

Abstract
Aims: To investigate the role of phosphorylation in SARS-CoV-2 infection, potential therapeutic targets and its harmful genetic sequences. Materials & Methods: Data mining techniques were employed to identify upregulated kinases responsible for proteomic changes induced by SARS-CoV-2. Spike and nucleocapsid proteins' sequences were analyzed using predictive tools, including SNAP2, MutPred2, PhD-SNP, SNPs&Go, MetaSNP, Predict-SNP and PolyPhen-2. Missense variants were identified using ensemble-based algorithms and homology/structure-based models like SIFT, PROVEAN, Predict-SNP and MutPred-2. Results: Eight missense variants were identified in viral sequences. Four damaging variants were found, with SNPs&Go and PolyPhen-2. Promising therapeutic candidates, including gilteritinib, pictilisib, sorafenib, RO5126766 and omipalisib, were identified. Conclusion: This research offers insights into SARS-CoV-2 pathogenicity, highlighting potential treatments and harmful variants in viral proteins.
Plain language summary
This study explores the process called phosphorylation, which involves adding phosphate groups to certain proteins, influences the way the SARS-CoV-2 virus causes disease. The virus manipulates host enzymes to help it spread and survive. Researchers used data analysis techniques to identify the proteins that play a role in this process, aiming to find potential targets for treatments. They analyzed genetic sequences of key virus proteins and used various tools to predict harmful mutations. The study found several promising compounds that could be used to target the virus. Further research and experiments are needed to confirm their effectiveness as COVID-19 treatments.
Tweetable abstract
This research explored the process called phosphorylation, which involves adding certain molecules to proteins, affects how the SARS-CoV-2 virus makes people sick. The virus uses our own cell's machinery to help it spread. Researchers used computer analysis to find out which proteins are involved in this process, hoping to find new ways to treat COVID-19. They studied the genetic code of important parts of the virus and used computer programs to predict if there were harmful changes in the code. They found some potential medicines that could be used to fight the virus and reduce its harm, but more research and testing are needed to be sure.
Graphical abstract
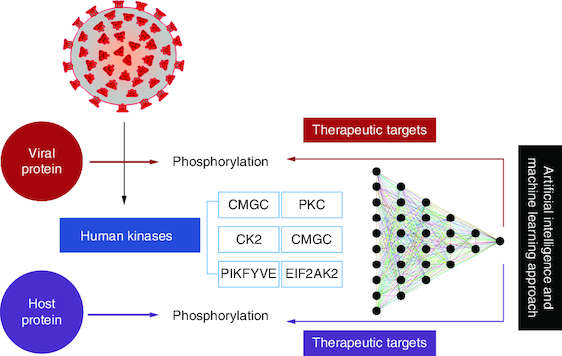
Phosphorylation, involving phosphate group addition to viral and host proteins, influences SARS-CoV-2 pathogenicity.
Study uses data mining to identify upregulated kinases responsible for proteomic changes induced by SARS-CoV-2.
Spike and nucleocapsid protein sequences of SARS-CoV-2 were analyzed using predictive and structure-based tools.
The tools categorized into groups such as protein structure analysis, consensus-based, homology-based, and supervised methods.
Eight missense variants identified in viral sequences, with one potentially damaging variant.
SNPs&Go and PolyPhen-2 predicted four distinct possibly damaging variants of SARS-COV-2.
Promising therapeutic candidates include gilteritinib, pictilisib, sorafenib, RO5126766 and omipalisib.
These compounds could mitigate SARS-CoV-2 virulence and pathogenicity by modulating phosphorylation pathways and addressing viral protein variants.
The current study is not but a comprehensive analysis crucial to understanding disruptions in spike and nucleocapsid protein's structure and function.
MutPred2 uses a grouping-based machine learning model trained on positive-unlabeled data.
MutPred2 demonstrates probabilities to interpret pathogenicity and mutation scores.
Enhances understanding of genetic variations' impact on protein structure and function.
SNAP2 employs a neural network-based classifier with 848 input nodes, 25 hidden nodes and two output nodes (neutral and deleterious).
The prediction analysis cover structural, functional, secondary structure, localization, transmembrane domains, catalytic functions, interactions, post-translational modifications, metal-binding and allosteric properties.
Drugs and kinase inhibitors mapped to specific phosphorylation sites.
Kinases and phosphatases are phospho-transferases, regulating cellular protein form and function.
Human genome contains approximately 156 phosphatases and 568 protein kinases, involved in biological regulation.
Dysregulation of kinases and phosphatases can have serious health consequences.
Mutations in these enzymes can lead to disorders, and inhibitors are used as pharmacological tools.
Protein Kinase Inhibitors (PKIs) and Phosphatase Inhibitors (PIs) are in experimental and clinical development, with various attributes and applications.
The use of multiple methods increases prediction robustness in the research.
Further investigation and experimental validation required to assess their efficacy as COVID-19 treatments.
Author contributions
Conceptualization, F Qamar, S Salman, Z Sharif; methodology, J Idrees, F Qamar; software, Z Sharif; validation, S Haider; formal analysis, J Idrees; investigation, S Haider; resources, A Wasim; data curation, A Wasim; writing – original draft preparation, F Qamar, writing – review and editing, S Haider; visualization, S Salman; supervision, S Haider, S Salman; project administration. All authors have read and agreed to the published version of the manuscript.
Financial disclosure
The authors have no financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations.
Acknowledgments
The authors acknowledge all the authors who have supported the data collection.