were observed for all non-covalent compounds C2–C5 (i.e. from 0.3 μM to 9.4 μM) with C5 exhibiting the highest potency. These values were comparable to the EC50 value of 1.3 μM observed for C7/nirmatrelvir under the same experimental conditions (Table S2). These findings demonstrate that our novel non-covalent, non-peptide 3CLpro inhibitors can act as pan-SARS-CoV-2 VOC antivirals with a potency comparable to that of C7/nirmatrelvir.
C5 is a broad-acting coronavirus inhibitor against the seasonal human coronavirus, HCoV-229E
The seasonal human coronavirus, HCoV-229E, causes a mild upper-respiratory tract infection and belongs to the alpha-coronavirus genera of the coronaviridae family, in contrast to SARS-CoV-2, which is a member of the beta-coronavirus genus [Citation46]. Although there is overall limited homology between SARS-CoV-2 and HCoV-229E 3CLpro, the active sites of both are highly conserved [Citation47]. To investigate the broad-spectrum coronavirus activity of our novel inhibitors, we tested C2–C5 and C7/nirmatrelvir against HCoV-229E in susceptible human Caco-2 cells [Citation48] and determined that the EC50 values were comparable to those for SARS-CoV-2 VOCs in Calu-3 cells (Table S3 and Figure S13). Using dsRNA as a marker of infection, we determined EC50 values of 5.6 ± 1.4 μM for C2; 9.6 ± 1.0 μM for C3; 7.9 ± 3.8 μM for C4; 4.2 ± 0.7 μM for C5; and 2.0 ± 1.2 μM for C7/nirmatrelvir against HCoV-229E. Overall, these results suggest that 3CLpro inhibitors, including our lead non-covalent, non-peptide inhibitor C5, will maintain antiviral activity against evolving SARS-CoV-2 VOCs and future emerging HCoVs.
C5a is a nanomolar inhibitor of SARS-CoV-2 Omicron BA.5, BQ.1.1, and XBB.1.5 in human lung (Calu-3) and colon (Caco-2) epithelial cell lines
While we were initially writing this manuscript, the Omicron subvariant BA.2 was dominating infections globally, but it has rapidly evolved into BA.5, BQ.1.1, and XBB.1.5 subvariants [Citation49,Citation50], thus, 3CLpro antivirals must be effective against all emerging Omicron lineages. We tested C5a, C5, and C7/nirmatrelvir on Calu-3 cells pre-treated with serially diluted compounds for 3 h before SARS-CoV-2 Omicron BA.5, BQ.1.1, and XBB.1.5 infection, and we compared the results using a second cell line, Caco-2 (colon) cells. Fluorescent imaging of viral nucleocapsid and dsRNA confirmed viral infection, and relative quantification of virally infected cells was used to calculate their EC50 values. C5 shows the same level of inhibition against Omicron BA.5, BQ.1.1, and XBB.1.5 as C7/nirmatrelvir, with EC50 values between 0.2–0.7 μM and 0.1–0.3 μM for C5 and 0.4–0.7 μM and 0.3–0.4 μM for C7/nirmatrelvir, using dsRNA and nucleocapsid staining, respectively ( and Table S4). Remarkably, C5a shows the highest potency for blocking the infection of Omicron BA.5, BQ.1.1, and XBB.1.5 with EC50 values between 30–69 nM using nucleocapsid staining and 28–95 nM using dsRNA staining ( and Table S4). The other non-peptide compounds C2–C4 and C5b–C5e were also tested, but they showed EC50 values in the micromolar range (i.e. from 0.3 to 2 μM) (Figure S14 and S15). The cytotoxicity and selectivity index (SIs) of our novel compounds were evaluated in Calu-3 and Caco-2 cells (Tables S2 and S4). Importantly, the SI for our lead antiviral, C5a, expressed as the ratio of CC50 over EC50, indicates high SI values in both human Calu-3 cells and human Caco-2 cells for all 3 SARS-CoV-2 Omicron subvariants tested (i.e. from 267 to 1690; Table S4). A high SI (>10) is preferable if a drug is to be viewed as having a favourable safety profile in human cells.
Figure 5. C5a blocks infection of the SARS-CoV-2 BA.5 variants of concern in human lung (Calu-3) and colon cells (Caco-2). (A) Representative fluorescent images of human Calu-3 and Caco-2 cells infected with SARS-CoV-2 BA.5, following treatment with the indicated concentrations of C5a, C5, and C7/nirmatrelvir. Control images of infected untreated (DMSO) and uninfected (Mock) cells are shown at the bottom. Hoechst is shown in blue, nucleocapsid in red, and dsRNA in green. Images were captured with a CellInsight CX7 High Content Screening System with a 10X objective lens. (B-G) Dose-response curves were generated for C5a, C5, and C7/nirmatrelvir in Calu-3 cells (B-D) and Caco-2 cells (E-G) infected with SARS-CoV-2 BA.5 using dsRNA (green square) and nucleocapsid (red circle) as infection markers (n = 3).
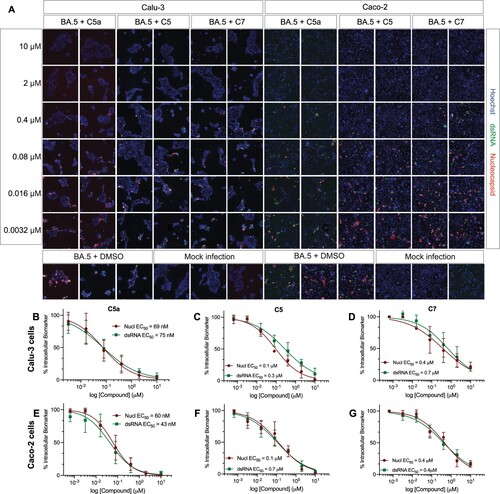
Figure 6. C5a blocks infection of the SARS-CoV-2 BQ.1.1 Omicron subvariant in human lung (Calu-3) and colon cells (Caco-2). (A) Representative fluorescent images of human Calu-3 and Caco-2 cells infected with SARS-CoV-2 BQ.1.1 following treatment with the indicated concentrations of C5a, C5, and C7/nirmatrelvir. Control images of infected untreated (DMSO) and uninfected (Mock) cells are shown at the bottom. Hoechst is shown in blue, nucleocapsid in red, and dsRNA in green. Images were captured with a CellInsight CX7 High Content Screening System with a 10X objective lens. (B-G) Dose-response curves were generated for C5a, C5, and C7/nirmatrelvir in Calu-3 cells (B-D) and Caco-2 cells (E-G) infected with SARS-CoV-2 BQ.1.1 using dsRNA (green square) and nucleocapsid (red circle) as infection markers (n = 3).
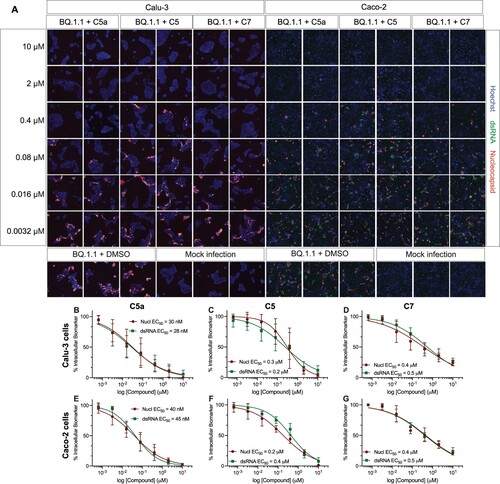
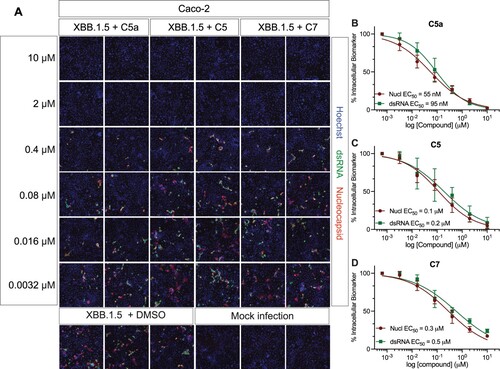
Figure 7. C5a blocks infection of the SARS-CoV-2 XBB.1.5 Omicron subvariant in human colon cells (Caco-2). (A) Representative fluorescent images of Caco-2 cells infected with SARS-CoV-2 XBB.1.5 following treatment with the indicated concentrations of C5a, C5, and C7/nirmatrelvir. Control images of infected untreated (DMSO) and uninfected (Mock) cells are shown at the bottom. Hoechst is shown in blue, nucleocapsid in red, and dsRNA in green. Images were captured with a CellInsight CX7 High Content Screening System with a 10X objective lens. (B–D) Dose-response curves were generated for C5a, C5, and C7/nirmatrelvir in Caco-2 cells infected with SARS-CoV-2 XBB.1.5 using dsRNA (green square) and nucleocapsid (red circle) as infection markers (n = 3).
Finally, we tested whether C5a can inhibit Omicron BA.5 replication in NHBE cells. NHBE cells were pre-treated with 1 µM of C7, C5, and C5a for 3 h prior to Omicron BA.5 infection (Figure S16). The 1 µM concentration used for this experiment was selected based on the 100% viral inhibition observed at that concentration for each compound in immortalized human cells (). The relative quantification of intracellular viral nucleocapsid signal was performed on the CellInsight CX7 system, and the relative intensity values were used to calculate the percentage of viral inhibition for each compound (see Materials and Methods). We observed that C5a robustly inhibited BA.5 infection in NHBE cells (84.8%). The inhibitory effect observed was comparable to the values determined for C7 (78.5%), C5 (73%), and remdesivir (77.7%). Together, these results underline the broad-spectrum nanomolar activity of C5a against Omicron subvariants (BA.5, BQ.1.1, and XBB.1.5) and C5a antiviral activity against Omicron BA.5 in patient-derived primary lung cells with a potency comparable to that of C7/nirmatrelvir and remdesivir.
SARS-CoV-2 antiviral synergy between C5a and host-directed antivirals in human lung cells (Calu-3)
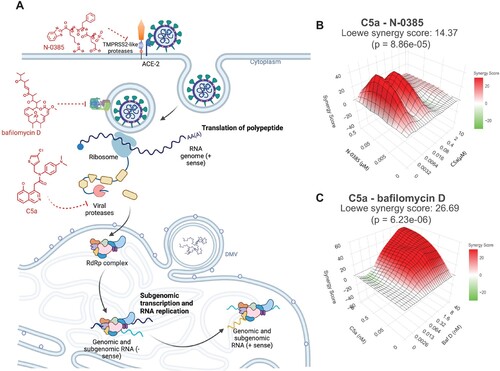
In order to reduce the potential for emerging mutations in SARS-CoV-2 infection, we investigated the synergistic effect of C5a in the presence of two other compounds that interfere with SARS-CoV-2 entry ((A) and Figure S17). N-0385 is a small-molecule protease inhibitor, which inhibits TTSP-dependent proteolytical activation of the virus spike protein, with nanomolar activity against SARS-CoV-2 infection in Calu-3 cells [Citation31]. Bafilomycin D has been described as an inhibitor of vacuolar-type H+ (V)- ATPases [Citation51], also with nanomolar activity against SARS-CoV-2 VOCs [Citation32]. Calu-3 cells infected with Omicron BA.5 were either treated with C5a, N-0385, or bafilomycin D as a single treatment or in combination. The inhibitory effects of the drug combinations were higher than those with a single compound. To analyze the drug synergy, we used the Loewe additive reference model. The synergy score of the interactions and the efficiency of the combinations were quantified with the open-source web application SynergyFinder [Citation33–35]. We found that combining C5a with N-0385 ((B) and Figure S17D-E) or bafilomycin D ((C) and Figure S17A-C) results in a synergistic effect with synergy scores of 14.37 and 26.69, respectively. Synergy scores above 10 are interpreted as synergistic [Citation32,Citation35].
Figure 8. Synergistic inhibition of SARS-CoV-2 Omicron BA.5 infection in human lung cells (Calu-3) by combined use of C5a and N-0385 or bafilomycin D. (A) Overview of drug target in virus cycle. (B-C) Dose-response curves of inhibition of single and combined treatment of C5a and N-0385 or bafilomycin D in SARS-CoV-2 infected Calu-3 cells were used for synergy analysis using the open-source web application SynergyFinder. 3D visualization synergy maps of C5a with N-0385 (B) or bafilomycin D (C), were calculated using the Loewe additive model. The surface is color-coded: red indicates synergistic interactions and green indicates antagonistic interactions (n = 2).
Discussion
This study describes interdisciplinary work that combined computer-aided drug design, co-crystallization, enzymatic assays, and cell-based antiviral investigations against SARS-CoV-2 Delta and Omicron VOCs to develop and characterize novel 3CLpro inhibitor analogues based on previously reported promising 3CLpro inhibitors [Citation12,Citation27,Citation36]. Our findings provide strong evidence that these compounds function by inhibiting 3CLpro activity, and these findings provide insight into their unique binding modes. Importantly, we confirm that our novel non-covalent, non-peptide molecules inhibit seasonal HCoV-229E, highly pathogenic SARS-CoV-2 Delta, and highly transmissible Omicron BA.1, BA.2, BA.5, BQ.1.1, and XBB.1.5 with micromolar to nanomolar EC50 values in human cells. Overall, our study confirms the feasibility and application of DAAs targeting 3CLpro for tackling the critical emergence of VOCs in the continuing SARS-CoV-2 pandemic [Citation52].
Of the designed analogues, C5a was the most potent against all viruses tested, and it demonstrated an EC50 with nanomolar activity comparable to or better than that observed for C7/nirmatrelvir [Citation12] across the previous and currently circulating Omicron SARS-CoV-2 subvariants tested. C7/nirmatrelvir was recently reported to have a Ki of 3 nM against 3CLpro and an EC50 of 74.5 nM against SARS-CoV-2 when used in combination with an efflux pump inhibitor [Citation12]. Although this pump inhibitor was not used under our assay conditions, the effectiveness of C5a appears better than that of C7/nirmatrelvir when tested in two different human cell lines and against highly transmissible Omicron BA.5, BQ.1.1, and XBB.1.5 (Table S4). The enhancement of C5a potency compared to C5 is supported by the increase in their affinity, resulting from the interaction’s potential changes.
To date, very few substitutions have been observed in the 3CLpro of VOCs; none have been reported in the Delta variant, and one mutation, P132H, is present in SARS-CoV-2 Omicron 3CLpro, which did not impact the effectiveness of the compounds on the 3CLpro enzymatic activity (data not shown). The active site of 3CLpro is also highly conserved across other coronaviruses, and we confirmed the effectiveness of C5 and C7/nirmatrelvir against the alpha-coronavirus HCoV-229E. Thus, 3CLpro inhibitors targeting the protease active site should be effective against future emerging coronavirus outbreaks [Citation23,Citation53,Citation54].
Interestingly, a correlation was not always observed between the in vitro IC50 values and the in cellulo EC50 values obtained for 3CLpro inhibitors. These discrepancies may be due to factors such as the stability of the compounds in cellulo and the potentially limited capacity of the compounds to cross the plasma membrane or interact with cellular transporters. These observations stress the importance of evaluating lead antiviral candidates in the context of cell-based infection assays.
Despite a multitude of studies investigating 3CLpro inhibitors, to date, few inhibitors of this protease have demonstrated nanomolar potency in the context of SARS-CoV-2-infected cells [Citation55]. GC-376 is one of the most effective compounds identified so far, but other promising studies have also identified 3CLpro inhibitors that all fall within a similar effective dose range [Citation20,Citation53,Citation56,Citation57]. This may indicate inherent difficulties in designing molecules that can efficiently access the 3CLpro within infected cells (replication organelles), or it may be due to the efflux of these compounds as suggested by studies with C7/nirmatrelvir [Citation12].
It is interesting to note that C5 presents a more robust antiviral activity (EC50 values > 10–30 times) for Omicron subvariants [BA.5 (EC50, 100 nM); BQ.1.1 (EC50, 300 nM); XBB.1.5 (EC50, 100 nM)] when compared to Delta VOCs (EC50, 3.2 μM). This effect could be a reflection of the attenuation or slow replication kinetics reported for the Omicron subvariants [Citation58], where 3CLpro inhibitors would be effective at lower concentrations when tested in Omicron-infected cells as compared to Delta-infected cells. This hypothesis is supported by the robust increase (EC50 values > 3–4 times) in the antiviral activity also observed for C7/nirmatrelvir in cells infected with Omicron subvariants [BA.5 (EC50, 400 nM); BQ.1.1 (EC50, 400 nM); XBB.1.5 (EC50, 300 nM) when compared to Delta VOC (EC50, 1.3 μM)] in human Calu-3 cells.
Given the non-covalent mode of binding and the different ways the functional groups engage with the protease subsites, 3CLpro active site mutations implicated in the emergence of resistance to covalent peptidomimetic inhibitors, including nirmatrelvir, (C7/nirmatrelvir) are likely to have different effects on C2–C5a (Figure S18). For example, Ip et al. described 3CLpro active site mutants, identified from the GISAID database, that had minimal impact on enzymatic activity but had a decreased sensitivity to nirmatrelvir, based on kinetic assays [Citation19]. Although some of the mutants implicated in potential drug resistance would also likely impact the binding of C2–C5a, e.g. those at Met165 or Glu166, others might have less impact due to their relative position to the C5 and C5a binding site (His172 or Gln189) ( and Figure S18).
Of note, the mutation of Ser144, which has been identified as a mutational hotspot, is located immediately adjacent to the catalytic cysteine and contributes to the oxyanion hole formed by residues 143–145, which stabilizes the transition state of the enzymatic reaction. It is possible that non-covalent binding inhibitors such as C5 and C5a would be less sensitive to Ser144 mutations compared to covalent acting peptidomimetics that are more reliant on productive interaction with the oxyanion hole. Interestingly, multiple mutations at Met49, Asn142, and Gln189 were observed that did not confer resistance to nirmatrelvir although their proximity to the S1 and S2 subsites suggests a potential structural impact. Flexibility in these regions of the enzyme may facilitate escape from resistance effects in these cases for C7/nirmatrelvir, C5, and C5a (Figure S16).
Combination-therapy approaches are critical to preventing antiviral resistance to the currently limited repertoire of treatment approaches against SARS-CoV-2 VOCs [Citation17,Citation59,Citation60]. Combining antiviral drugs with different modes of action may ultimately lead to synergistic activity with a reduced likelihood of sensitivity to emerging mutations. In addition, the combination of treatments can be administered at lower doses whereas the higher doses of single drugs would lead to intolerable dose ranges if used as monotherapies. Thus, the combination would reduce treatment side effects. We tested the effect of C5a with two of our newly discovered HDAs: N-0385 (a highly potent inhibitor of human TMPRSS2 protease) and bafilomycin D (a human V-ATPase inhibitor) [Citation31,Citation32]. We demonstrated a synergistic action of C5a in combination with bafilomycin D and N-0385 against SARS-CoV-2 Omicron BA.5 (). These results suggest that a multi-targeted treatment can lead to therapeutic benefits both by enhancing treatment efficacy and by avoiding the development of monotherapy resistance.
Overall, our study provides insight into the binding modes and mechanism of action of several promising 3CLpro inhibitors, it confirms their utility against SARS-CoV-2 VOCs and seasonal HCoV-229E, and it can be used to inform future investigations into developing lead antiviral molecules against SARS-CoV-2. The co-crystal structures provide useful insight into how to optimize inhibitory and pharmacokinetic profiles for further development. Although our compounds were comparable to C7/nirmatrelvir in our assays, our future optimization and design studies will focus primarily on increasing affinity with 3CLpro while monitoring pharmacokinetic properties. Later-stage designs will focus more heavily on pharmacokinetic and safety improvements as a means of boosting oral efficacy. Practically, as demonstrated by clinical trials with remdesivir and Paxlovid, DAAs should be administered as soon as possible following exposure to SARS-CoV-2 for maximal effect [Citation10]. Importantly, DAAs such as C5a will be most effective when used in combination with other antivirals such as HDAs to help reduce the risk of antiviral resistance mutations, as is the case with other common viral infections such as HIV and HCV [Citation5,Citation6].
Author contributions
Isolation, sharing, and sequencing of viral strains: SE, MK, PNL, NP, MP, MN;
Conceptualization: FJ, AC, NCJS, RNY
Methodology: CADD – ATT, JRS, FB, AC; 3CLpro cloning, protein production for crystallography and enzymatic assays – JL, MV, LJW, MP, NCJS; Enzymatic assays – FJ, IV, JPV, SD, JDG, SK, JRS, MB; Cellular and viral assays – FJ, ODA, CAHT, JPV, SE, SD, TS, MN; Compound synthesis – RNY, MB, JRS; Cell imaging – FJ, CAHT, JPV.
Investigation: CADD – AT, JRS, FB, AC; Cellular and viral assays – JPV, SE, SD, JDG, TS, MN, OA, FJ; Enzyme assays – IV, SD, SG, SK, JRS, MB, RNY, JL, MV, NCJS, FJ; Compound synthesis – JRS, MB, RNY; 3CLpro cloning, protein production for crystallography and enzymatic assays – JL, MV, LJW, CK, MP, NCJS; Crystallographic data collection, data processing, structure refinement – LJW, JL, CK, MP, NCJS; General structure analysis – LJW, JL, CK, CH, MP, AT, CK, NCJS; Image acquisition and analysis (CX7) – CAHT, JPV, FJ; Tissue culture – CAHT, JPV, SD, FJ.
Visualization: Cell culture, enzyme assay, and infection figures – ADO, JPV, IV, CAHT, MB, DG, FJ; Crystallographic structure figures – LJW, CK, CH, NCJS; CADD structure figures – ATT, JRS, AC; Cell imaging figures – AO, CAHT, JPV, AC, DG, FJ; Medicinal chemistry figures – JRS, MB, RNY.
Funding acquisition: NCJS, AC, FJ.
Project administration: AO, JRS, NCJS, AC, FJ.
Supervision: CADD – AC; Enzyme assays – RNY, FJ; Cellular and viral assays – FJ; Protein production and crystallography – NCJS; Medicinal chemistry – RNY; Cell imaging – FJ.
Writing: Original draft: CADD – AC; Enzyme and viral assays – JPV, AO, FJ; Medicinal chemistry – JRS, MB, RNY; Crystallography – LJW, JL, MP, NCJS; Cell imaging – JPV, AO, FJ; Abstract, Introduction, Discussion – JPV, AO, LJW, FJ
Supplemental Material
Download MS Word (23 MB)Acknowledgments
The authors acknowledge the support of the CL3 facility (Facility for Infectious Disease and Epidemic Research, FINDER) of the Life Sciences Institute of the University of British Columbia founded by Dr. François Jean and its biosafety support staff including Dr. Bintou Ahidjo (Research Platform Manager) and T. Dean Airey (FINDER Senior Research Technician). We thank the LSI imaging facility of the Life Sciences Institute of the University of British Columbia funded by grants from the Canadian Foundation of Innovation, BC Knowledge Development Fund, Natural Sciences and Engineering Research Council (NSERC) Research Tools and Instruments (RTI) grant and the Strategic Investment Fund (Faculty of Medicine, University of British Columbia). We further thank Dr. Alex Ball, Jr., MD, Senior Scientist (GeneTex), for supplying the SARS-CoV-2 (COVID-19) nucleocapsid antibody [HL344] (GTX635679). We also acknowledge a generous donation towards the purchase of the CellInsight CX7 HCS system provided by the Vancouver General Hospital Foundation to Dr. Jean. We also thank Dr. Jill Kelly for proofreading the manuscript, and Dr. Carl Perez for his administrative and logistic support during this study. Ms. Olivia Garland is thanked for her assistance in generating figures. We thank GM/CA beamline staff at beamline 23-ID-B at the APS for access and support. GM/CA@APS has been funded in whole or in part with federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources from the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The Eiger 16 M detector at GM/CA-XSD was funded by NIH grant S10 OD012289. Part of the research described in this paper was performed using beamline CMCF-BM at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Callaway E. Heavily mutated coronavirus variant puts scientists on alert. Nature. 2021;600(7887):21–21. doi:10.1038/D41586-021-03552-W
- Planas D, Saunders N, Maes P, et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2022;602:671–675. doi:10.1038/s41586-021-04389-z
- Takashita E, Yamayoshi S, Simon V, et al. Efficacy of antibodies and antiviral drugs against omicron BA.2.12.1, BA.4, and BA.5 subvariants. N Engl J Med. 2022;387:468–470. doi:10.1056/NEJMc2207519
- Miller J, Hachmann NP, Collier AY, et al. Substantial neutralization escape by SARS-CoV-2 Omicron variants BQ.1.1 and XBB.1. N Engl J Med. 2023;388:662–664. doi:10.1056/NEJMc2214314
- Holmes JA, Rutledge SM, Chung RT. Direct-acting antiviral treatment for hepatitis C. Lancet. 2019;393:1392–1394. doi:10.1016/S0140-6736(18)32326-2
- Walensky RP, Paltiel AD, Losina E, et al. The survival benefits of AIDS treatment in the United States. J Infect Dis. 2006;194:11–19. doi:10.1086/505147
- Chitalia VC, Munawar AH. A painful lesson from the COVID-19 pandemic: the need for broad-spectrum, host-directed antivirals. J Transl Med. 2020;18:390. doi:10.1186/s12967-020-02476-9
- Murgolo N, Therien AG, Howell B, et al. SARS-CoV-2 tropism, entry, replication, and propagation: considerations for drug discovery and development. PLoS Pathog. 2021;17:e1009225. doi:10.1371/journal.ppat.1009225
- Shagufta AI. The race to treat COVID-19: potential therapeutic agents for the prevention and treatment of SARS-CoV-2. Eur J Med Chem. 2021;213:113157. doi:10.1016/j.ejmech.2021.113157
- Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19 – final report. N Engl J Med. 2020;383:1813–1826. doi:10.1056/NEJMoa2007764
- Arch B, Kovacs D, Scott J, et al. Evaluation of the effectiveness of remdesivir in treating severe COVID-19 using data from the ISARIC WHO clinical characterisation protocol UK: a prospective, national cohort study. medRxiv. 2021; 2021.06.18.21259072.
- Owen DR, Allerton CMN, Anderson AS, et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi:10.1126/science.abl4784
- Ledford H, Maxmen A. African clinical trial denied access to key COVID drug Paxlovid. Nature. 2022;604:412–413. doi:10.1038/d41586-022-01069-4
- Bartlett JA, DeMasi R, Quinn J, et al. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS. 2001;15:1369–1377. doi:10.1097/00002030-200107270-00006
- Hussain M, Galvin HD, Haw TY, et al. Drug resistance in influenza A virus: the epidemiology and management. Infect Drug Resist. 2017;10:121–134. doi:10.2147/IDR.S105473
- Stanciu C, Muzica CM, Girleanu I, et al. An update on direct antiviral agents for the treatment of hepatitis C. Expert Opin Pharmacother. 2021;22:1729–1741. doi:10.1080/14656566.2021.1921737
- Jochmans D, Liu C, Donckers K, et al. The substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. mBio. 2023. doi:10.1128/MBIO.02815-22/SUPPL_FILE/MBIO.02815-22-S0004.DOCX
- Ranganath N, O’Horo JC, Challener DW, et al. Rebound phenomenon after nirmatrelvir/ritonavir treatment of coronavirus disease-2019 in high-risk persons. Clin Infect Dis. 2023;76:e537–e539. doi:10.1093/cid/ciac481
- Ip JD, Wing-Ho Chu A, Chan W-M, et al. Global prevalence of SARS-CoV-2 3CL protease mutations associated with nirmatrelvir or ensitrelvir resistance. EBioMedicine. 2023;91:104559. doi:10.1016/j.ebiom.2023.104559
- Mody V, Ho J, Wills S, et al. Identification of 3-chymotrypsin like protease (3CLpro) inhibitors as potential anti-SARS-CoV-2 agents. Communications Biol. 2021;4(1):1–10. doi:10.1038/s42003-020-01566-0
- Shi J, Wei Z, Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors*. J Biol Chem. 2004;279:24765–24773. doi:10.1074/jbc.M311744200
- Silvestrini L, Belhaj N, Comez L, et al. The dimer-monomer equilibrium of SARS-CoV-2 main protease is affected by small molecule inhibitors. Sci Rep. 2021;11:1–16. doi:10.1038/s41598-020-79139-8
- Anand K, Ziebuhr J, Wadhwani P, et al. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi:10.1126/science.1085658
- Yang H, Yang M, Ding Y, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003;100:13190–13195. doi:10.1073/pnas.1835675100
- Xue X, Yu H, Yang H, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82:2515–2527. doi:10.1128/JVI.02114-07
- Kneller DW, Phillips G, O’Neill HM, et al. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun. 2020;11(1):1–6. doi:10.1038/s41467-019-13872-1
- Lee J, Worrall LJ, Vuckovic M, et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat Commun. 2020;11:1–9. doi:10.1038/s41467-019-13993-7
- Callaway E. Beyond Omicron: what’s next for COVID’s viral evolution. Nature. 2021;600:204–207. doi:10.1038/d41586-021-03619-8
- Indari O, Jakhmola S, Manivannan E, et al. An update on antiviral therapy against SARS-CoV-2: how far have We come? Front Pharmacol. 2021;0:133.
- Douangamath A, Fearon D, Gehrtz P, et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat Commun. 2020;11:1–11. doi:10.1038/s41467-019-13993-7
- Shapira T, Monreal IA, Dion SP, et al. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature. 2022;605:340–348. doi:10.1038/s41586-022-04661-w
- Pérez-Vargas J, Shapira T, Olmstead AD, et al. Discovery of lead natural products for developing pan-SARS-CoV-2 therapeutics. Antiviral Res. 2023;209:105484. doi:10.1016/j.antiviral.2022.105484
- Tang J, Wennerberg K, Aittokallio T. What is synergy? The Saariselkä agreement revisited. Front Pharmacol. 2015;6:1–6. doi:10.3389/fphar.2015.00181
- Yadav B, Wennerberg K, Aittokallio T, et al. Searching for drug synergy in complex dose–response landscapes using an interaction potency model. Comput Struct Biotechnol J. 2015;13:504–513. doi:10.1016/j.csbj.2015.09.001
- Zheng S, Wang W, Aldahdooh J, et al. Synergyfinder plus: toward better interpretation and annotation of drug combination screening datasets. Genomics Proteomics Bioinformatics. 2022. doi:10.1016/j.gpb.2022.01.004
- Sacco MD, Ma C, Lagarias P, et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci Adv. 2020: 6. doi:10.1126/SCIADV.ABE0751
- Hamill P, Hudson D, Kao RY, et al. Development of a red-shifted fluorescence-based assay for SARS-coronavirus 3CL protease: identification of a novel class of anti-SARS agents from the tropical marine sponge Axinella corrugata. Biol Chem. 2006;387:1063–1074. doi:10.1515/BC.2006.131
- Meyer B, Chiaravalli J, Gellenoncourt S, et al. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat Commun. 2021;12:1–16. doi:10.1038/s41467-020-20314-w
- Jean F, Basak A, Dimaio J, et al. An internally quenched fluorogenic substrate of prohormone convertase 1 and furin leads to a potent prohormone convertase inhibitor. Biochem J. 1995;307:689–695. doi:10.1042/bj3070689
- Yaron A, Carmel A, Katchalski-Katzir E. Intramolecularly quenched fluorogenic substrates for hydrolytic enzymes. Anal Biochem. 1979;95:228–235. doi:10.1016/0003-2697(79)90210-0
- Froggatt HM, Heaton BE, Heaton NS. Development of a fluorescence-based, high-throughput SARS-CoV-2 3CLpro reporter assay. J Virol. 2020;94; doi:10.1128/JVI.01265-20
- Citarella A, Scala A, Piperno A, et al. SARS-CoV-2 mpro: a potential target for peptidomimetics and small-molecule inhibitors. Biomolecules. 2021;11:607. doi:10.3390/biom11040607
- Cons BD, Twigg DG, Kumar R, et al. Electrostatic complementarity in structure-based drug design. J Med Chem. 2022;65:7476–7488. doi:10.1021/acs.jmedchem.2c00164
- Hou N, Shuai L, Zhang L, et al. Development of highly potent noncovalent inhibitors of SARS-CoV-2 3CLpro. ACS Cent Sci. 2022. doi:10.1021/ACSCENTSCI.2C01359/ASSET/IMAGES/LARGE/OC2C01359_0006.JPEG
- Plante JA, Mitchell BM, Plante KS, et al. The variant gambit: COVID-19’s next move. Cell Host Microbe. 2021;29:508–515. doi:10.1016/j.chom.2021.02.020
- Poutanen SM. Human Coronaviruses. Principles and Practice of Pediatric Infectious Diseases. 2012:1117–1120.e4. doi:10.1016/B978-1-4377-2702-9.00224-5.
- Shitrit A, Zaidman D, Kalid O, et al. Conserved interactions required for inhibition of the main protease of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Sci Rep. 2020;10:20808. doi:10.1038/s41598-020-77794-5
- Chang FY, Brady SF. Discovery of indolotryptoline antiproliferative agents by homology-guided metagenomic screening. Proc Natl Acad Sci USA. 2013;110:2478–2483. doi:10.1073/pnas.1222125110
- Parums DV. Editorial: The XBB.1.5 (‘Kraken’) subvariant of Omicron SARS-CoV-2 and its rapid global spread. Med Sci Monit. 2023;29:e939580-1.
- Lewnard JA, Hong V, Kim JS, et al. Increased vaccine sensitivity of an emerging SARS-CoV-2 variant. medRxiv. 2023. 2023.03.11.23287148.
- Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci USA. 1988;85:7972–7976. doi:10.1073/pnas.85.21.7972
- Greasley SE, Noell S, Plotnikova O, et al. Structural basis for the in vitro efficacy of nirmatrelvir against SARS-CoV-2 variants. J Biol Chem. 2022;298:101972. doi:10.1016/j.jbc.2022.101972
- Drayman N, DeMarco JK, Jones KA, et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science. (1979) 2021;373:931–936. doi:10.1126/science.abg5827
- Kim Y, Shivanna V, Narayanan S, et al. Broad-Spectrum inhibitors against 3C-like proteases of feline coronaviruses and feline caliciviruses. J Virol. 2015;89:4942–4950. doi:10.1128/JVI.03688-14
- Tzou PL, Tao K, Nouhin J, et al. Coronavirus antiviral research database (CoV-RDB): an online database designed to facilitate comparisons between candidate anti-coronavirus compounds. Viruses. 2020;12:1006. doi:10.3390/v12091006
- Dampalla CS, Zheng J, Dinali Perera K, et al. Postinfection treatment with a protease inhibitor increases survival of mice with a fatal SARS-CoV-2 infection. Proc Natl Acad Sci USA. 2021. doi:10.1073/pnas.2101555118
- Su H, Yao S, Zhao W, et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat Commun. 2021;12:1–12. doi:10.1038/s41467-020-20314-w
- Singh J, Anantharaj A, Panwar A, et al. BA.1, BA.2 and BA.2.75 variants show comparable replication kinetics, reduced impact on epithelial barrier and elicit cross-neutralizing antibodies. PLoS Pathog. 2023;19:e1011196. doi:10.1371/journal.ppat.1011196
- Li P, Wang Y, Lavrijsen M, et al. SARS-CoV-2 Omicron variant is highly sensitive to molnupiravir, nirmatrelvir, and the combination. Cell Res. 2022;32:322–324. doi:10.1038/s41422-022-00618-w
- Schultz DC, Johnson RM, Ayyanathan K, et al. Pyrimidine inhibitors synergize with nucleoside analogues to block SARS-CoV-2. Nature. 2022;604:134–140. doi:10.1038/s41586-022-04482-x