Abstract
In the twentieth century, NAD+ research generated multiple discoveries. Identification of the important role of NAD+ as a cofactor in cellular respiration and energy production was followed by discoveries of numerous NAD+ biosynthesis pathways. In recent years, NAD+ has been shown to play a unique role in DNA repair and protein deacetylation. As discussed in this review, there are close interactions between oxidative stress and immune activation, energy metabolism, and cell viability in neurodegenerative disorders and ageing. Profound interactions with regard to oxidative stress and NAD+ have been highlighted in the present work. This review emphasizes the pivotal role of NAD+ in the regulation of DNA repair, stress resistance, and cell death, suggesting that NAD+ synthesis through the kynurenine pathway and/or salvage pathway is an attractive target for therapeutic intervention in age-associated degenerative disorders. NAD+ precursors have been shown to slow down ageing and extend lifespan in yeasts, and protect severed axons from degeneration in animal models neurodegenerative diseases.
Keywords:
Introduction
Current thinking regarding the importance of NAD (including NAD+ and NADH) metabolism in health and disease stems from the original discovery that niacins were efficacious for the treatment of pellagra.Citation1 In 1937, ElvehjemCitation2 showed that nicotinic acid (NA) and nicotinamide (NM) (the breakdown product of NAD+) were effective agents for both the treatment and prevention of black tongue in dogs, and the human equivalent, pellagra. While the disappearance of pellagra in the developed world reduced interest in this life-threatening deficiency disease, a recent observation that several disorders mimic pellagra in its range of symptoms (e.g. dermatitis, diarrhoea, dementia, and death) has re-ignited considerable interest on the mechanism of action of NAD+ and its key metabolites.Citation3–Citation7
Intriguingly, with the understanding of the pharmacological role of niacins was the almost simultaneous discovery of the structure and function of NAD+ by four Nobel laureates.Citation8 In 1904, Sir Arthur Harden separated Buchner's yeast juice into a high- and low-molecular-weight fraction, neither of which could undergo fermentation individually. However, recombination of both fractions allowed fermentation to take place. Harden inferred the existence of high-molecular-weight ferment (enzyme) and a low-molecular-weight coferment or ‘cozymase’.Citation8
The involvement of cozymase in fermentation, respiration, and glycolysis was identified in a variety of organisms in subsequent years. However, due to the low-molecular-weight nature of cozymase, it was difficult to isolate.Citation8 It was Hans von Euler-ChelpinCitation9 who eventually succeeded in isolating cozymase from yeast extracts in the late 1920s. He determined the dinucleotide structure of NAD+ and two other mononucleotides, adenosine monophosphate (AMP) and nicotinamide mononucleotide (NMN).Citation9 The central redox function of NAD+ was determined by Otto Warburg and Christian in the mid-1930s.Citation10 He identified the capability of NAD+ to transfer hydrogen from one molecule to another.Citation10 Since then, ongoing investigations have been directed towards the identification of the enzymatic pathways involved in its synthesis and metabolism.Citation11 In 1954, Arthur KornbergCitation12 discovered the NAD phosphorylase reaction, the crucial step of NAD+ synthesis. He detected the enzymatic activity in yeast extracts which catalysed the condensation of ATP with NMN to form NAD.Citation12 While this reaction is catalysed by nicotinamide mononucleotide adenylyltransferase (NMNAT) [EC 2.7.7.1], it took another 55 years for the primary structure of this enzyme to be discovered.Citation13
Another important milestone was the discovery of the NAD+-consuming activity associated with the transfer of ADP-ribosyl moieties to protein acceptors which occurred in the 1960s.Citation14 In the same period, the term ‘pyridine nucleotide cycle’ was introduced to define several enzymatic reactions involved in the biosynthesis and catabolism of NAD+, although the significance of NAD+ metabolism remained unclear.Citation15 Rechsteiner and Catanzarite (1974)Citation16 showed that NAD+ turnover was strongly suppressed in enucleated yeast cells, suggesting that the nucleus is the major compartment responsible for the anabolism and breakdown of NAD+. Since then, studies on the enzymology of mammalian NAD+ metabolism have increased in line with those investigating the potential involvement of NAD-related metabolites in cellular physiology.Citation11,Citation17,Citation18 The involvement of NAD+ in several key cellular processes has suggested to some the possibility that NAD+ metabolism may be an attractive therapeutic target.Citation19,Citation20
Changes in NAD+ metabolism have been associated with several pathologies, including neurodegenerative diseases, cancer, cardiovascular disease, and normal ageing. A number of comprehensive papers have appeared in the literature, which provide a generalized overview of the information in this field.Citation11,Citation17,Citation18,Citation21 Beginning with an overview of relevant background research by others, this review provides additional insight into the involvement of NAD+ metabolism in neurodegenerative disorders and ageing.
NAD+ biosynthesis and salvage pathways
The kynurenine pathway and de novo NAD+ synthesis
In mammals, the kynurenine pathway (KP) represents the de novo synthesis of NAD+ from dietary tryptophan (TRP) (). The KP begins with the oxidative cleavage of TRP to N-formylkynurenine by the two enzymes tryptophan 2,3-dioxygenase (TDO) [EC 1.13.1.2] and indoleamine 2,3-dioxygenase (IDO) [EC 1.13.11.17].Citation22,Citation23 Both IDO and TDO are haem-requiring enzymes and are considered rate limiting for this pathway. TDO has been localized predominantly in the liver, but has also been found in the brain, and is induced by a number of factors including TRP, glucocoticoids, hydrocortisone, and fasting.Citation22,Citation23 IDO, on the other hand, has been identified in several extrahepatic tissues including the brain, and is up-regulated by various cytokines and inflammatory molecules such as lipopolysaccharides,Citation24 amyloid peptides,Citation25 human immunodeficiency virus (HIV) proteins,Citation26 and tumour cells.Citation27 TDO uses l-TRP exclusively as the substrate, whereas IDO can metabolize l- and d-TRP as well as serotonin and other related indoleamines.
Figure 1. NAD+ metabolism in higher eukaryotes. The de novo pathway represents the catabolism of the amino acid l-tryptophan to quinolinic acid through the KP. Quinolinic acid is then converted to nicotinic acid mononucleotide (NaMN) which connects to the salvage pathway. The three different salvage pathways start either from nicotinamide (Nam), nicotinic acid (Na), or nicotinamide riboside (NR). Nicotinamide is a by-product of NAD metabolism which gets converted into nicotinamide mononucleotide (NMN by nicotinamide phosphoribosyl transferase (NamPRT)) and then into NAD by the action of nicotinamide mononucleotide adenylyl transferases (Na/NMNAT1, 2, and 3).
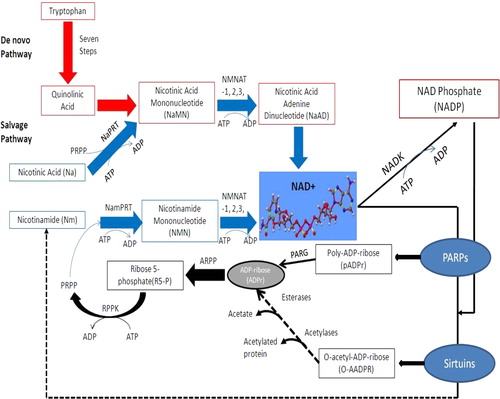
The catabolite of TRP, formylkynurenine is then hydrolysed to form kynurenine (KYN) by the action of kynurenine formamidase (KFase) [EC 3.5.1.9].Citation28 KYN can be transformed to kynurenic acid (KYNA) by the action of kynurenine-oxoglutarate transaminase [EC 2.6.1.7].Citation29 The remaining enzymes of the pathway include kynurenine 3-hydroxylase (K3H) [EC 1.14.13.9];Citation29 and kynureninase [EC 3.7.1.3].Citation30 K3H can convert KYN to 3-hydroxykynurenine (3-HK) by the hydroxylation of the aromatic ring. Kynureninase produces anthranilic acid (AA) by the cleavage of the alanine side chain from KYN. AA can also undergo hydroxylation to 5- and 3-hydroxyanthranilic acid (3-HAA) via non-specific microsomal hydroxylating enzymes. Kynureninase is also involved in the formation of 3-HAA from 3-HK. Furthermore, 3-hydroxyanthranilic acid 3,4-dioxygenase [EC 1.13.11.6] converts 3-HAA to the unstable intermediate, α-amino-β-carboxymuconic-∑-semialdehyde which cyclizes spontaneously to form quinolinic acid (QUIN). 3-HAA can also be converted to picolinic acid (PIC) by aminocarboxymuconate-semialdehyde decarboxylase [EC 4.1.1.45].Citation31,Citation32 Quinolinic acid phosphoribosyltransferase (QPRT) [EC 2.4.2.19] catalyses the formation of nicotinic acid mononucleotide (NAMN) using phosphoribosyl pyrophosphate (PRPP), which is then transformed spontaneously to NAD+.Citation33
In recent years, the KP has generated considerable interest following the observation that some intermediates are endowed with neuroactive properties. Among them is the selective N-methyl-d-aspartate (NMDA) receptor agonist and free radical generator, QUIN;Citation34 KYNA, a non-selective antagonist at both the NMDA site and the glycine strychnine-resistant site of the same receptor, which can antagonize the cytotoxic effects of QUIN;Citation35,Citation36 PIC, an endogenous metal chelator;Citation37 3-HK and 3-HAA, which can generate free radicals.Citation38,Citation39 Alterations in the endogenous levels of these metabolites has been implicated in the pathogenesis of several inflammatory brain diseases, including AD,Citation40–Citation42 AIDS dementia complex,Citation43,Citation44 cerebral malaria,Citation45 amyotrophic lateral sclerosis,Citation46 MS,Citation47 Huntington's disease,Citation48,Citation49 schizophrenia,Citation50 and Parkinson's disease (PD).Citation51 The inflammatory state can induce IDO, mainly by interferon gamma (IFN-γ).Citation52 IDO induction in immune-activated astrocytes and microglia appears to be a primary site for TRP catabolism via the KP, leading to increased production of the neurotoxic TRP metabolites, whose accumulation in the brain has been implicated in the pathogenesis of the aforementioned disorders.Citation51
Our group has identified a direct involvement of the KP in NAD+ synthesis in human brain cells, and that reduced intracellular NAD+ synthesis leads to reduced activity of NAD+-dependent histone deacetylase function. We have shown that the KP metabolite and NMDA receptor agonist, QUIN is a substrate of NAD+ synthesis in nanomolar concentrations in both astrocytes and neurons but is cytotoxic at higher amounts in both cell types. These results also show a clear neuroprotective effect of NMDA receptor antagonism and nitric oxide (NO) synthase inhibition against QUIN-mediated neuronal and glial cytoxicity, strongly suggesting that QUIN-induced excitotoxicity in astrocytes and neurons is mediated by overactivation of the NMDA receptor. Recently, we showed that natural polyphenolic compounds can attenuate QUIN-mediated neurotoxicity by a number of different mechanisms.
Also, the KP represents a defence mechanism following IDO induction during an immune-mediated response. Up-regulation of IDO expression in infected tissue can deprive the infected area of the essential amino acid, TRP, and together with the cytotoxic effects of the released KP intermediates, can exert antimicrobial activity.Citation53 Enhanced IDO expression has been reported in rats during endotoxin shock,Citation54 or during malarial infection,Citation55 suggesting that IDO may be involved in the host response following systemic infections. Moreover, increased IDO expression in dendritic cells and macrophages can deplete TRP levels from the surrounding microenvironment, which can selectively affect surrounding T-cells and inhibit their replication or induce apoptosis. It is well established that IDO induction is associated with the development of T-cell-mediated immune tolerance.Citation53 Indeed, increased IDO expression is involved in the inhibition of T-cell-mediated rejection of allogenic foetuses,Citation56 allografted pancreas islets in mice,Citation57 as well as suppression of T-cell-mediated experimental asthma.Citation58 IDO induction in tumour cells can also suppress T-cell immunity in the tumour microenvironment leading to tumoural immune resistance.Citation59,Citation60 The KP therefore represents a potential target for the development of novel therapies in several disorders associated with immune function.Citation53 However, the involvement of the KP in maintaining cellular NAD+ homeostasis in various tissues, and its potential role in neurodegenerative disease and ageing, remains unclear.
Since 2007, another enzyme with IDO-1like activity, IDO-like-protein 1 (INDOL1, IDO2), has been described in both mice and humans. In an exploratory application, Maiwald et al. (2011) substantial differences exist between the expression of IDO1 and IDO2 by professional antigen-presenting cells and MSCs (mesenchymal stromal cells) under the influence of IFN-γ and T lymphocyte media (TCM), although the exact mechanisms remain unclear. Further research is needed to delineate the functional differences between IDO1 and IDO2 in degenerative diseases and ageing.
Some studies have observed that IDO activity is directly linked to the maintenance of NAD+ levels in diverse cell types. IDO induction due to IFN-γ resulted in a significant increase in de novo NAD+ synthesis in a mouse macrophage/monocyte cell line RAW 264.7.Citation61 This suggests that KP activation may play a key role in maintaining NAD+ levels during the immune response, mainly due to the activation of poly(ADP-ribose) polymerase (PARP), which consumes a majority of intracellular NAD+.Citation62 Similarly, IDO induction has been shown to boost NAD+ concentrations and reduce cell death following PARP activation in murine astrocytes treated with hydrogen peroxide.Citation62 Conversely, inhibition of IDO has been shown to significantly reduce NAD+ levels in primary human astrocytes.Citation63 Together, these results suggest that IDO and KP metabolites are an important source of NAD+ synthesis particularly under conditions of increased NAD+ turnover. Therefore, the use of KP inhibitors as potential therapeutic targets needs to be reconsidered since they can profoundly reduce cellular NAD+ levels. Another study reported a decline in intracellular NAD+ levels in primary murine macrophages following IDO induction via tumour necrosis factor.Citation64 This study shows that activation of primary murine macrophages increases NAD+ turnover due to increased oxidative stress.
NAD+ salvage pathway
Apart from the de novo biosynthesis pathway, NAD+ can also be synthesized by the NAD+ salvage pathway (). In this pathway, NAD+ can be synthesized by one of two routes from NM, the breakdown product of NAD+.Citation18,Citation65 Firstly, the enzyme nicotinamide phosphoribosyl transferase (NMPRT) [EC 2.4.4.12] converts NM to NMN using PRPP as a cosubstrate, and subsequently to NAD+ by the action of NMNAT.Citation66 Alternatively, the enzyme nicotinamide deamidase (NM deamidase) [EC 3.5.19] can convert NM to NA, and then to NAMN by the enzyme nicotinic acid phosphoribosyl transferase [EC 2.4.2.11].Citation67 NAMN can also be directly converted to nicotinic acid adenine dinucleotide via NMNAT. A further amidation reaction catalysed by NAD synthetase [EC 6.3.5.1] is necessary to achieve the effective NAD+ form using glutamine as the nitrogen donor. Genomic analysis suggests that these two pathways are often exclusive: many organisms contain either NMPRT or NM deamidase.Citation65
Nicotinamide riboside (NR) represents another precursor for NAD+ synthesis. The phosphorylation of NR to NMN is catalysed by nicotinamide riboside kinase (NRK) [EC 2.7.1.22].Citation68 In mammalian cells, NR is formed from NMN in a reaction catalysed by NMN 5′-nucleotidase [EC 3.1.3.5].Citation68 Apart from its role in NAD+ synthesis, NRK is also involved in the phosphorylation of the compounds tiazofurin and, benzamide riboside, which is necessary to enable their further conversion into NAD+ analogues (TAD and BAD) by NMNAT.Citation68 These analogues have been previously shown to inhibit the action of inosine mononucleotide dehydrogenase (IMPDH), the rate-limiting enzyme involved in guanine nucleotide biosynthesis. Up-regulation of IMPDH activity has been reported in cancer and tiazofurin has been approved as an orphan drug for the treatment of chronic myelogenous leukaemia.Citation69 However, since the active forms of these compounds are NAD+ analogues, administration of these drugs is associated with some amount of toxicity in the clinic.Citation66
It is important to note that both NM and NA appear to be involved in numerous physiological processes. NM can enhance energy metabolism by inhibiting PARP and a new class of NAD+-dependent histone deacetylase enzymes known as sirtuins.Citation70 Numerous studies have highlighted the potential neuroprotective role of NM in multiple diseases such as cerebral ischaemia.Citation70 NA has been shown to significantly affect brain function by inducing glutamate release.Citation71 Additionally, NR has been shown to extend the replicative lifespan of yeast.Citation72
Oxidative stress in cellular degeneration
Ageing is defined as the time-dependent progressive decline of biochemical and physiological functions, and is associated with increased risk of mortality and morbidity.Citation73 There is a growing awareness that oxidative stress plays a key role not only in the ageing process but also in various other clinical conditions including neurodegenerative diseases such as Alzheimer's disease and PD, cancer, diabetes, chronic inflammation and ischaemia reperfusion injury.Citation74 Harman (1956)Citation75 was the first to propose the free-radical theory which suggested that age-related biochemical and physiological decline is associated with an accumulation of reactive oxygen species (ROS) that is generated as a result of normal cellular metabolic processes. These free radicals are thought to have deleterious side effects on the cellular constituents and play an essential role in ageing.Citation76 In the last 50 years, Harman's hypothesis has been modified to include other forms of activated oxygen such as aldehydes and peroxides which also contribute to cellular damage. The contemporary version of this tenet is now known as the oxidative stress theory of ageing. While there are several hypotheses that explain how ageing occurs, the oxidative stress theory of ageing is considered a key player during the ageing process and cell senescence.
The oxidative stress theory of ageing suggests that there is an imbalance between the amount of ROS (such as singlet oxygen, superoxide anion, hydrogen peroxide, and NO) that is generated during normal oxidative metabolism and the complex system of endogenous antioxidants (i.e. glutathione, pyridine nucleotides, and retinoic acid), and damage repair mechanisms which counter-act the deleterious effects generated by the antioxidants.Citation77 These antioxidants are thus essential to maintain the cells in their balanced state of redox. The gradual shift in the net disparity between the ROS generation and antioxidant system is thought be an important driving force behind the ageing process.Citation77,Citation78
Although current information from the studies performed on selected in-vitro model organisms are in support of the oxidative stress theory of ageing, its exact role in some unicellular organisms is still debatable.Citation79 Previous studies in clonal ageing using Saccharomyces cerevisiae (a unicellular fungus) found no significant evidence to support the limiting role oxidative stress in reducing lifespan.Citation80 However, a unicellular organism would seem a poor choice as a model system for ageing cell division leaves two identical daughter cells, which would result in either population immortality or extinction. Oxidative stress also appears to be unambiguously limited in hyphae senescence in P. Anserine.Citation81 Considering the strong dose-response between lifespan and oxidative stress in Drosophilla melanogaster, a provisional case can be made that oxidative stress appears to play a major role in influencing lifespan, even in wild-type flies under normal physiological conditions. Further studies are necessary to evaluate the role of oxidative stress and ROS production in an ageing human population.
Sources of ROS
Cells are vulnerable to ROS insult from a large variety of both exogenous and endogenous sources.Citation82 The central nervous system (CNS) is extremely vulnerable to free radical-mediated destruction due to significant oxidative metabolic activity, lower levels of antioxidants and protective enzymes and an abundance of polyunsaturated fatty membranes.Citation82 Since most neurons do not proliferate, the CNS neural network can be readily disrupted.Citation83 Importantly, the aged have an increased susceptibility to oxidative stress due to generally negative changes in dietary patterns, absorption or utilization of nutrients, or underlying infections, which can disrupt the normal antioxidant defence system.Citation83
It is generally accepted that the mitochondrial electron transport chain (ETC) is the major site for the generation of ROS.Citation77 The ETC is continuously involved in reducing molecular oxygen to water in a four electron reduction process.Citation77 However, only a small percentage of the oxygen consumed escapes the ETC as superoxide anion (O2−), which can generate other endogenous ROS, therefore posing a great threat to aerobic organisms, and their intracellular constituents.Citation77 The main forms of ROS are summarized in .
Table 1. Main ROS and their effects
Superoxide is relatively labile which means it is rapidly dismutated to H2O2 in the mitochondrial membrane by superoxide dismutase. H2O2 is detoxified by catalases to H2O and O2. Alternatively, transition metals such as ferrous and cuprous ions can reduce H2O2 into hydroxyl anion (OH).Citation84 O2− is unable to cross the mitochondrial membrane whereas H2O2 can easily diffuse across the membrane, and is thought to have important effects on intracellular signalling pathways in the cytosol, which can affect redox balance, cellular responses, and energy metabolism.Citation84
Over the last decade, new insights on the potential role of mitochondrion H2O2 have been uncovered. In particular, these studies have shown that H2O2 may regulate mitochondrial ROS production through activation of mild mitochondrial uncoupling.Citation85 Furthermore, O2−, NO, and other ROS may play a role as important mediators of cell signalling processes at moderate concentrations.Citation84 For instance, O2− can be involved in oxidative stress responses and maintenance of redox homeostasis. These studies support the notion that low levels of ROS are vital to the maintenance of various cell functions. However, detrimental effects on cells are likely if ROS are in excess.Citation86
A major exogenous source of ROS in living organisms is exposure to ionizing and non-ionizing irradiation. Exposure of tissue and cells to γ-irradiation results in the formation of several free radical species following the ionization of intracellular water.Citation87 ROS such as OH, O2−·, and H2O2 can also be produced following exposure to non-ionizing irradiation such as ultraviolet B (UV-B).Citation88 Haemolytic cleavage of H2O2 by UV radiation produces the ·OH radical. Air pollutants such as cigarette smoke, car exhaust, and industrial waste containing NO derivatives are another major source of ROS capable of damaging the organism by either inhalation into the lungs or direct interaction with the skin.Citation88 Certain drugs may also produce ROS.Citation89,Citation90 For instance, the mechanism of action of the antibiotic bleomycin is mediated by ROS production.Citation90 Several xenobiotics, such as pesticides and herbicides, may also form ROS as by-products of their metabolism in vivo.Citation91 Additionally, pathogenic organisms may also induce free radical production either by direct release from the pathogen or as an immune response by phagocytes and neutrophils.Citation92
Oxidative damage targets and types
Acute and chronic exposure to ROS from exogenous and endogenous sources can lead to the accumulation of oxidative damage to cellular components, and a significant impairment in normal cellular function. Among the biological targets vulnerable to oxidative damage are proteins, lipid membranes, and DNA.Citation86
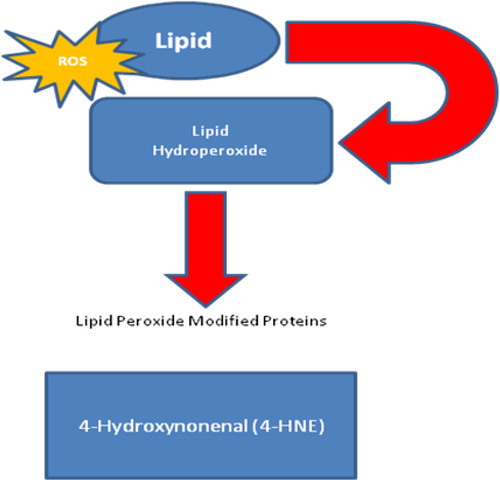
The phospholipid bilayer of all cellular membranes is extremely vulnerable to oxidation due to their higher concentrations of polyunsaturated fatty acids.Citation93 The general process of oxidative damage to lipids consists of three stages: initiation, propagation, and termination. The first phase involves hydrogen ion abstraction. Several ROS species are capable of abstracting a hydrogen atom from the methylene group in the lipid. Following hydrogen abstraction, the remaining fatty acid radical retains a single electron and is stabilized following the rearrangement of the molecular structure of the newly formed conjugate diene. The fatty acid can react to form the peroxyl radical (ROO−) in the presence of oxygen in the surrounding microenvironment during the propagation phase. The ROO− can become a lipid hydroperoxide that can further breakdown to form reactive aldehyde products, including malondialdehyde, 4-hydroxy-2-nonenal, 4-hydroxy-2-hexenal, and acrolein in the presence of reduced metals or ascorbate ().Citation92 Lipid peroxidation can disrupt the normal assembly of the cellular membrane leading to alterations in fluidity and permeability, disregulation of ion transport, and inhibition of normal metabolic processes. Lipid peroxidation is one of the major outcomes of ROS-mediated damage to cells and tissues.Citation86,Citation92,Citation93
Figure 2. Formation of lipid hydroperoxides. A great variety of compounds are formed during lipid peroxidation of membrane phospholipids.
Proteins, which also represent major components of the cellular membrane, are also potential targets for ROS-mediated damage. H2O2 appears to have a weak effect on proteins.Citation86,Citation94,Citation95 However, proteins containing the sulfhydryl (-SH) group can undergo oxidation following interaction with H2O2. Protein oxidation products can include aldehydes, ketone compounds, and carbonyls.Citation86,Citation94,Citation95 A major adduct that can be easily detected as a marker for protein oxidation is 3-nitrotyrosine (3-NT). 3-NT is formed following the interaction of the peroxynitrite (ONOO−) and other reactive nitrogen species with the amino acid tyrosine.Citation96 Proteins can undergo direct and indirect damage following exposure to ROS, including peroxidation, damage to amino-acid residues, denaturation of the tertiary protein structure, and fragmentation, leading to loss of function.Citation86,Citation94,Citation95
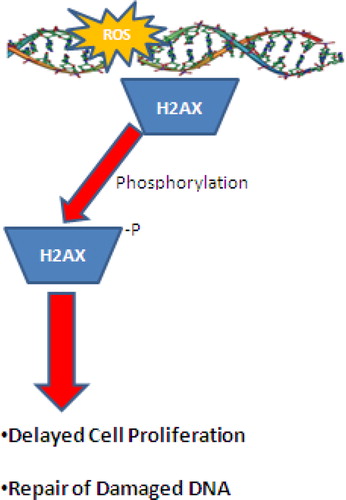
A major factor associated with age-related diseases is the increase in oxidative DNA damage. ROS can interact with DNA leading to the modification of DNA bases, loss of purines, and single- and double-strand DNA breaks. The main ROS capable of causing damage to DNA is the ·OH radical.Citation86,Citation97,Citation98 Following exposure of DNA to OH, several adducts, such as the oxidation product, 8-hydroxydeoxyguanosine are formed at the C-8 position.Citation99 ·NO and O2− can also lead to the formation of ONOO−, which can also cause DNA damage similar to the ·OH radical. More recently, ROS induced DNA double-stranded breaks have been shown to induce histone H2AX phosphorylation on serine 139 ().Citation100,Citation101 DNA damage may affect the expression of a variety of genes involved in the regulation of cell proliferation or inhibit expression of other genes associated with DNA repair. Many cell types do not show time-related degeneration due to rapid turnover. However, some mammalian cells, particularly neurons change considerably with age.Citation102 Elevated levels of ·OH mediated DNA and RNA damage are consistently observed in mutations, carcinogenesis, degenerative and other diseases, inflammation, ageing and during development.Citation86,Citation97,Citation98
Figure 3. Phosphorylation of H2AX at Serine 139 following ROS damage to DNA. Under mild-to-moderate oxidative damage, phosphorylation of H2AX can lead to temporary cessation of cellular function and DNA repair. However, under extreme conditions, hyperphosphorylation of H2AX can lead to cell death via an apoptotic mechanism.
Importance of maintaining intracellular NAD+ levels
NAD+ has now been identified as a ubiquitous molecule which plays a critical role in several biological processes, including cellular respiration, DNA repair, and transcriptional regulation.Citation103 NAD+ and its reduced form NADH have enormous importance in cell biology, and are generally considered core components in redox reactions.Citation104 NAD+ plays a critical role in an increasingly diverse range of cellular processes, including signal transduction, DNA repair, and post-translational protein modifications and apoptosis.Citation104 NAD+ and NADH are used during cellular respiration during the process of oxidative phosphorylation and ATP production. Therefore, ATP synthesis and redox potential is directly proportional to intracellular NAD+ concentrations.Citation104 In addition to its role in cellular metabolism, NAD+ is also important for a number of ADP-ribosylation reactions associated with cell regulation and repair mechanisms which are discussed later in this paper.
NAD+ in energy metabolism
As mentioned previously, NAD+ and NADH serve as key regulators of glycolysis by acting as important cofactors for GAPDH in the cytosol.Citation105 Cytosolic NAD+ and NADH also mediate other energy metabolism-related reactions in the cytosol, including the lactate dehydrogenase-catalysed lactate–pyruvate conversions and the PDHC, which converts pyruvate to acetyl-CoenzymeA, a substrate for the tricarboxylic cycle (TCA).Citation106 Moreover, cytosolic NADH may also affect oxidative phosphorylation, since the reducing equivalents of NADH can enter the mitochondria through NADH shuttles.Citation107
NAD+ and NADH also play pivotal roles in the TCA and mitochondrial electron transport chain (NAD+ and NADH serve as cofactors for at least three rate-limiting enzymes in the TCA).Citation108 NADH is also one of the major electron donors in the electron transport chain, mediated by cytochome c and mitochondrial cytochrome oxidase.Citation109 Cytosolic NADH is transferred to cytochrome c by the NADH-cytochrome b5 oxido-reductase complex on the external mitochondrial membrane, and the cytochrome c transfers the electron to mitochondrial complex IV (cytochrome oxidase) at the respiratory site. Subsequently, molecular oxygen is reduced with generation of the electrochemical membrane potential. This process has been shown to occur in the early stage of apoptosis, and may also occur under physiological conditions, as constitutive release of mitochondrial cytochrome c to the cytosol does occur.Citation109,Citation110 This biological process may act not only to enhance removal of excessive NADH but also to promote cell survival when the first three respiratory complexes are impaired.
Poly(ADP-ribose) polymerase and NAD+ depletion
In genomic DNA repair, NAD+ is the sole substrate for the DNA nick sensor, PARP. The PARP family of enzymes is DNA-binding enzymes activated by ROS-initiated breaks to the DNA and is critical to the base excision repair (BER) process.Citation111 While many proteins are involved in repairing DNA damage, the majority of lesions are repaired by BER.Citation111 For many years, PARP1 was thought to be the only enzyme catalysing poly-ADP-ribosylation. PARPs now constitute a new class of protein, consisting of six members in humans.Citation112
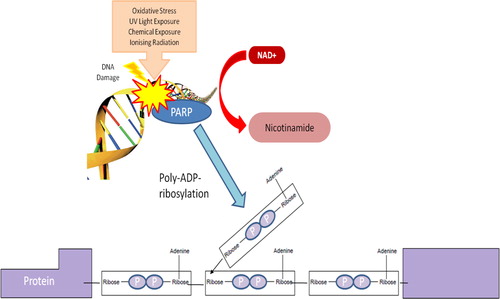
Several reports indicate that PARP activity represents the main NAD+ catabolic process in the cell, thereby forcing the cell to continuously synthesize NAD+ from the de novo or salvage pathway to maintain cellular viability following oxidative damage.Citation113–Citation115 The fast recycling is consistent with the short half-life of NAD+, which is estimated to be around 1–2 hours.Citation116 Activation of PARP catalyses the cleavage of NAD+ into adenosine 5′-diphosphoribose (ADPR) and nicotinamide, and the covalent attachment of polymers of ADP-ribose to histones and other nuclear proteins, including PARP itself ().Citation117 PARP1, which accounts for the majority of the ADPR synthesized, is found at the highest concentrations in the nucleus.
Figure 4. Schematic representation of poly(ADP-ribose) synthesis. Poly(ADP-ribose) polymerases breaks the bond between nicotinamide and ribose in NAD+ leading to the formation of ADP-ribosyl moiety. Repeated reaction triggers the formation of PAR chains.
PARP1 activation leads to DNA repair and recovery of normal cellular function. Experimental studies have shown that PARP1 is activated in response to free radical-mediated injury to DNA after brain ischaemia and reperfusion.Citation118 Hyperactivation of PARP1 following DNA strand breaks can rapidly consume intracellular NAD+ pools, resulting in a loss of ability to synthesize ATP, and the cessation of all energy-dependent functions and consequent cell death ().Citation112,Citation119,Citation120
Figure 5. PARP-1-dependent signalling in apoptosis. Excessive oxidative stress leads to hyperactivation of PARP-1 in the nucleus. PAR formation and NAD+ depletion can trigger a cascade of events which can lead to the release of apoptotic inducing factor (AIF). AIF can migrate to the nucleus where it can trigger cell death. AIF can also promote release of cytochrome c (cyt c) through upregulation of Beclin-2 protein (Bcl-2) which can cause apoptosis via a downstream signalling through caspase activation.
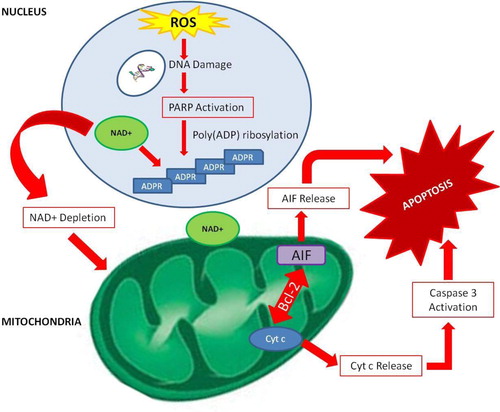
PARP-mediated NAD+ depletion has been implicated in the pathogenesis of AD, with one study showing that poly(ADP-ribose) (PAR) polymers accumulate at higher concentrations in the temporal and frontal cortex in the brains of AD patients compared with control brains.Citation121 This indicates that PARP1 is over expressed in the AD brain and implies an excessive NAD+ turnover in susceptible neuronal cells.Citation121 Over-activation of PARP1 has also been reported in diabetes, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism, traumatic brain injury, hypoglycaemic brain disease, and shock.Citation73,Citation112,Citation122–Citation124
Sirtuin activity
In addition to its role in PARP activity, another essential factor that is greatly altered by changes in intracellular NAD+ levels are the sirtuins, or the silent information regulators of gene transcription.Citation125 Sirtuins are a highly conserved family of class III deacetylase proteins which catalyse a unique reaction in which NAD+ is used to remove an acetyl group from the lysine residue, releasing nicotinamide and acetyl-ribose as end products ().Citation126 At least seven classes of sirtuins (SIRT1–7) have been identified, each of which exhibit a wide range of fascinating biological functions, including the control of gene expression, cell cycle regulation, apoptosis, DNA repair, metabolism, and ageing.Citation127,Citation128 SIRT1 is located in the nucleus and is thought to play a vital role in ageing and cell longevity.
Figure 6. Sirtuin enzymatic activities. Sirtuins display two different NAD+ consuming activities, both of which render NM as a product.
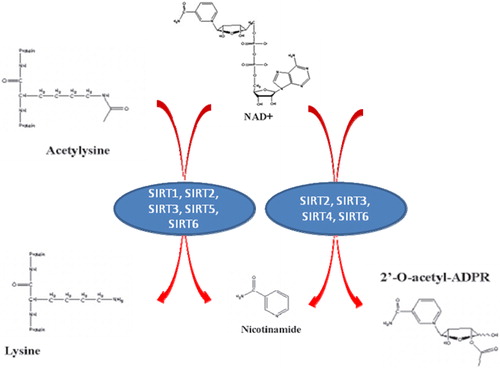
In mammals, seven homologues of Sir2 have been described, namely SIRT1–7, which are ubiquitously expressed. SIRT1, SIRT6, and SIRT7 are nuclear proteins involved in the regulation of chromatin structure and gene expression.Citation129 In contrast, SIRT2 is localized in the cytoplasm where it mediates gene expression by deacetylating transcription factors which shuttle from the cytoplasm to the nucleus.Citation130 The remaining members of the sirtuin family (SIRT3, SIRT4, and SIRT5) are mitochondrial protein.Citation129
SIRT1 is the most characterized mammalian sirtuin,Citation20 and is activated in response to energy stress, such as fasting,Citation131 exercise,Citation132 or low glucose availability which also serves to increase intracellular NAD+ levels.Citation133 As well, SIRT1 modulates the acetylation status of a number of important transcription factors, including the peroxisome proliferator-activated receptor-γ (PPARγ), tumour suppressor protein (p53), and the FOXO forkhead family of transcription factors, all of which are key metabolic regulators.Citation134 Consistent with observations in lower-order organisms, a number of beneficial effects have been observed in mice that over-express SIRT1.Citation127,Citation128 Bordone and colleagues (2007) found that these transgenic mice displayed phenotypes similar to mice on a calorie-restricted diet, including reduced body weight, greater metabolic activity, reduced blood cholesterol, adipokines, insulin and fasted glucose, and were more glucose tolerant.Citation127,Citation128 Not surprisingly, the life-enhancing properties of sirtuins go hand in hand with those of NAD+ metabolism, suggesting a causal relationship where SIRT1 translates alterations of NAD+ levels into transcriptional events.Citation103,Citation135
Although less is known about the cellular roles of other sirtuins on NAD+ levels, some interesting functions have been associated with these sirtuins which are worth mentioning. SIRT2 is a tubulin deacetylase which is down-regulated in gliomas.Citation136 Ectopic expression of SIRT2 in glioma cell lines has been shown to decrease colony formation, suggesting that SIRT2 may have a tumour suppressor role.Citation136 Furthermore, SIRT2 also acts as a mitotic checkpoint which maintains chromosomal stability in the early metaphase.Citation137 Recently, SIRT2 has been shown to promote myelination in oligodendrocytes.Citation138
SIRT2 and SIRT1 have been thoroughly investigated in regard to ageing. Activation can mimic the effect of calorie restriction (CR) and extend the lifespan of yeast, worms, and flies. Moreover, overexpression of SIRT2 has been shown to extend lifespan in Caenorhabditis elegans.Citation139 Recently, Pearson et al. (2008)Citation140 showed that resveratrol (a putative SIRT1 activator) improved metabolism and significantly enhanced the health and survival of mice on high-calorie diet. In addition, both overexpression of SIRT1 and administration resveratrol have been shown to have neuroprotective effects.Citation140–Citation142 Collectively, these studies raise the possibility that activation of human sirtuins may slow down the ageing process. However, additional studies in the human population are required in order to elucidate the involvement of sirtuins in human population health.
Impaired SIRT1 activity due to PARP-1-mediated NAD+ depletion stimulates the activity of several apoptotic effectors such as p53,Citation143 therefore, sensitizing cells to apoptosis. Both human and mouse SIRT1 are thought to promote cell survival by deacetylating and thus deactivating p53 tumour suppressor gene hence enhancing p53 degradation.Citation20,Citation144 Adequate NAD+ levels are therefore critical to maintaining SIRT1 activity which can delay apoptosis and provide vulnerable cells with additional time to repair even after the repeated oxidative stress insult.
SIRT3 has been linked to adaptive thermogenesis,Citation145 mitochondrial function,Citation145 energy homeostasis,Citation146 and cellular viability following genotoxic insult.Citation147 On the other hand, despite the presence of a conserved sirtuin domain, SIRT4 does not appear to exhibit deacetylase activity in vitro. Instead, SIRT4 appears to target protein activities through ADP-ribosylation.Citation148 Like SIRT3 and SIRT4, SIRT5 is a mitochondrial sirtuin with deacetylase activity, although the exact role in maintaining cellular homeostasis remains unknown.Citation149 Upcoming research will shed some light in our understanding of SIRT5 biology.
While SIRT6 was initially suggested to possess ADP-ribosylation activity only,Citation150 it recently has been shown to deacetylate histones and DNA polymerase β, a DNA repair enzyme.Citation151 Several lines of evidence suggest that SIRT6 regulates genomic stability and DNA repair. SIRT6-deficient mice die prematurely, and exhibit severe defects, such as lymphopenia, decreased bone mineral density, and impaired glucose homeostasis. This phenotype mimics multiple pathologies observed in elderly humans, suggesting that SIRT6 could play an essential role in maintaining organ integrity during ageing and development.Citation150,Citation151
Finally, SIRT7 is localized in the nucleolus where it positively regulates transcription of ribosomal DNA during elongation, which can account for up to 60% of total transcription in metabolically active states.Citation152,Citation153 Over-expression of SIRT7 increases RNA polymerase transcription in an NAD-dependent manner, whereas down-regulation of SIRT7 can reduce cell proliferation and trigger apoptosis.Citation152,Citation153 In addition, the tumorigenic potential of several cell lines inversely correlates with SIRT7 expression.Citation154
Interactions between PARPs and sirtuins
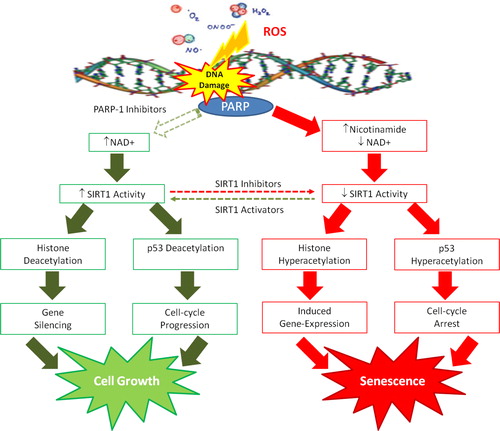
There is now emerging evidence indicating an association between the PARP-1 and SIRT1 pathways, which have been previously independently studied. As previously described, overactivation of PARP-1 following extensive DNA damage can lead to cell death, since prolonged PARP-1 activation can deplete the essential substrate NAD+, leading to a subsequent increase in the byproduct, nicotinamide. The decline in NAD+ and the rise in nicotinamide may downregulate the SIRT1 deacetylase activity.Citation155 It is conceivable, therefore, that poly(ADP)-ribosylation by PARP-1, as induced by DNA damage, can modulate SIRT1 protein deacetylation via the NAD+/nicotinamide connection ().
Figure 7. NAD+/nicotinamide levels may serve as converging points for interaction of PARP-1 and SIRT1 pathways. It is conceivable that poly(ADP-ribose) metabolism can downregulate SIRT1 through NAD+ depletion and nicotinamide production during oxidative stress. Reciprocally, regulation by SIRT1 deacetylation for expressing genes related to apoptosis or longevity may depend on PARP-1 activity. PARP-1 inhibitors maintain NAD+ levels and suppress the nicotinamide surge, and therefore may indirectly serve as SIRT1 enhancers. The interactions of PARP-1/SIRT1 pathways provide a network for multicellular eukaryotes to effectively deal with nutritional supply and oxidative stress.
Subcellular distribution of NAD+ and its metabolism
Compartmentation of NAD+
A major complication associated with the assessment of intracellular NAD+ levels is the subcellular compartmentation of NAD+ in mammalian cells. There are two major independent NAD+ pools: cytosolic and mitochondrial.Citation119 It is estimated that the relative number of mitochondria within a tissue corresponds to the share of mitochondrial NAD+. The mitochondrial fraction of NAD+ is therefore significantly higher in the liver and other mitochondria-rich tissue such as the brain and heart. The cytosolic pool of NAD+ appears to be freely exchangeable with the nucleus. As no physiological means of exchange have been identified between the mitochondria and cytosolic NAD+ compartments, these two pools are considered independent of each other.Citation119,Citation156,Citation157 di Lisa and Ziegler (2001),Citation119 however, suggested that mitochondrial permeability transition (MPT) pore opening can lead to mitochondrial NAD+ release that is metabolized by NAD-dependent dehydrogenases in the heart. However, it is unclear as to whether MPT-dependent NAD+ release also takes place in neurons and glial cells.
The NAD+:NADH ratio
The NAD+:NADH ratio plays a pivotal role in regulating the intracellular redox state, and is hence considered to be a measure of the metabolic state.Citation144 As previously mentioned, several enzymes appear to be regulated by the NAD+:NADH ratio, such as GAPDH and dehydrogenase reactions.Citation158 Several studies have shown that the NAD+:NADH ratio fluctuates in response to a change in metabolism.Citation159–Citation163
If NAD+ is a metabolic regulator, then the ratio of the intracellular NAD+ to NADH is close to 1. Therefore, the NAD+:NADH is regulated by small changes in the NAD+ concentration.Citation161 For instance, if the ratio is very high (e.g. 600), then one would assume that the NAD+:NADH ratio will be more sensitive to a change in the NADH concentration, and not NAD+.Citation144 shows the reported ratios of total intracellular NAD+:NADH. These values suggest that NAD+ is a key metabolic regulator of the NAD+:NADH ratio in a variety of tissues.
Table 2. Baseline ratio of cytosolic NAD+:NADH ratio in various species
Compartmentation of NAD+ metabolism in mammalian cells
Interestingly, the key enzymes involved in the biosynthesis of NAD+ are localized within the nucleus.Citation164 In humans, the predominant isoform of NMNAT (NMNAT1) is located within the nucleus (.17). The recent discovery of nuclear NAD+-dependent signalling pathways such as poly-ADP-ribosylation and sirtuin-mediated histone deacetylation suggests that nuclear NAD+ production is necessary to compensate for the high rate of NAD+ turnover. Indeed, the inhibitory effect of NMNAT1 on PARP activity has been previously described to counteract NAD+ and ATP depletion following hyperactivation of PARP in response to oxidative stress.Citation165
Recent studies have indicated the presence of two additional isoforms of human NMNATs – NMNAT2 and NMNAT3, which are located in the Golgi complex and mitochondria, respectively.Citation166,Citation167 These findings, together, with discovery of NAD-dependent tankyrases in Golgi complex and NAD+-consuming enzymes in the mitochondria, provide further evidence to support the existence of independent NAD+ synthesizing machinery in the nucleus, Golgi complex, and mitochondria.Citation168
Therapeutic potential of NAD+ metabolism
NAD+ in neurodegeneration
Considering the importance of NAD+ in energy metabolism, DNA repair and transcriptional regulation, maintaining intracellular NAD+ reserves emerges as a major therapeutic target for the treatment of several age-related degenerative diseases, including AD.Citation169 In particular, increased nuclear NAD+ biosynthesis and consequent activation of SIRT1 has been shown to protect mouse neurons from mechanical and chemical injury.Citation170 Restoring cellular NAD+ levels have been shown to protect against axonal degeneration in the experimental autoimmune encephalomyelitis animal model for MS in humans.Citation19
Axonopathy is a critical feature of several neurodegenerative diseases and often precedes the death of neuronal bodies in AD, PD, and MS.Citation171 As axonal deficits are central to the patient's neurological disability, therapies that prevent axonal degradation are of great therapeutic importance.Citation172 Increased NMNAT1 activity has also been shown to protect against axonal degeneration in Wallerian degeneration slow (Wlds) mice. Exogenous administration of NAD+ prior to axotomy also delayed axonal degeneration, but to a lesser extent in NMNAT1 expressed mice, further indicating the importance of maintaining intracellular NAD+ pools as a preventive measure against axonal degradation. In the absence of exogenous NAD+, PARP inhibition increased the survival of dorsal root ganglion cultures following mechanical injury. No protective effect on Wlds mice was observed following PARP inhibition in the presence of exogenous NAD+.Citation170,Citation172 This suggests that maintaining adequate intracellular NAD+ levels can promote neuronal survival.
Another link between neurodegeneration and NAD+ metabolism lies in the fact that many neurodegenerative disorders, including AD, PD, and MS, are associated with mitochondrial dysfunction due to extensive oxidative damage.Citation173 Therefore, it is possible that activation of PARP1 can mediate the neuronal cell death observed in these pathological states.Citation73 Indeed, increased PARP expression has been reported in the brains and peripheral cells in post-mortem patients with AD, PD, and MS.Citation121,Citation174,Citation175 Further work is necessary to identify the mechanism(s) underlying the major differences in PARP expression in these neurodegenerative diseases.
As previously mentioned, the KP represents the major route of TRP catabolism leading to the synthesis of a number of neuroreactive compounds.Citation176 Among those, QUIN appears to be involved in the pathogenesis of several neuroinflammatory disorders.Citation177 Therefore, KP inhibitors have been developed to reduce QUIN levels in the CNS.Citation178 It remains unresolved whether KP inhibition will also result in decreased NAD+ biosynthesis in the brain.
NAD+ in ageing
As chronic oxidative stress and associated nuclear damage can promote NAD+ catabolism, the NAD+ metabolic pathway has been implicated as a potential therapeutic target to promote longevity.Citation113,Citation114,Citation179 Owing to the importance of oxidative stress in ageing, it is highly likely that PARPs may play major roles in ageing by promoting NAD+ depletion.Citation73,Citation123 Grube and Burkle (1992)Citation124 showed that PARP-1 activity in mononuclear blood cells increases with ageing in at least thirteen mammalian species, and this is likely to be associated with its important DNA repair function. Consistent with its role as the major player in the OS theory of ageing, the mitochondrial dysfunction which can occur in the ageing process is associated with changes in NAD+ levels.Citation180 Although a number of studies have demonstrated elevated levels of oxidative stress-mediated damage in aged tissue, to our knowledge, no study has yet reported on changes in NAD+ levels during the human ageing process. Braidy et al. (2011)Citation181 recently reported a parallel increase in p53 acetylation with age, closely correlating with increased levels of lipid peroxidation, protein cross-linking, and DNA damage in the heart, lung, liver, and kidney, or aged female wistar rats. However, the potential for increased NAD+ catabolism appears to be involved in ageing, the impact of this process and its contribution on NAD+ in extending lifespan needs to be clarified.
It has been well established that CR is one of the best strategies to improve lifespan in several species. The hypothesis that increased mitochondrial function upon CR is associated with the beneficial effects on lifespan has been recently extended to humans, in which a general increase in metabolism occurred during CR.Citation20 The beneficial effects of CR appear to be NAD+ dependent and partly mediated by SIRT1/Sir2 activity.Citation182 In C. elegans, extra copies of the SIRT2 orthologue sir-2.1 extended lifespan.Citation139 Both human and mouse SIRT2 have been shown to function as NAD-dependent p53 deacetylases, and deacetylation of p53 by SIRT2 can promote cell survival under stress.Citation20,Citation144 Recently, Pearson et al. (2008)Citation140 showed that activation of SIRT1 using resveratrol in mice fed a high-calorie diet improved metabolism and significantly enhanced the lifespan of these animals. Previous work from our laboratory showed for the first time that resveratrol does have a novel function in specifically upregulating NAD+ synthesis. Additionally, SIRT3, SIRT6, and SIRT7 have been linked with a longer lifespan in mice,Citation151,Citation154,Citation183 while SIRT1 gene polymorphisms can affect lifespan by stimulating energy release.Citation184 Additional human studies are required to clarify the involvement of sirtuins in human population health.
However, Anderson et al. (2003)Citation147 suggested an alternative mechanism by which CR and Sir2 may mediate longevity in yeast. Pyrazinamide/nicotinamidase (PNC1) encodes an enzyme which converts NM to NA, thereby promoting Sir2 activity and depleting the endogenous Sir2 inhibitor, NM.Citation147 PNC1 appears necessary for the lifespan extension mediated by CR. Within the NAD+ biosynthesis pathway NMPRT has been shown to regulate the ageing process.Citation185 Reduced NMPRT expression resulted in premature senescence in ageing human smooth muscle cells, whereas over-expression of NMPRT delayed senescence while promoting enhanced antioxidant defence.Citation185
Due to the important role of oxidative stress in ageing, it is conceivable that PARPs may play significant roles in ageing by increasing demand for its substrate, NAD+.Citation73,Citation123 PARP1 activities in mononuclear blood cells strongly correlate with longevity in at least 13 mammalian species, which may be associated with its important DNA repair function.Citation124 Moreover, PARP1 has been shown to inhibit the catalytic activities of the protein of Werner syndrome, a human disease of premature ageing.Citation186 While the NAD+ metabolic pathway appears to be involved in ageing, the exact role of NAD+ in extending lifespan is not so clear-cut. Further research in this field is necessary to determine whether NAD+ is an effective target to increase lifespan.
Conclusion
Pellegra, a syndrome caused by a diet deficient in either NA or TRP can lead to psychotic symptoms leading to presenile dementia likely due to upregulation of IDO, which can deplete neurons of the essential amino acid, TRP causing neurodegeneration.Citation6 Administration of the NAD+ precursors, NA or NM previously improved the neurological state of dementia patients in the 1930s.Citation6 Pharmacological doses of either NA and NM have also provided dramatic therapeutic benefits for other diseases, including rheumatoid arthritis, type I diabetes, colitis, MS, and schizophrenia in animal models and in the clinical setting.Citation4 Among these precursors, NA appears to specifically activate the G-protein coupled receptor, GPR109, leading to the release of the prostaglandins, PGE2 and PGD2.Citation187 These prostaglandins exert potent anti-inflammatory effects through endogenous signalling mechanisms involving PPARγ.Citation187 While NM can prevent MS in animal models, it is also an inhibitor of sirtuins, and may therefore prove detrimental on long-term cell survival and longevity.Citation147,Citation188
There is growing evidence suggesting that NAD+ administration may also reduce cellular injury in multiple diseases.Citation115 NAD+ treatment has been shown to reduce PARP1-induced astrocyte death.Citation189 It has been also shown to prevent PARP1-mediated NAD+ depletion in cardiac myocytes in the presence of H2O2.Citation190 PARP1 has been implicated in the pathogenesis of several diseases including diabetes, AD and PD (Ying, 2006, 2008).Citation115 Since supplementation with NAD+ can protect against PARP1 mediated cell death, NAD+ administration may improve cell viability in these diseases by at least partially ameliorating PARP1 toxicity. In vitro studies have shown that NAD+ remains protective even when administered at 3–4 hours following PARP1 activation, suggesting that NAD+ administration has a long window period for reducing cellular injury.Citation189 In addition, NAD+ may also improve cell viability by enhancing sirtuin activities and/or improving energy metabolism (Ying, 2006, 2008).Citation115
Resveratrol is a polyphenol with major health benefits that is thought to operate through direct activation of the ‘antiageing’ enzyme SIRT1.Citation191 However, recent reports have challenged this ‘direct-activation’ hypothesis,Citation192 suggesting that the mechanism by which resveratrol increases SIRT1 function is still unknown. Previous work from our group has shown for the first time that resveratrol induces a dose-dependent increase in activity of the NAD+ synthetic enzyme nicotinamide mononucleotide adenyl transferase (NMNAT1) (unpublished data). As SIRT1 requires NAD+ as a substrate to perform its gene-silencing function, higher NAD+ levels will enhance SIRT1 activity.Citation172 This finding suggests that resveratrol may promote SIRT1 function by enhancing NAD+ synthesis in whole cell systems without requiring direct activation.
Our observation that resveratrol increases NAD+ levels in primary human brain cells by acting on NMNAT, together with the neuroprotective effects of green tea polyphenols against QUIN-mediated excitotoxicity, supports the view that polyphenols have considerable therapeutic potential, particularly for the treatment of neurodegenerative diseases.Citation193 As NMNAT can accelerate NAD+ synthesis from all three substrates, QUIN, NA, and NM (Berger et al., 2005),Citation17,Citation167 NMNAT activation by resveratrol represents an ideal natural therapeutic to replenish NAD+ levels. Maintenance of higher cellular NAD+ will enhance SIRT1 activity and other NAD+-dependent pathways, impacting positively on cell viability and longevity. This work has therefore formed the basis of relevant patent applications.
While the potential involvement of NAD+ metabolic pathways in energy metabolism and mitochondrial function have been known for quite some time, suggestions of the involvement of NAD+ in DNA repair and longevity have grown at a rapid rate in the last decade.Citation8 Characterization of the NAD+ synthetic pathways has not only made these advancements possible, but also contributed extensively to the understanding of the diverse roles of pyridine nucleotides in cellular biology.Citation8 Despite this, information regarding the fundamental roles of NAD+ in neurodegeneration and ageing remains limited. Further investigations are necessary in this increasingly interesting field. Research in the following areas may be of particular interest.
Firstly, maintaining intracellular NAD+ levels in human brain cells such as astrocytes and neurons is crucial for the retention of cellular viability during conditions of chronic oxidative stress and immune activity through the promotion of oxidative phosphorylation (ATP production), DNA repair (PARP activity), and gene expression (sirtuin activity). Therefore, characterization of the effect of varying degrees of immune activation on de novo NAD+ synthesis in selected brain cells is necessary to validate the role of NAD+ metabolism as a primary contributor to neuronal dysfunction and cell death during neuroinflammatory disorders.
Secondly, as NAD+ is an essential molecule for all living organisms, it is not surprising that numerous cell types may possess a number of different strategies to generate NAD+, particularly under conditions of acute and chronic oxidative stress and NAD+ depletion.Citation194 For instance, post-mitotic cells such as neurons may rely on different pathways than those actively in dividing cells such as astrocytes.Citation194 Further studies may therefore need to be aimed at identifying the preferred substrate for NAD+ synthesis in neuronal cells under various conditions.
Thirdly, additional NAD+ regulates diverse pathways which may control lifespan. The importance of NAD+ is further underscored by recent work providing genetic evidence for the existence of several pathways necessary for NAD+ synthesis. For example, Belenky et al. (2007)Citation72 recently demonstrated that a newly identified NAD+ precursor, NR, can contribute to NAD+ synthesis by at least two unique pathways in the yeast S. cerevisiae. Both pathways require the nicotinamide ring for entry into the previously established pathways for NAD+ synthesis.Citation72 Future studies are required to address the importance of NR in human health and disease, and whether it can be effectively used to replenish lowered NAD+ levels in age-related diseases, such as AD.
Fourthly, the essential cofactor PRPP is an important regulator for the de novo NAD+ synthetic enzyme, QPRT, which catalyses the conversion of the excitotoxin QUIN to NAMNCitation11,Citation18,Citation166. PRPP concentration has been positively correlated with cytosolic NAD+ and ATP levels in whole animals.Citation195 Therefore, the availability of PRPP for QPRT activity may be compromised during increased NAD+ turnover. This may occur in neurodegenerative disorders and ageing due to ROS-mediated DNA damage.Citation196 Increased QUIN secretion into the CSF may be due to increased flux through the KP parallel to reduced QPRT activity associated with an increased demand for PRPP for NAD+ synthesis in damaged cells. Additional studies are required to investigate the effect of PRPP on de novo NAD+ synthesis during neuroinflammation and ageing. Fifthly, the roles of different sirtuins in brain function need to be further investigated under different physiological and pathological conditions. Numerous studies have highlighted the importance of sirtuins as key regulators in ageing.Citation197 However, the precise roles of different sirtuin isoforms (SIRT1-7) and their response to varying NAD+ concentrations in brain function remain unclear. Future work may be aimed at establishing the roles of sirtuins in brain function as a therapeutic target for the treatment of a variety of brain disorders and increasing lifespan.
Sixthly, NAD+ plays a key role in regulating intracellular calcium homeostasis by acting as the substrate for NAD-dependent glycohydrolase (NADase) (see Section 1.4.5). While PARP1 is a major NAD-consuming enzyme in the cell, recent studies have raised the possibility that NADase may play a key role in NAD+ metabolism under physiological conditions.Citation8 For instance, increased NAD+ levels have been reported in the brain, lung, and kidney in NADase-deficient mice.Citation198 Moreover, NADase activity was absent in the plasma membranes, mitochondria, sarcoplasmic reticulum, and nuclei in NADase-deficient mice.Citation199 These studies suggest that NADase is a key regulator of cellular NAD+ levels under physiological conditions, while PARP1 is a key factor determining intracellular NAD+ levels when significant oxidative stress and DNA damage occurs.Citation115 Owing to the critical roles of NADase and calcium in cellular function, it is warranted to further examine the roles of NAD+-dependent changes in calcium homeostasis not only in normal brain function but also in brain ageing and neurological disorders, in general.
Seventhly, while the current investigations reported herein focussed on PARP1 in cellular degeneration, the role of other PARPs such as tankyrases in cellular function remains largely unknown. Since NAD-dependent tankyrases are main mediators of telomerase activity, it is highly likely that NAD+ may also affect the ageing process through regulation of tankyrase activity.Citation200 It would therefore be intriguing to study the effects of tankyrases and telomerases on certain biological functions, including neurogenesis, which might affect the ageing brain.
Acknowledgements
Hassina Massudi is a reciepient of the Australian Postgraduate Award (APA) at the University of New South Wales. Nady Braidy is the recipient of an Alzheimer's Australia Viertel Foundation Postdoctoral Research Fellowship at the University of New South Wales.
References
- Elvehjem C, Madden R, Strong F, Woolley D. Relation of nicotinic acid and nicotinic acid amide to canine black tongue. J Am Chem Soc 1937;59:1767–8.
- Elvehjem C. Pellagra – a deficiency disease. Proc Am Philos Soc 1949;93:335–9.
- Broer S, JA C, Rasko J. Neutral amino acid transport in epithelial cells and its malfunction in Hartnup disorder. Biochem Soc Trans 2005;33:233–6.
- Monteiro J, da Cunha D, Filho D, SilvaVergara M, dos Santos V, da Costas J , et al. Niacin metabolite excretion in alcholic pellagra and AIDS patients with and without diarrhea. Nutrition 2004;20:778–82.
- Comaish J, Felix R, McGrath H. Topically applied niacinamide in ioniazid-induced pellagra. Arch Dermatol 1976;112:70–2.
- Penberthy WT. Pharmacological targeting of IDO-mediated tolerance for treating autoimmune disease. Curr Drug Metab 2007;8:245–66.
- Brown RR, Ozaki Y, Datta SP, Borden EC, Sondel PM, Malone DG. Implications of interferon-induced tryptophan catabolism in cancer, auto-immune diseases and AIDS. Adv Exp Med Biol 1991;294:425–35.
- Berger F, Ramirez-Hernandez MH, Ziegler M. The new life of a centenarian: signalling functions of NAD(P). Trends Biochem Sci 2004;29:111–8.
- Schlenk F, von Euler H. Cozymase. Naturwissenschaften 1936;24:794–5.
- Warburg O, Christian W. Pyridin, der wasserstoffubertragende Bestandteil von Garungsfermenten (Pyridin-Nucleotide). Biochem Z 1936;287:291–328.
- Magni G, Orsomando G, Raffelli N, Ruggieri S. Enzymology of mammalian NAD metabolism in health and disease. Front Biosci 2008;13:6135–54.
- Kornberg A. The participation of inorganic pyrophosphate in the reversible enzymatic synthesis of diphosphopyridine nucleotide. J Biol Chem 1948;176:1475–6.
- Rizzi M, Schindelin H. Structural biology of enzymes involved in NAD and molybdenum cofactor biosynthesis. Curr Opin Struct Biol 2002;12:709–20.
- Chambon P, Weill J, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesising nuclear enzyme. Biochem Biophys Res Commun 1963;11:39–43.
- Gholson RK. The pyridine nucleotide cycle. Nature 1966;212:933–4.
- Rechsteiner M, Catanzarite V. The biosynthesis and turnover of nicotinamide adenine dinucleotide in enucleated culture cells. J Cell Physiol 1974;84:409–22.
- Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Structure and function of nicotinamide mononucleotide adenylyltransferase. Curr Med Chem 2004;11:873–85.
- Magni G, Amici A, Emanuelli M, Raffaelli N, Ruggieri S. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol 1999;73:135–82, xi.
- Khan JA, Forouhar F, Tao X, Tong L. Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery. Expert Opin Ther Targets 2007;11:695–705.
- Houtkooper R, Canto C, Wanders R, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 2010;31:194–223.
- Braidy N, Guillemin G, Grant R. Promotion of cellular NAD+ anabolism: therapeutic potential for oxidative stress in ageing and Alzheimer's disease. Neurotox Res 2008;13:173–84.
- Mackay GM, Forrest CM, Stoy N, Christofides J, Egerton M, Stone TW, et al. Tryptophan metabolism and oxidative stress in patients with chronic brain injury. Eur J Neurol 2006;13:30–42.
- Ruddick JP, Evans AK, Nutt DJ, Lightman SL, Rook GA, Lowry CA. Tryptophan metabolism in the central nervous system: medical implications. Expert Rev Mol Med 2006;8:1–27.
- Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, et al. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur J Immunol 2001;31:2313–8.
- Guillemin GJ, Smith DG, Williams K, Smythe GA, Dormont D, Brew BJ. β amyloid peptide 1–42 induces human macrophages to produce the neurotoxin quinolinic acid. J Neuroimmunol 2001;118:112, A:336.
- Boasso A, Herbeuval JP, Hardy AW, Anderson SA, Dolan MJ, Fuchs D, et al. HIV-1 inhibits CD4+ T cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood 2006;109:3351–9.
- Frumento G, Piazza T, Di Carlo E, Ferrini S. Targeting tumor-related immunosuppression for cancer immunotherapy. Endocr Metab Immune Disord Drug Targets 2006;6:233–7.
- De Castro F, Price J, Brown R. Reduced triphophopyridine-nucleotide requirement for the enzymatic formation of 3-hydroxykynurenine from L-kynurenine. J Am Chem Soc 1956;78:2904–5.
- Mason M. The kynurenine transaminase of rat kidney. J Biol Chem 1954;211:839–44.
- Wiss O, Fuchs H. Uber den Abbau von Kynurenin, Oxykynurenin und verwandten Substanzen durch Rattenleberenzym. Experientia (Basel) 1950;6:472–3.
- Bokman AH, Schweigert BS. 3-Hydroxyanthranilic acid metabolism. IV. Spectrophotometric evidence for the formation of an intermediate. Arch Biochem Biophys 1951;33:270–6.
- Mehler A, May F. Studies with carboxyl-labelled 3-hydroxyanthranilic acid and picolinic acid in vivo and in vitro. J Biol Chem 1956;223:449–55.
- Cao H, Pietrak BL, Grubmeyer C. Quinolinate phosphoribosyltransferase: kinetic mechanism for a type II PRTase. Biochemistry 2002;41:3520–8.
- Connick JH, Stone TW. Quinolinic acid effects on amino acid release from the rat cerebral cortex in vitro and in vivo. Br J Pharmacol 1988;93:868–76.
- Bergeron R, Meyer TM, Coyle JT, Greene RW. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc Natl Acad Sci USA 1998;95:15730–4.
- Boegman RJ, el-Defrawy SR, Jhamandas K, Beninger RJ, Ludwin SK. Quinolinic acid neurotoxicity in the nucleus basalis antagonized by kynurenic acid. Neurobiol Aging 1985;6:331–6.
- Coggan S, Smythe G, Bilgin A, Grant R. Age and circadian influences on picolinic acid concentrations in human cerebrospinal fluid. J Neurochem 2009;108:1220–5.
- Guidetti P, Schwarcz R. 3-Hydroxykynurenine and quinolinate: pathogenic synergism in early grade Huntington's disease? Adv Exp Med Biol 2003;527:137–45.
- Morita T, Saito K, Takemura M, Maekawa N, Fujigaki S, Fujii H, et al. 3-Hydroxyanthranilic acid, an L-tryptophan metabolite, induces apoptosis in monocyte-derived cells stimulated by interferon-gamma. Ann Clin Biochem 2001;38:242–51.
- Guillemin GJ, Brew BJ, Noonan CE, Knight TG, Smythe GA, Cullen KM. Mass spectrometric detection of quinolinic acid in microdissected Alzheimer's disease plaques. In: , Takai K(ed.) International Congress Series. 2007. Elsevier B.V., Amsterdam. p. 404–8.
- Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer's disease hippocampus. Neuropathol Appl Neurobiol 2005;31:395–404.
- Guillemin GJ, Brew BJ. Implications of the kynurenine pathway and quinolinic acid in Alzheimer's disease. Redox Rep 2002;7:199–206.
- Guillemin GJ, Brew BJ. Chronic HIV infection leads to an Alzheimer's disease like illness. Involvement of the kynurenine pathway. In: , Takai K(ed.) International Congress Series. 2007. Elsevier B.V., Amsterdam. p. 324–34.
- Guillemin GJ, Kerr SJ, Brew BJ. Involvement of quinolinic acid in AIDS dementia complex. Neurotox Res 2005;7:103–23.
- Clark CJ, Mackay GM, Smythe GA, Bustamante S, Stone TW, Phillips RS. Prolonged survival of a murine model of cerebral malaria by kynurenine pathway inhibition. Infect Immun 2005;73:5249–51.
- Guillemin G, Meininger V, Brew B. Implications for the kynurenine pathway and quinolinic acid in amyotrophic lateral sclerosis. Neurodegener Dis 2006;2:166–76.
- Chiarugi A, Cozzi A, Ballerini C, Massacesi L, Moroni F. Kynurenine 3-mono-oxygenase activity and neurotoxic kynurenine metabolites increase in the spinal cord of rats with experimental allergic encephalomyelitis. Neuroscience 2001;102:687–95.
- Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington's disease. J Neurosci 1991;11:1649–59.
- Beal MF, Matson WR, Storey E, Milbury P, Ryan EA, Ogawa T, et al. Kynurenic acid concentrations are reduced in Huntington's disease cerebral cortex. J Neurol Sci 1992;108:80–7.
- Chess AC, Simoni MK, Alling TE, Bucci DJ. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophrenia Bull 2007;33:797–804.
- Schwarcz R. The kynurenine pathway of tryptophan degradation as a drug target. Curr Opin Pharmacol 2004;4:12–7.
- Grant RS, Naif H, Thuruthyil SJ, Nasr N, Littlejohn T, Takikawa O, et al. Induction of indolamine 2,3-dioxygenase in primary human macrophages by human immunodeficiency virus type 1 is strain dependent. J Virol 2000;74:4110–5.
- Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol 2003;81:247–65.
- Takikawa O, Yoshida R, Kido R, Hayaishi O. Tryptophan degredation in mice initiated by indoleamine 2,3-dioxygenase. J Biol Chem 1986;261:3648–53.
- Hansen AM, Ball HJ, Mitchell AJ, Miu J, Takikawa O, Hunt NH. Increased expression of indoleamine 2,3-dioxygenase in murine malaria infection is predominantly localised to the vascular endothelium. Int J Parasitol 2004;34:1309–19.
- Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998;281:1191–3.
- Alexander A, Crawford M, Bertera S, Rudert W, Takikawa O, Robbin P, et al. Indoleamine 2,3-dioxygenase expression in transplanted NOD Islets prolongs graft surviva; after adoptive transfer of diabetogenic splenocytes. Diabetes 2002;51:356–65.
- Hayashi T, Beck L, Rossetto C, Gong X, Takikawa O, Takabayashi K, et al. Inhibition of experimental asthma by indoleamine 2,3-dioxygenase. J Clin Invest 2004;114:270–9.
- Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003;9:1269–74.
- Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med 2004;10:15–8.
- Grant RS, Passey R, Matanovic G, Smythe G, Kapoor V. Evidence for increased de novo synthesis of NAD in immune-activated RAW264.7 macrophages: a self-protective mechanism? Arch Biochem Biophys 1999;372:1–7.
- Grant RS, Naif H, Espinosa M, Kapoor V. IDO induction in IFN-gamma activated astroglia: a role in improving cell viability during oxidative stress. Redox Rep 2000;5:101–4.
- Grant R, Kapoor V. Inhibition of indoleamine 2,3-dioxygenase activity in IFN-gamma stimulated astroglioma cells decreases intracellular NAD levels. Biochem Pharmacol 2003;66:1033–6.
- Iqbal J, Zaidi M. TNF regulates cellular NAD+ metabolism in primary macrophages. Biochem Biophys Res Commun 2006;342:1312–8.
- Rongvaux A, Andris F, Van Gool F, Leo O. Reconstructing eukaryotic NAD metabolism. Bioessays 2003;25:683–90.
- Khan JA, Tao X, Tong L. Molecular basis for inhibition of human NMPRTase, a novel target for anticancer agents. Nat Struct Mol Biol 2006;13:582–8.
- Gross J, Rajavel M, Grubmeyer C. Kinetic mechanism of nicotinic acid phosphoribosyltransferase: implications for energy coupling. Biochem 1998;37:4189–99.
- Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Priess-Handler independent route to NAD+ in fungi and humans. Cell 2004;117:495–502.
- Grifantini M. Tiazofurine ICN Pharmaceuticals. Curr Opin Invest Drugs 2000;1:257–62.
- Klaidman L, Morales M, Kem S, Yang J, Chang M, Adams J Nicotinamide offers multiple protective mechanisms in stroke as a precursor for NAD+, as a PARP inhibitor and by partial restoration of mitochondrial function. Pharmacology 2003;69:150–7.
- Wang B, Liao W, Chang C, Wang S. Facilitation of glutamate release by nicotine involves the activation of Ca2+/calmodulin signaling pathway in rat prefrontal cortex nerve terminals. Synapse 2006;59:491–501.
- Belenky P, Racette F, Bogan KL, McClure J, Smith J, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 2005;129:473–84.
- Burkle A, Beneke S, Muiras ML. Poly(ADP-ribosyl)ation and aging. Exp Gerontol 2004;39:1599–1601.
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 2004;3:205–214.
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956;11:298–300.
- Bechman K, Ames B. The free radical theory of aging matures. Physiol Rev 1998;78:547–81.
- Shi Y, Buffenstein R, Pulliam D, Van R. Comparative studies of oxidative stress and mitochondrial function in aging. Integr Comp Biol 2010;50:869–79.
- Oliveira M, Schoffen J. Oxidative stress action in cellular aging. Braz Arch Biol Tech 2010;53:1333–42.
- Buffenstein R, Edrey Y, Yang T, Mele J. The oxidative stress theory of aging:embattled or invincible? Insights from non-traditional model organisms. AGE 2008;30:99–109.
- Koc A, Gasch A, Rutherford J, Kim H, Gladyshev V. Methionine sulfoxide reductase regulation of yeast lifespan reveals reactive oxygen species-dependent and independent components of aging. Proc Natl Acad Sci 2004;101:7999–8004.
- Osiewacz H. Aging in fungi: role of mitochondria in Podospora anserina. Mech Ageing Dev 2002;123:755–64.
- Mates JM. Effects of antioxidant enzymes in molecular control of reactive oxygen species toxicology. Toxicology 2000;153:83–104.
- Martin R, Chan C, Veurink G, Laws S, Croft K, Dharmarajan A. β-Amyloid and oxidative stress in the pathogenesis of Alzheimer's disease. In: , Basu T, Temple N, Garg M(eds.) Antioxidants in human health and disease. Oxford: CABI Publishing; 1999.
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev 2002;82:47–95.
- Murphy M. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13.
- Halliwell B, Cutteridge J. Free radicals in biology and disease. Oxford: Oxford Science Pub; 1999.
- Pentland A. Active oxygen mechanisms of UV inflammation. Adv Exp Med Biol 1994;366:87–97.
- Koren H. Association between criteria air pollutants and asthma. Environ Health Perspect 1995;103:235–42.
- Naito Y, Yoshikawa M, Yoshida A, Kondo M. Role of oxygen radical and lipid peroxidation in indomethacin-induced gastric mucosal injury. Dig Dis Sci 1998;43: 30S–4S.
- Rav R, Mehrotra S, Shanker U, Babu G, Joshi P, Hanss R. Evaluation of UV-induced superoxide radical generation potential of some common antibiotics. Drug Chem Toxicol 2001;24:191–200.
- Obata T, Yamanaka Y, Kinemuchi H, Oreland L. Release of dopamine by perfusion with 1-methyl-4-phenylpyridinium ion (MPP(+)) into the striatum is associated with hydroxyl free radical production generation. Brain Res 2001;906:170–5.
- Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol 2001;30:620–50.
- Meral A, Tuncel P, Surmen-Gur E, Ozbek R, Ozturk E, Gunay U. Lipid peroxidation and antioxidant status in beta-thalassemia. Pediatr Hematol Oncol 2000;17:687–93.
- Davis K. Protein damage and degradation by oxygen radicals. J Biol Chem 1987;262:9895–901.
- Grune T, Reinheckei T, Davies K. Degradation of oxidised proteins in mammalian cells. FASEB J 1997;11:526–34.
- Ryberg H, Soderling AS, Davidsson P, Blennow K, Caidahl K, Persson LI. Cerebrospinal fluid levels of free 3-nitrotyrosine are not elevated in the majority of patients with amyotrophic lateral sclerosis or Alzheimer's disease. Neurochem Int 2004;45:57–62.
- Halliwell B. Oxygen radicals as key mediators in neurological disease: Fact or fiction? Ann Neurol 1992;32:S10–15.
- Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 2001;18:685–716.
- Iida T, Furuta A, Kawashima M, Nishida J, Nakabeppu Y, Iwaki T. Accumulation of 8-oxo-2′-deoxyguanosine and increased expression of hMTH1 protein in brain tumors. Neuro-oncol 2001;3:73–81.
- Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int J Oncol 2006;29:495–501.
- Tanaka T, Huang X, Halicka H, Zhao H, Traganos F, Albino A, et al. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A 2007;71:648–61.
- Miquel M. The rate of DNA damage and ageing. In: , Emerit I, Button C(eds.) Free radicals in ageing. Basel, Switzerland: Birhauser Verlag; 1992.
- Anderson RM, Bitterman KJ, Wood JG. Manipulation of nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J Biol Chem 2002;277:18881–90.
- Hassa P, Haenni S, Elser M, Hottiger M. Nuclear ADP-ribosylation reactions in mammalian cells: Where are we today and where are we going? Microbiol Mol Biol Rev 2006;70:789–829.
- Kletzein R, Harris P, Foellmi L. Glucose-6-phosphate dehydrogenase: a ‘housekeeping’ enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J 1994;8:174–81.
- Berg JM, Tymockzo JL, Stryer L. Biochemistry. 6th ed. New York: Freeman; 2007.
- Tzagoloff A. Mitochondria. New York: Plenum Press; 1982.
- McCormack J, Denton R. The role of of Ca2+ in the regulation of intramitochondrial energy production in heart. Biomed Biochim Acta 1987;46:S487–92.
- Marzulli D, La Piana G, Fransvea E, Lofrumento N. Modulation of cytochrome c-mediated extramitochondrial NADH oxidation by contact site density. Biochem Biophys Res Commun 1999;259:325–30.
- La Piana G, Fransvea E, Marzulli D, Lofrumento N. Mitochondrial membrane potential supported by exogenous cytochrome c oxidation mimics the early stages of apoptosis. Biochem Biophys Res Commun 1998;246:556–61.
- Bouchard V, Rouleau M, Poirier GG. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol 2003;31:446–54.
- Meyer R, Meyer-Ficca M, Jacobsen E, Jacobsen M. Enzymes in poly(ADP-Ribose) metabolism. In: , Burkle A(ed.) Poly(ADP-Ribosyl)ation. New York: Springer-Landes Bioscience; 2006.
- Grant RS, Kapoor V. Murine glial cells regenerate NAD, after peroxide-induced depletion, using either nicotinic acid, nicotinamide, or quinolinic acid as substrates. J Neurochem 1998;70:1759–63.
- Furukawa A, Tada-Oikawa S, Kawanishi S. H2O2 accelerates cellular senescence by accumulation of acetylated p53 via decrease in the function of SIRT1 by NAD+ depletion. Cell Physiol Biochem 2007;20:45–54.
- Ying W. NAD+ and NADH in brain functions, brain diseases and brain aging. Front Biosci 2007;12:1863–88.
- Wielckens K, Schmidt A, George E, Bredehorst R, Hilz H. DNA fragmentation and NAD depletion. Their relation to the turnover of endogenous mono(ADP-ribosyl) and poly(ADP-ribosyl) proteins. J Biol Chem 1982;257:12872–77.
- de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Nat Acad Sci USA 1997;94:7303–7.
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly (ADP-ribose) synthetase in neurotoxicity. Science 1994;263:687–9.
- di Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD+ metabolism. FEBS Lett 2001;492:4–8.
- Wang H, Schimoji M, Yu SW, Dawson TM, Dawson VL. Apoptosis inducing factor and PARP mediated injury in the MPTP mouse model of Parkinson's disease. Ann NY Acad Sci 2003;991:132–9.
- Love S, Barber R, Wilcock GK. Increased poly(ADP-ribosyl)ation of nuclear proteins in Azheimer's Disease. Brain 1999;122:247–53.
- Beneke S, Diefenbach J, Burkle A. Poly(ADP-ribosyl)ation inhibitors: promising drug candidates for a wide variety of pathophysiologic conditions. Int J Cancer 2004;111:813–8.
- Burkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+. Febs J 2005;272:4576–89.
- Grube K, Burkle A. Poly(ADP-ribose) polymerase activity in mononuclear cell lines of 13 mammalian species correlates with species specific lifespan. Proc Nat Acad Sci USA 1992;89:11759–63.
- Milne J, Denu JM. The Sirtuin family: therapeutic targets to treat diseases of aging. Curr Pharm Des 2008;12:11–7.
- Anastasiou D, Krek W. SIRT1: linking adaptive cellular responses to aging-associated changes in organismal physiology. Physiology (Bethesda) 2006;21:404–10.
- Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, et al. SIRT1 trangenic mice show phenotypes resembling calorie restriction. Aging Cell 2007;6:759–67.
- Bordone L, Motta MC, Picard F. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol 2006;4:e31.
- Michishita E, Park J, Burneskis J, Barrett J, Horikawa I. Evolutionary conserved and nonconserved cellular localisations and functions of human SIRT proteins. Mol Biol Chem 2005;16:4623–35.
- Jing E, Gesta S, Kahn C. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab 2007;6:105–14.
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005;434:113–8.
- Canto C, Gerhart-Hines Z, Feige J, Lagouge M, Noriega L, Milne J, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009;458:1056–60.
- Fuclo M, Cen Y, Zhao P, Hoffman E, McBurney M, Sauve A, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell 2008;14:661–73.
- Fiege J, Auwerx J. Transcriptional targets of sirtuins in the coordination of mammalian physiology. Curr Opin Cell Biol 2008;20:303–9.
- Porcu M, Chiarugi A. The emerging therapeutic potential of sirtuin-interacting drugs: from cell death to lifespan extension. Trends Pharmacol Sci 2005;26:94–103.
- Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, Kamitani H, et al. Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem Biophys Res Commun 2003;309:558–66.
- Inoue T, Hiratsuka M, Osaki M, Yamada H, Kishimoto I, Yamaguchi S, et al. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007;26:945–57.
- Li W, Zhang B, Tang J, Cao Q, Wu Y, Wu C, et al. Sirtuin2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating α-tubulin. J Neurosci 2007;27:2606–16.
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem 2004;73:417–35.
- Pearson K, Baur J, Lewis K, Peshkin L, Price N, Labinskyy N, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending lifespan. Cell Metab 2008;8:157–68.
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. Embo J 2007;26:3169–79.
- Sinclair D. Toward a unified theory of calorie restriction and longevity regulation. Mech Ageing Dev 2005;126:987–1002.
- Sablina A, Budanov A, Iilinskaya J, Agapova L, Kravchenko E, Chumakov M. The antioxidant function of the p53 tumour suppressor. Nat Med 2005;11:1306–13.
- Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol 2003;15:241–6.
- Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem 2005;280:13560–67.
- Ahn B, Kim H, Song S, Lee I, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. PNAS 2008;105:14447–52.
- Anderson RM, Bitterman KJ, Wood JG. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 2003;423:181–5.
- Haigis M, Mostoslavsky R, Haigis K, Fahie K, Christodoulou D, Murphy A, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells. Cell 2006;126:941–54.
- Nakagawa T, Lomb D, Haigis M, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 2009;137:560–70.
- Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem 2005;280:21313–20.
- Mostoslavsky R, Chua K, Lombard D, Pang W, Fischer M, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006;124:315–29.
- Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sirt2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev 2006;20:1075–80.
- Grummt I, Pikaard C. Epigenetic silencing of RNA polymerase I transcription. Nat Rev Mol Cell Biol 2003;4:641–9.
- Vakhrusheva O, Braeuer D, Liu Z, Braun T, Bober E. Sirt7-dependent inhibition of cell growth and proliferation might be instrumental to mediate tissue integrity during aging. J Physiol Pharmacol 2008;59:201–12.
- Zhang J. Are poly(ADP-ribosyl)ation by PARP-1 and deacetylation by Sir2 linked? Bioessays 2003;25:808–14.
- de Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD+ metabolism. FEBS Lett 2001;492:4–8.
- Douce R, Neuburger M. The uniqueness of plant mitochondria. Ann Rev Plant Physiol Plant Mol Biol 1989;40:371–414.
- Mathew C, Van Holde K, Ahern K. Biochemistry. 3rd ed. Boston, MA: Addison-Wesley; 2000.
- Sanni LA, Rae C, Maitland A, Stocker R, Hunt N. Is ischemia involved in the pathogenesis of cerebral malaria? Am J Pathol 2001;159:1105–12.
- Gaikwad A, Long DI, Stringer J, Jaiswal A. In vivo role of NAD(P)H: quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J Biol Chem 2001;276:22559–64.
- Ramasamy R, Trueblood N, Schaefer S. Metabolic effects of aldose reductase inhibition during low-flow ischemia and reperfusion. Am J Physiol 1998;275:H195–203.
- Mongan P, Capacchione J, West S, Karaian J, Dubois D, Keneally R, et al. Pyruvate improves redox status and decreases indicators of hepatic apoptosis during hemorrhagic shock in swine. Am J Physiol Heart Circ Physiol 2002;384:143–53.
- MacDonald M, Marshall L. Mouse lacking NAD+ linked glycerol phosphate dehydrogenase has normal pancreatic beta cell function but abnormal metabolite pattern in skeletal muscle. Arch Biochem Biophys 2000;384:143–53.
- Schweiger M, Hennig K, Lerner F, Niere M, Hirsch-Kauffmann M, Specht T, et al. Characterization of recombinant human nicotinamide mononucleotide adenylyl transferase (NMNAT), a nuclear enzyme essential for NAD synthesis. FEBS Lett 2001;492:95–100.
- Berger F, Lau C, Ziegler M. Regulation of poly(ADP-ribose) polymerase 1 activity by the phosphorylation state of the nuclear NAD biosynthetic enzyme NMN adenylyl transferase 1. Proc Natl Acad Sci USA 2007;104:3765–70.
- Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri A. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci 1999;61:19–34.
- Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, Magni G. Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem Biophys Res Commun 2002;297:835–40.
- Ziegler M. NAD: Metabolism and Regulatory Functions. In: , Burkle A(ed.) Poly(ADP-ribosyl)ation. Georgetown, TX: Landes Bioscience; 2006.
- Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci 2007;32:12–9.
- Bedalov A, Simon DA. NAD to the rescue. Sci Mag 2004;305:954–8.
- Raff MC, Whitemore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science 2002;296:868–71.
- Arraki T, Sasaki A, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004;305:1010–3.
- Lin M, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–95.
- Kauppinen TM, Suh SW, Genain C, Swanson RA. Poly(ADP-ribose) polymerase-1 activation in a primate model of multiple sclerosis. J Neurosci Res 2005;81:190–8.
- Cosi C, Colpaert F, Koek W, Degryse A, Marien M. Poly (ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res 1996;729:264–9.
- Stone TW, Behan WM, Jones PA, Darlington LG, Smith RA. The role of kynurenines in the production of neuronal death, and the neuroprotective effect of purines. J Alzheimers Dis 2001;3:355–66.
- Stone TW. Kynurenines in the CNS: from endogenous obscurity to therapeutic importance. Prog Neurobiol 2001;64:185–218.
- Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov 2002;1:609–20.
- Ying W, Sevigny MB, Chen Y, Swanson RA. Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc Natl Acad Sci USA 2001;98:12227–32.
- Berger SJ, Sudar DC, Berger NA. Metabolic consequences of DNA damage: DNA damage induces alterations in glucose metabolism by activation of Poly(ADP-ribose) polymerase. Biochem Biophys Res Commun 1986;134:227–32.
- Braidy N, Guillemin G, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism, oxidative stress and Sirt1 activity in Wistar Rats. PLOSONE 2011;6:e19194.
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol 2005;6:298–305.
- Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, et al. A novel VTNR enhancer within the SIRT3 gene, a human homologue of SIR2 is associated with survival at oldest ages. Genomics 2005;85:258–63.
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006;127:1109–22.
- van der Veer E, Ho A, O'Neil C, Barbosa N, Scott R, Cregan S, et al. Extension of human lifespan by nicotinamide phosphoribosyltransferase. Am Soc Biochem Mol Biol. 2007;282:10841–5.
- von Kobbe C, Harrigan J, Schreiber V, Stiegler P, Piotrowski J, Dawut L, et al. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res 2004;32:4003–14.
- Gille A, Bodor E, Ahmad K, Offermanns S. Nicotinic acid: pharmacological effects and mechanisms of action. Annu Rev Pharmacol Toxicol 2008;48:79–106.
- Penberthy WT, Tsunoda I. The importance of NAD in multiple sclerosis. Curr Pharm Des 2009;15:64–99.
- Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1 mediated cell death in astrocytes required NAD+ depletion and mitochondrial permeability transition. J Biol Chem 2004;279:18895–902.
- Pillai JB, Isbatan A, Imai SI, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sirt2 deacetylase activity. J Biol Chem 2005;280:43121–30.
- Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem 2005;280:17187–95.
- Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci USA 2007;104:7217–22.
- Braidy N, Grant R, Adams S, Guillemin G. Neuroprotective effects of naturally occurring polyphenols on quinolinic-acid induced excitotoxicity in human neurons. FEBS J 2010;277:368–82.
- Denu JM. Vitamins and aging: pathways to NAD+ synthesis. Cell 2007;129:453–4.
- Chikenji T, Asai T, Tatibana M. Protein-diet-induced elevation of 5-phosphoribosyl 1-disphosphate concentrations in mouse liver associated with increased syntheses of various nucleotides and the possible involvement of glucagon. Biochim Biophys Acta 1984;802:274–81.
- Adlebiassette H, Bell J, Creange A, Sazdovitch V, Authier F, Gray F, et al. DNA breaks detected by in situ end-labelling in dorsal root ganglia of patients with AIDS. Neuropathol Appl Neurobiol 1998;24:373–80.
- Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the ‘magnificent seven', function, metabolism and longevity. Ann Med 2007;39:335–45.
- Young G, Choleris E, Lund F, Kirkland J. Decreased cADPR and increased NAD+ in the CD38−/− mouse. Biochem Biophys Res Commun 2006;346:188–92.
- Aksoy P, White T, Thompson M, Chini E. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun 2006;345:1386–92.
- Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T. Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 2005;7:25–37.