
Abstract
Purpose
Viruses are a common cause of exacerbations in chronic obstructive pulmonary disease (COPD). They activate toll-like receptors (TLRs) 3, 7, and 8, leading to a pro-inflammatory response. We have characterized the responses of TLR3 and TLR7/8 in lung tissue explants from COPD patients and control smokers.
Methods
We prepared lung whole tissue explants (WTEs) from patients undergoing surgery for confirmed or suspected lung cancer. In order to mimic the conditions of viral infection, we used poly(I:C) for TLR3 stimulation and R848 for TLR7/8 stimulation. These TLR ligands were used alone and in combination. The effects of tumor necrosis factor α (TNFα) neutralization and dexamethasone on TLR responses were examined. Inflammatory cytokine release was measured by enzyme-linked immunosorbent assay and gene expression by quantitative real-time polymerase chain reaction.
Results
WTEs from COPD patients released higher levels of pro-inflammatory cytokines compared with WTEs from smokers. Activation of multiple TLRs led to a greater than additive release of TNFα and CCL5. TNFα neutralization and dexamethasone treatment decreased cytokine release.
Conclusion
This WTE model shows an enhanced response of COPD compared with controls, suggesting an increased response to viral infection. There was amplification of innate immune responses with multiple TLR stimulation.
Keywords:
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by poorly reversible airflow limitation and airway inflammation that arise in response to the inhalation of noxious particles. COPD patients can suffer with exacerbations; these events are an acute increase in symptoms beyond normal day-to-day variation. The majority of exacerbations are due to viral or bacterial respiratory infections and result in increased levels of airway and systemic inflammation. COPD exacerbations are often treated with high doses of anti-inflammatory corticosteroids.Citation1
Viruses such as rhinovirus, adenovirus, and influenza cause COPD exacerbations.Citation2 The viral genome is recognized by pattern recognition receptors, including toll-like receptors (TLRs) such as TLR3, TLR7, and TLR8, that are expressed on the cell surface and in the endosomes of immune and structural cells. Stimulation of these TLRs activates transcription factors, such as nuclear factor-κB and interferon (IFN) regulatory factors, which cause increased secretion of pro-inflammatory cytokines and chemokines (such as tumor necrosis factor α [TNFα] and CCL5) as well as an antiviral response with increased secretion of type I and III IFNs.Citation3
The effects of TLR activation have traditionally been studied using synthetic ligands of these receptors, such as poly(I:C) that targets TLR3, using single-cell culture systems. However, whole tissue explants (WTEs) offer the opportunity to investigate TLR-induced responses in a complex system with multiple cell types. Previous studies have demonstrated that COPD lung tissue readily responds to TLR3 and TLR4 stimulation.Citation4,Citation5 Viruses can activate multiple TLRs; for example, rhinovirus is recognized by both TLR3 and TLR7.Citation6,Citation7 Previous studies using COPD lung tissue have not assessed simultaneous stimulation of multiple TLRs.
In this study, we characterized the response of lung WTE of COPD patients to TLR3 and TLR7/8 ligands, poly(I:C), and R848, respectively, as a model of viral infection. The primary aim was to compare the inflammatory response of samples from COPD patients with those from controls and to investigate the effect of simultaneous stimulation with TLR3 and TLR7/8 on the inflammatory response. The secondary aim was to investigate the effect of TNFα inhibition and corticosteroids on these TLR-induced responses.
Materials and methods
Patient information
Lung samples were obtained from 52 patients undergoing surgery for confirmed or suspected lung cancer. All the patients were current or ex-smokers; the complete demographic details are presented in . COPD patients (n=33) were diagnosed according to the Global Initiative for Chronic Obstructive Lung Disease guidelinesCitation1 and had forced expiratory volume in 1 second % predicted <80% and ratio of forced expiratory volume in 1 second to forced vital capacity <70%. Patients without COPD were classified as smokers with normal lung function (n=19). Ex-smokers were defined as individuals who had not smoked for at least 1 year. All the patients gave written informed consent, and the study was approved by the local research ethics committee (03/SM/396, NRES Committee North West – Greater Manchester South). The demographics of patient samples used in various experiments are summarized in Tables S1 and S2.
Table 1 Demographics of patients used in this study
Preparation of whole tissue explants
Human lung tissue was prepared as previously described.Citation4 Tissue that was distal from the tumor was used. Briefly, using two sterile scalpels, 1–5 g of tissue was cut into small (~1 mm3) fragments – WTEs. WTEs were rinsed with RPMI-1640 medium (Sigma-Aldrich, Dorset, UK) supplemented with 10% (v/v) fetal bovine serum (Invitrogen, Paisley, UK), 100 U penicillin/0.1 mg streptomycin (Sigma-Aldrich), and 2 mM l-glutamine (Invitrogen). WTEs (30±3 mg) were placed in 24-well plates with 800 μL supplemented RPMI medium per well and incubated at 37°C, 5% (v/v) CO2 overnight.
Tissue stimulation with TLR ligands
TLR3 (poly(I:C)) and TLR7/8 ligand (R848) stock solutions (Invivogen, San Diego, CA, USA) were diluted in supplemented RPMI-1640 medium to obtain final concentrations ranging from 0.01 to 1,000 μg/mL, depending on the experiment. Following overnight incubation of WTE, the medium was replaced with 800 μL of fresh supplemented RPMI medium, containing medium only, poly(I:C) only, R848 only, or poly(I:C) and R848 combined. Plates were then incubated at 37°C, 5% (v/v) CO2 for 1, 6, 24, or 48 hours. Supernatants were collected at a set time point and stored at −20°C, and tissue was stored in RNAlater (Invitrogen) at −80°C. All the conditions were performed in triplicate.
TNFα neutralization
Human TNFα Antibody (Clone #28401) and Mouse IgG1 Isotype (Clone #11711) were purchased from R&D Systems Europe (Abingdon, UK), and the stock solutions were prepared according to the manufacturers’ instructions. Following overnight incubation of WTE, the medium was replaced with 784 μL of fresh supplemented RPMI medium, containing medium only, neutralizing TNFα antibody only (1 μg/mL), or control immunoglobulin G (IgG) antibody only (1 μg/mL). Plates were then incubated at 37°C, 5% (v/v) CO2 for 1 hour. Then, 16 μL of medium only, poly(I:C) only, or R848 only was added to the wells, and the plates were incubated for further 24 hours. Supernatants were collected and stored at −20°C, and tissue was stored in RNAlater (Invitrogen) at −80°C. All the conditions were performed in triplicate.
Effect of dexamethasone
Dexamethasone was diluted in dimethyl sulfoxide (DMSO) (Sigma-Aldrich) to stock concentration of 10 mM. Following overnight incubation of WTE, the medium was replaced with 784 μL of fresh supplemented RPMI medium, containing DMSO only (controls) or dexamethasone only (final concentration 1 μM). Plates were then incubated at 37°C, 5% (v/v) CO2 for 1 hour. Then, 16 μL of supplemented RPMI medium containing poly(I:C) only, R848 only, or poly(I:C) and R848 was added to the wells, and the plates were incubated for further 24 hours. Supernatants were collected and stored at −20°C and tissue was stored in RNAlater (Invitrogen) at −80°C. All the conditions were performed in triplicate. Final DMSO concentration present in all conditions was 0.01% (v/v).
Enzyme-linked immunosorbent assay
TNFα, CCL5, and IL-6 protein levels in supernatants were measured by enzyme-linked immunosorbent assay (ELISA; R&D Systems Europe, Abingdon, UK) according to the manufacturer’s protocol. All data were adjusted for tissue weight and were presented as pg/mL/mg of tissue.
Quantitative real-time polymerase chain reaction
The levels of TNFα, CCL5, and IL-6 mRNA following TLR stimulation were measured using Taqman polymerase chain reaction (PCR) (see Supplementary materials for the detailed method).
Statistics
All the data were analyzed and plotted in GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA). Normality was assessed using the Kolmogorov–Smirnov test. All TNFα and IL-6 ELISA data presented were parametric, whereas CCL5 ELISA data and PCR data were nonparametric. In the time course experiments, cytokine release or relative gene expression in response to a TLR ligand was compared with time-matched unstimulated controls using paired t-tests (or Wilcoxon signed-rank test for nonparametric data). In the experiments with different concentrations of TLR ligand, cytokine release was analyzed by one-way analysis of variance (ANOVA) with Dunnett’s posttest. CCL5 was analyzed using Friedman test with Dunn’s posttest. Cytokine levels after simultaneous TLR3 and TLR7/8 stimulation were compared with the cytokine levels using a single TLR agonist using paired t-tests or Wilcoxon signed-rank test for CCL5. The combination effect was also compared with the predicted sum (where absolute levels of cytokines released by single ligands were added together) using paired t-tests or Wilcoxon signed-rank test. The effects of TNFα neutralization were analyzed using multiple repeated ANOVA with Dunnett’s posttest and Friedman with Dunn’s posttest for nonparametric data. The effect of dexamethasone on cytokine release was compared with time-matched control using paired t-tests (or Wilcoxon signed-rank test for CCL5 data).
Results
Time course of poly(I:C)- and R848-induced cytokine release and mRNA expression
Initial experiments were performed to establish the time course of poly(I:C)- and R848-induced inflammatory response in WTE from COPD patients (n=7) and smokers (n=6). It was decided a priori to pool the data from both the groups for the primary aim of characterizing the time course of cytokine release () and mRNA production (). Poly(I:C) (100 μg/mL) and R848 (10 μg/mL) caused time-dependent increases in TNFα, CCL5, and IL-6 release. In general, cytokine secretion was relatively low at 1 and 6 hours with higher levels at 24 and 48 hours; the only exception was high levels of TNFα release at 6 hours after R848 stimulation. Poly(I:C) induced higher levels of CCL5 and IL-6 compared with R848 reaching statistical significance at 24 and 48 hours, respectively. However, R848 was much more potent at TNFα induction throughout the time course (Figure S1 shows a direct comparison of R848 and poly(I:C)). On the basis of these results, the time point of 24 hours for maximal cytokine release was used for the subsequent experiments.
Figure 1 Time course of cytokine release from poly(I:C)- and R848-stimulated lung tissue.
Notes: The release of TNFα (A and D), CCL5 (B and E), and IL-6 (C and F) from poly(I:C)- (100 μg/mL) (A–C) and R848-stimulated (10 μg/mL) (D–F) whole tissue explants from smokers and COPD patients (pooled, n=13) were measured after 1, 6, 24, and 48 hours. Data presented as mean with SEM (TNFα and IL-6) or median with range (CCL5). *, **, ***Refer to significantly above time-matched unstimulated control (P<0.05, 0.01, 0.001, respectively). Results were obtained using paired t-tests (TNFα and IL-6) and Wilcoxon matched-pairs test (CCL5).
Abbreviations: TNFα, tumor necrosis factor α; IL-6, interleukin 6; COPD, chronic obstructive pulmonary disease; SEM, standard error of the mean; h, hour(s).
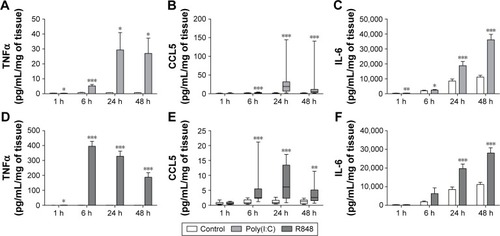
Figure 2 Time course of cytokine mRNA expression in poly(I:C)- and R848-stimulated lung tissue.
Abbreviations: TNFα, tumor necrosis factor α; IL-6, interleukin 6; COPD, chronic obstructive pulmonary disease; h, hour(s).
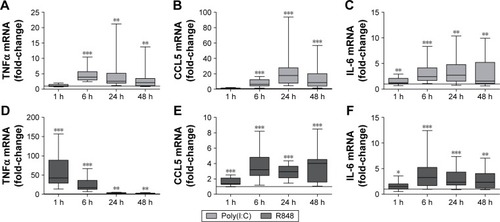
Changes in cytokine release were preceded or correlated with changes at the mRNA level. Thus, both poly(I:C) and R848 significantly upregulated cytokine mRNA expression from 6 to 48 hours after stimulation for all cytokines that were analyzed (). There was some evidence of increased mRNA expression at 1 hour, most notably for TNFα after R848 stimulation. Poly(I:C) caused significantly lower TNFα and higher CCL5 mRNA expression than R848 (Figure S2).
Because of the limited sample size, we were unable to observe any consistent differences between COPD patients (n=7) and smokers (n=6) for cytokine release (Figure S3) and mRNA production (Figure S4).
Poly(I:C) and R848 stimulation: comparison of COPD patients and smokers
The response of WTE from COPD patients (n=13) and smokers (n=13) was compared. Cytokine secretion was TLR ligand concentration-dependent with maximal effects at 100–1,000 μg/mL of poly(I:C) and 1–10 μg/mL of R848 (). There was evidence across the concentration response curve of significantly increased CCL5 release from COPD patients compared with smokers after both poly(I:C) and R848 stimulation. There was a similar pattern for TNFα secretion, although significant differences were observed at lower concentrations of TLR ligands. There were no differences for IL-6 release.
Figure 3 Concentration-dependent induction of cytokine release from poly(I:C)- and R848-stimulated lung tissue.
Abbreviations: TNFα, tumor necrosis factor α; IgG, immunoglobulin G; IL-6, interleukin 6; COPD, chronic obstructive pulmonary disease; SEM, standard error of the mean.
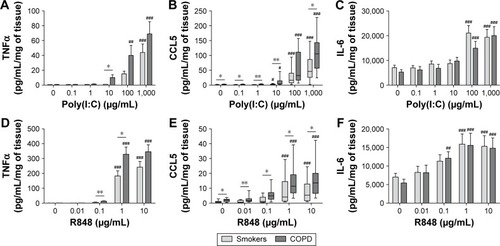
Current smoking caused lower TNFα release in the smokers group, but there was no effect in COPD patients (Figure S5). Current smoking had no effect on CCL5 or IL-6 release.
Simultaneous activation of TLR3 and TLR7/8
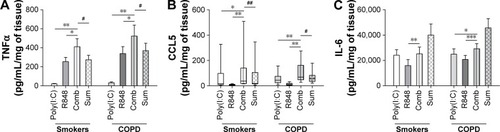
Poly(I:C) (100 μg/mL) and R848 (10 μg/mL) when combined caused TNFα and CCL5 secretion that was greater than when the ligand was used alone in WTE from COPD patients (n=12) and smokers (n=10) (). Furthermore, TNFα and CCL5 secretion was at a level that was greater than the sum of the levels induced by the ligands used alone (predicted fully additive effect). The release of IL-6 in response to poly(I:C) and R848 was greater than that when the TLR ligands were used alone but lower than the predicted fully additive effect induced by these ligands. Cytokine levels induced after the addition of poly(I:C) and R848 combined were similar in WTE from COPD patients and smokers.
Figure 4 Effect of simultaneous activation of TLR3 and TLR7/8 on pro-inflammatory cytokine release from lung tissue.
Abbreviations: TNFα, tumor necrosis factor α; IL-6, interleukin 6; COPD, chronic obstructive pulmonary disease; SEM, standard error of the mean; Comb, combination.
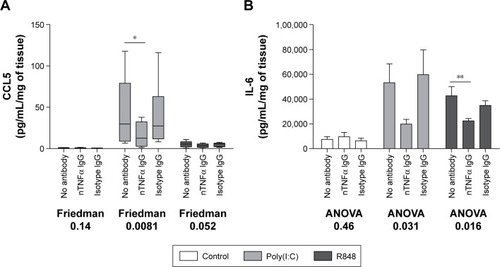
TNFα amplifies subsequent TLR3- and TLR7/8-induced pro-inflammatory response
A TNFα neutralizing antibody was preincubated with COPD WTE (n=6). TNFα neutralization caused a significant reduction in poly(I:C)-induced CCL5 and R848-induced IL-6 levels (). Poly(I:C)-induced IL-6 and R848-induced CCL5 levels were also reduced after TNFα neutralization, but these changes were not statistically significant. A control antibody had no effect on cytokine release.
Figure 5 Effect of TNFα neutralization on pro-inflammatory cytokine release from lung tissue.
Abbreviations: TNFα, tumor necrosis factor α; IgG, immunoglobulin G; IL-6, interleukin 6; COPD, chronic obstructive pulmonary disease; SEM, standard error of the mean; ANOVA, analysis of variance.
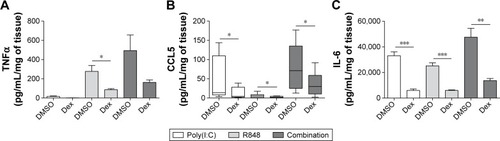
Corticosteroid inhibition of cytokines
WTE from COPD patients (n=6) were treated with dexamethasone (1 μM) prior to stimulation with poly(I:C) alone, R848 alone, or both TLR ligands simultaneously (). Dexamethasone significantly decreased cytokine release in the majority of conditions, ranging from 52% to 82% at 24 hours.
Figure 6 Effect of dexamethasone on pro-inflammatory cytokine release from TLR-stimulated lung tissue.
Abbreviations: TNFα, tumor necrosis factor α; IL-6, interleukin 6; COPD, chronic obstructive pulmonary disease; SEM, standard error of the mean; Dex, dexamethasone; DMSO, dimethyl sulfoxide.
Discussion
We have characterized the cytokine response of COPD WTE to synthetic viral TLR stimuli. Poly(I:C) and R848 both caused increased TNFα and CCL5 secretion in COPD patients compared with smokers. This increased inflammatory response could contribute to the pathophysiology of COPD exacerbations after viral infection. Combination of these TLR ligands produced greater responses than either ligand alone. TNFα had an important role in promoting the secretion of other cytokines, and we show that these responses are partially inhibited by corticosteroids.
Increased cytokine secretion has been reported in COPD patients compared with controls using TLR4-stimulated WTE.Citation4 We observed similar findings using different TLR ligands relevant to viral infection. Interestingly, differences in cytokine release between smokers and COPD patients were not observed when both the TLR agonists were used simultaneously. This might signify near maximal inflammation being achieved in this tissue model, making it difficult to observe differences between groups.
Viral infections are likely to trigger the activation of multiple pattern recognition receptors. The combination of poly(I:C) and R848 caused TNFα and CCL5 secretion that was greater than the predicted fully additive effect of combining the TLR ligands. This raises the possibility that this combination of TLR ligands caused a synergistic pro-inflammatory effect. However, IL-6 was not released in such a synergistic manner. This may be due to IL-6 levels being already high when stimulated by one ligand. Perhaps using lower ligand concentrations for this experiment would reveal a different pattern for IL-6, and it would be important to perform experiments using different TLR ligand concentrations in combination in order to confirm this possibility of synergism. Others have also examined multiple TLR activation in different models, eg, monocyte-derived dendritic cells and macrophages, and also have observed synergistic release of various cytokines (including TNFα and IL-6).Citation8–Citation13 However, synergy in cytokine release was not observed in monocyte-derived macrophages.Citation10
TLR3 and TLR7/8 use different molecular adaptors to activate downstream signaling pathways: TLR3 signals via TRIF, whereas TLR7 and TLR8 use MyD88.Citation14 Previous studies examining simultaneous activation of different TLRs have demonstrated that activation of both MyD88- and TRIF-dependent pathways results in synergistic cytokine release.Citation10,Citation15 The exact mechanisms are still to be elucidated, but the crosstalk between multiple pro-inflammatory pathways downstream of TLRs appears to play a role in this process.Citation10
Viral infections are a common cause of COPD exacerbations and are associated with increased airway inflammation in COPD patients.Citation16–Citation18 The additive, and possibly synergistic, cytokine production reported here are mechanisms that are likely to contribute to the excessive airway inflammation during viral infections, predisposing to other harmful pathophysiological effects such as tissue damage. On the other hand, the host immune response is required for the recruitment of cells responsible for pathogen clearance. A variety of mechanism are involved in viral clearance,Citation19 and our investigations focus more on immune pathways that could cause excessive inflammation.
There is evidence of attenuation of TLR-induced cytokine release in cells pretreated with cigarette smoke extract or cells derived from smokers.Citation20–Citation22 For example, Metcalfe et alCitation20 showed that acute cigarette smoke extract exposure decreased TLR-induced cytokine release from alveolar macrophages while Chen et alCitation22 observed attenuation in TLR2- and TLR4-induced cytokine release from alveolar macrophages from smokers compared with nonsmokers. We observed that the TLR7/8- and TLR3-induced TNFα response was attenuated by current smoking in the smokers group but that there was no effect in COPD patients. Furthermore, there was no effect of current smoking on CCL5 and IL-6. One should be cautious when interpreting this subanalysis with small sample sizes. Overall, we feel that current smoking had a minimal impact on the data presented.
TNFα is implicated in inflammatory responses in COPD.Citation23–Citation25 Multiple cell types are able to release and respond to TNFα, including epithelial cells and macrophages.Citation26–Citation28 We found that a neutralizing TNFα antibody attenuated TLR3- and TLR7/8-induced release of CCL5 and IL-6. Similar observations were made by Hackett et al using lung tissue exposed to lipopolysaccharide (LPS).Citation4 These data suggest that TNFα is required for maximal pro-inflammatory responses in the lung tissue. However, anti-TNFα treatment did not have any benefit in COPD patients.Citation29,Citation30 It is possible that TNFα plays a role as an amplifier of TLR signaling during infection in COPD patients, rather than having a significant role in the stable clinical state.
Corticosteroids are commonly used in COPD therapy, either as regular inhaled treatment or as short courses of oral treatment during exacerbations.Citation1 We showed that dexamethasone partially inhibited the release of all three pro-inflammatory cytokines measured. Similarly, other in vitro studies have shown that corticosteroids partially suppress pro-inflammatory cytokine release from different cell types, including alveolar macrophages, monocyte-derived macrophages, and primary bronchial epithelial cells.Citation26,Citation31–Citation33 Our observations suggest a partial anti-inflammatory effect by corticosteroids on COPD lung tissue during viral TLR stimulation.
Activation of TLR7 is being investigated as a therapeutic option for asthma, as there is evidence that this skews the immune response away from a Th2 profile and toward a Th1 profile, which may be useful for suppressing allergic inflammation.Citation34,Citation35 Our research was not focused on this question, but we show how TLR7 stimulation may affect COPD lung immune responses. It is possible that TLR antagonists may be beneficial in COPD patients to limit inflammation associated with smoking, as various TLRs are implicated in pro-inflammatory responses to cigarette smoke.Citation36–Citation40 However, since TLRs play a role in infection, using TLR antagonists during infection could lead to increased pathogen proliferation.Citation41 A balanced approach to TLR therapeutics is required to limit tissue-destructive inflammation without compromising pathogen clearance.Citation42
There were some practical limitations to our study. The vast majority of patients undergoing cancer surgery were current or ex-smokers, so it was difficult to recruit lifelong nonsmokers as a control group. Additionally, it is possible that the presence of cancer may influence the immune response, even though we used tumor-free tissue.
In summary, we have demonstrated that COPD WTE showed a greater pro-inflammatory response to TLR3 or TLR7/8 activation than control smokers. Interestingly, simultaneous stimulation of these TLRs caused innate immune responses that were at least additive. The model described here can be further used to model and dissect the COPD immune response during viral infection, for the purpose of identifying molecular pathways suitable for pharmacological intervention.
Acknowledgments
This study was funded by the Biotechnology and Biological Sciences Research Council and Pfizer. This report is an independent research supported by the National Institute for Health Research Respiratory and Allergy Clinical Research Facility at University Hospital of South Manchester NHS Foundation Trust. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
Disclosure
AP, SL, SH, and ML report no conflicts of interest in this work. DS has received sponsorship to attend international meetings, honoraria for lecturing and attending advisory boards, and research grants from various pharmaceutical companies, including Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, CIPLA, Forest, Genetech, GlaxoSmithKline, Merck, Novartis, Pfizer, and Takeda.
References
- Global Initiative for Chronic Obstructive Pulmonary DiseaseGlobal Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease2015 Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2015_Sept2.pdfAccessed October 10, 2015
- WedzichaJARole of viruses in exacerbations of chronic obstructive pulmonary diseaseProc Am Thorac Soc20041211512016113423
- O’NeillLAJGolenbockDBowieAGThe history of toll-like receptors – redefining innate immunityNat Rev Immunol201313645346023681101
- HackettT-LHollowayRHolgateSWarnerJDynamics of pro-inflammatory and anti-inflammatory cytokine release during acute inflammation in chronic obstructive pulmonary disease: an ex vivo studyRespir Res200894718510721
- CooperPRLambRDayNDTLR3 activation stimulates cytokine secretion without altering agonist-induced human small airway contraction or relaxationAm J Physiol Lung Cell Mol Physiol20092973L530L53719542247
- HewsonCAJardineAEdwardsMRLaza-StancaVJohnstonSLToll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cellsJ Virol20057919122731227916160153
- TriantafilouKVakakisERicherEAJEvansGLVilliersJPTriantafilouMHuman rhinovirus recognition in non-immune cells is mediated by toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune responseVirulence201121222921224721
- BohnenkampHRPapazisisKTBurchellJMTaylor-PapadimitriouJSynergism of toll-like receptor-induced interleukin-12p70 secretion by monocyte-derived dendritic cells is mediated through p38 MAPK and lowers the threshold of T-helper cell type I responsesCell Immunol20072472728417927969
- MäkeläSMOsterlundPJulkunenITLR ligands induce synergistic interferon-beta and interferon-lambda1 gene expression in human monocyte-derived dendritic cellsMol Immunol201148450551521040977
- MäkeläSMStrengellMPietiläTEÖsterlundPJulkunenIMultiple signaling pathways contribute to synergistic TLR ligand-dependent cytokine gene expression in human monocyte-derived macrophages and dendritic cellsJ Leukoc Biol200985466467219164128
- MorrisGEParkerLCWardJRCooperative molecular and cellular networks regulate toll-like receptor-dependent inflammatory responsesFASEB J200620122153215516935934
- RoelofsMFJoostenLAAbdollahi-RoodsazSThe expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cellsArthritis Rheum20055282313232216052591
- Suet Ting TanRLinBLiuQThe synergy in cytokine production through MyD88-TRIF pathways is co-ordinated with ERK phosphorylation in macrophagesImmunol Cell Biol201391537738723567895
- YamamotoMSatoSHemmiHRole of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathwayScience200330164064312855817
- OuyangXNegishiHTakedaRFujitaYTaniguchiTHondaKCooperation between MyD88 and TRIF pathways in TLR synergy via IRF5 activationBiochem Biophys Res Commun200735441045105117275788
- DaiMYQiaoJPXuYHFeiGHRespiratory infectious phenotypes in acute exacerbation of COPD: an aid to length of stay and COPD Assessment TestInt J Chron Obstruct Pulmon Dis2015102257226326527871
- WedzichaJAMechanisms of chronic obstructive pulmonary disease exacerbationsAnn Am Thorac Soc201512Suppl 2S157S159
- HewittRFarneHRitchieALukeEJohnstonSLMalliaPThe role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthmaTher Adv Respir Dis2015 Epub 2015 Nov 26
- YooJ-KKimTSHuffordMMBracialeTJViral infection of the lung: host response and sequelaeJ Allergy Clin Immunol201313261263127623915713
- MetcalfeHJLeaSHughesDKhalafRAbbott-BannerKSinghDEffects of cigarette smoke on toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophagesClin Exp Immunol2014176346147224528166
- TodtJCFreemanCMBrownJPSmoking decreases the response of human lung macrophages to double-stranded RNA by reducing TLR3 expressionRespir Res2013143323497334
- ChenHCowanMJHasdayJDVogelSNMedvedevAETobacco smoking inhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-κB in alveolar macrophages stimulated with TLR2 and TLR4 agonistsJ Immunol200717996097610617947684
- PedrozaMSchneiderDJKarmouty-QuintanaHInterleukin-6 contributes to inflammation and remodeling in a model of adenosine mediated lung injuryPLoS One201167e2266721799929
- VernooyJHKüçükaycanMJacobsJALocal and systemic inflammation in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200216691218122412403691
- YasudaNGotohKMinatoguchiSAn increase of soluble Fas, an inhibitor of apoptosis, associated with progression of COPDRespir Med19989289939999893764
- ArmstrongJSargentCSinghDGlucocorticoid sensitivity of lipopolysaccharide-stimulated chronic obstructive pulmonary disease alveolar macrophagesClin Exp Immunol20091581748319737233
- SchallTJLewisMKollerKJMolecular cloning and expression of a receptor for human tumor necrosis factorCell19906123613702158863
- WarkPABBucchieriFJohnstonSLIFN-γ-induced protein 10 is a novel biomarker of rhinovirus-induced asthma exacerbationsJ Allergy Clin Immunol2007120358659317628646
- van der VaartHKoëterGHPostmaDSKauffmanHFten HackenNHTFirst study of infliximab treatment in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2005172446546915937294
- RennardSIFogartyCKelsenSThe safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2007175992693417290043
- LeaSPlumbJMetcalfeHThe effect of peroxisome proliferator-activated receptor-γ ligands on in vitro and in vivo models of COPDEur Respir J201443240942023794466
- KentLMSmythLJCPlumbJInhibition of lipopolysaccharide-stimulated chronic obstructive pulmonary disease macrophage inflammatory gene expression by dexamethasone and the p38 mitogen-activated protein kinase inhibitor N-cyano-N′-(2-^#x0003C;8-(2,6-difluorophenyl)-4-(4-fluoro-2-methylphenyl)-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidin-2-yl]amino}ethyl)guanidine (SB706504)J Pharmacol Exp Ther2009328245846819004925
- HeijinkIvan OosterhoutAKliphuisNOxidant-induced corticosteroid unresponsiveness in human bronchial epithelial cellsThorax201369151323980116
- ZuoLLucasKFortunaCAChuangCCBestTMMolecular regulation of toll-like receptors in asthma and COPDFront Physiol2015631226617525
- DrakeMGKaufmanEHFryerADJacobyDBThe therapeutic potential of toll-like receptor 7 stimulation in asthmaInflamm Allergy Drug Targets201211648449123078048
- PaceEFerraroMSienaLCigarette smoke increases toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cellsImmunology2008124340141118217953
- KarimiKSarirHMortazEToll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophagesRespir Res200676616620395
- DroemannDGoldmannTTiedjeTZabelPDalhoffKSchaafBToll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patientsRespir Res200566816004610
- MortazEAdcockIMItoKKraneveldADNijkampFPFolkertsGCigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptorEur Respir J20103651143115419840968
- WorthamBWEppertBLFluryJLMorgado GarciaSBorchersMTTLR and NKG2D signaling pathways mediate CS-induced pulmonary pathologiesPLoS One2013810e7873524130907
- WielandCWFlorquinSMarisNAThe MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable Haemophilus influenzae from the mouse lungJ Immunol200517596042604916237099
- BezemerGFSagarSvan BergenhenegouwenJDual role of toll-like receptors in asthma and chronic obstructive pulmonary diseasePharmacol Rev201264233735822407613