

Abstract
A novel virtual screening approach is implemented herein, which is a further improvement of our previously published “target-bound pharmacophore modeling approach”. The generated pharmacophore library is based only on highly contributing amino acid residues, instead of arbitrary pharmacophores, which are most commonly used in the conventional approaches in literature. Highly contributing amino acid residues were distinguished based on free binding energy contributions obtained from calculation from molecular dynamic (MD) simulations. To the best of our knowledge; this is the first attempt in the literature using such an approach; previous approaches have relied on the docking score to generate energy-based pharmacophore models. However, docking scores are reportedly unreliable. Thus, we present a model for a per-residue energy decomposition, constructed from MD simulation ensembles generating a more trustworthy pharmacophore model, which can be applied in drug discovery workflow. This work is aimed at introducing a more rational approach to the field of drug design, rather than comparing the validity of this approach against those previously reported. We recommend additional computational and experimental work to further validate this approach. This approach was used to screen for potential reverse transcriptase inhibitors using the pharmacophoric features of compound GSK952. The complex was subjected to docking, thereafter, MD simulation confirmed the stability of the system. Experimentally determined inhibitors with known HIV-reverse transcriptase inhibitory activity were used to validate the protocol. Two potential hits (ZINC46849657 and ZINC54359621) showed a significant potential with regard to free binding energy. Reported results obtained from this work confirm that this new approach is favorable in the future of the drug design industry.
Supplementry materials
Figure S1 Two key binding interactions exist between GSK952 and the backbone NH and C=O groups of Lys103 of HIV-1 RT.
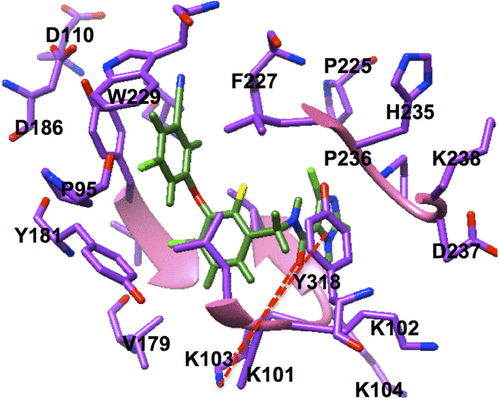
Figure S2 PRED-based pharmacophore model.
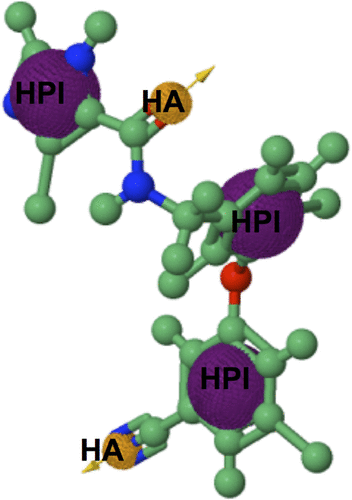
Figure S3 MD simulation results of ZINC54359621 and ZINC46849657.
Abbreviations: MD, molecular dynamic; RMSD, root-mean-square deviation; RMSF, root-mean-square fluctuation; Rg, radius of gyration.
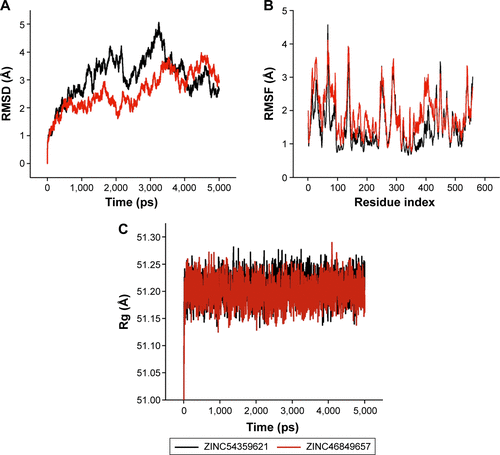
Table S1 PRED of highly interacting residues
Acknowledgments
Ethics committee approval was not required by our institute for this study because it did not involve human nor animal participants. The authors would like to acknowledge the School of Health Science, University of KwaZulu-Natal for financial support. Computational resources granted by the Center for High Performance Computing (http://www.chpc.ac.za) are highly appreciated.
Disclosure
The authors report no conflicts of interest in this work.