
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Naringenin (NRG), a flavonoid compound, had been reported to exhibit extensive pharmacological effects, but its water solubility and oral bioavailability are only~46±6 µg/mL and 5.8%, respectively. The purpose of this study is to design and develop NRG-loaded solid lipid nanoparticles (NRG-SLNs) to provide prolonged and sustained drug release, with improved stability, involving nontoxic nanocarriers, and increase the bioavailability by means of pulmonary administration. Initially, a group contribution method was used to screen the best solid lipid matrix for the preparation of SLNs. NRG-SLNs were prepared by an emulsification and low-temperature solidification method and optimized using an orthogonal experiment approach. The morphology was examined by transmission electron microscopy, and the particle size and zeta potential were determined by photon correlation spectroscopy. The total drug content of NRG-SLNs was measured by high-performance liquid chromatography, and the encapsulation efficiency (EE) was determined by Sephadex gel-50 chromatography and high-performance liquid chromatography. The in vitro NRG release studies were carried out using a dialysis bag. The best cryoprotectant to prepare NRG-SLN lyophilized powder for future structural characterization was selected using differential scanning calorimetry, powder X-ray diffraction, and Fourier transform infrared spectroscopy. The short-term stability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay, cellular uptake, and pharmacokinetics in rats were studied after pulmonary administration of NRG-SLN lyophilized powder. Glycerol monostearate was selected to prepare SLNs, and the optimal formulation of NRG-SLNs was spherical in shape, with a particle size of 98 nm, a polydispersity index of 0.258, a zeta potential of −31.4 mV, a total drug content of 9.76 mg, an EE of 79.11%, and a cumulative drug release of 80% in 48 hours with a sustained profile. In addition, 5% mannitol (w/v) was screened as a cryoprotectant. Fourier transform infrared spectroscopy, differential scanning calorimetry, and powder X-ray diffraction studies confirmed that the drug was encapsulated into SLNs in an amorphous form. The lyophilized powder was stable at both refrigeration (4°C) and ambient temperature (25°C) for 3 months, and the MTT assay demonstrated that the SLNs were nontoxic. The cellular uptake of fluorescein isothiocyanate-labeled SLNs in A549 cells was highly time dependent over a period of 3 hours, while the pharmacokinetic study in Sprague Dawley rats showed that the relative bioavailability of NRG-SLNs was 2.53-fold greater than that of NRG suspension after pulmonary administration. This study shows that SLNs offer a promising pulmonary delivery system to increase the bioavailability of the poorly water-soluble drug NRG.
Introduction
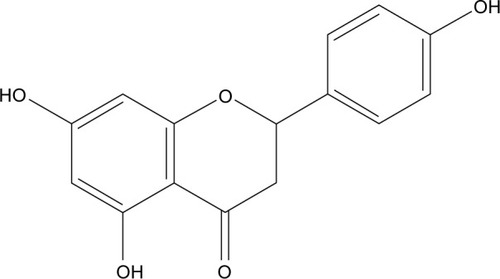
Naringenin (NRG; 5,7,4′-trihydroxyflavanone, molecular weight (MW) =272.3; ), a traditional Chinese medicine, contains a large amount of natural plants, including citrus fruits, tomatoes, cherries, grapefruit, and cocoa.Citation1 As a flavonoid compound, it has been reported to exhibit extensive pharmacological effects on biological systems,Citation2,Citation3 such as anti-inflammatory, anticancer, antiatherogenic, antifibrogenic, and antioxidant actions. Although it exhibits a wide range of effects, it has yet to be approved as a therapeutic agent. Physicochemical analysis suggests that NRG is a crystalline compound and is released slowly from oral dosage forms, thereby restricting its therapeutic applications. Moreover, NRG is a hydrophobic compound (its water solubility is 46±6 µg/mL) with a poor oral bioavailability (~5.81%).Citation4 Therefore, to increase its solubility, in vivo bioavailability, and therapeutic index and maintain a steady plasma concentration, there is a need to improve both its preparation and administration.
Figure 1 The structure of naringenin.
Up until now, a great deal of effort has been made to improve the solubility and bioavailability of NRG using a variety of approaches, including nanoparticles,Citation1 β-cyclodextrin inclusion complexes,Citation2 solid dispersions,Citation4 self-nanoemulsifying drug delivery systems,Citation5 submicron carriers,Citation6 and polymeric nanoparticles.Citation7 Unquestionably, nanotechnology is the most promising drug delivery system, and it has been widely applied to hydrophobic drugs. The nanoscale drug delivery systems include liposomes, solid lipid nanoparticles (SLNs), nanocrystals, nanoemulsions, cubosomes, and polymerosomes. Compared with polymers, SLNs have the benefits of low toxicity, no need for organic solvents, and no residual contamination and inexpensive excipients.Citation3 However, nanocrystals have the disadvantages of low drug solubility in biological fluids and instability. Compared with liposomes and emulsions, SLNs have the advantages of controlled drug release, the ability to incorporate lipophilic and hydrophilic drugs, reduced chemical degradation, increased physical stability, and low cost.Citation8 In addition, SLNs have other benefits such as the potential for large-scale production, targeting options, controlled release, improved solubility, good bioavailability and stability, the possibility of reducing the dose and dose frequency, increasing patient compliance, and delaying the onset of resistance.Citation9 Hence, nanotechnology SLNs have been used to develop a new drug delivery system for NRG.
SLNs were first described by Müller et al in 1991, as an alternative carrier system to traditional colloidal carriers with submicron-sized particles (50–1,000 nm).Citation10 They involve a physiological and biocompatible solid lipid matrix at both room and body temperatures and can be stabilized with nontoxic emulsifiers.Citation11 SLNs can be prepared for many different routes of administration including oral, pulmonary, ocular, dermal, parenteral, and nasal delivery.Citation12 The acidic environment in the stomach may result in the failure of oral drug delivery by causing the aggregation or degradation of SLNs. A further “bench to bedside” translation study may involve a new drug delivery mode of SLNs, apart from direct oral administration. Phospholipids are usually applied as multifunctional biocompatible, biodegradable excipients, which are found in lung tissue. In addition, after pulmonary administration, SLNs exhibit rapid absorption and degradation, together with sustained release. All these characteristics make SLNs especially favorable for pulmonary delivery. The benefits of direct delivery of a drug to the lung include providing a very large absorptive area (almost 140 m2), with a thin (0.1–0.5 µm) alveolar epithelium membrane, a high number of capillaries, low extracellular and intracellular enzyme activity, the absence of first-pass metabolism and no adverse reactions involving gastrointestinal irritation, reduced systemic toxicity, high bioavailability, and rapid absorption followed by rapid onset of action.Citation13–Citation15
The purpose of this study is to design and develop NRG-SLNs for prolonged and sustained drug release, offering improved stability, with nontoxic nanocarriers, and increased pulmonary bioavailability. In this work, the group contribution method (GCM) was used to select the most suitable lipid material. NRG-SLNs were first prepared using the emulsification and low-temperature solidification method after optimization with an orthogonal experimental approach.Citation16,Citation17 The physicochemical properties were characterized in terms of the shape, size, zeta potential, entrapment efficiency, and in vitro release behavior. Then, screening was carried out to identify suitable cryoprotectants, followed by lyophilization into dry powders to prevent any instability and further characterization by differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD), and Fourier transform infrared (FT-IR) spectroscopy to investigate changes in crystal structure or to detect any chemical interaction between the ingredients. Also, the short-term stability of NRG-SLN lyophilized powders was studied, and the in vitro cytotoxicity of SLNs in A549 cells was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay. Fluorescein isothiocyanate (FITC) was used as a fluorescent probe and incorporated into SLNs to evaluate the cellular uptake of NRG-SLNs in A549 cells using confocal microscopy. Finally, a bioavailability assessment was carried out in male Sprague Dawley (SD) rats. The pharmacokinetic parameters were calculated according to the plasma concentration data obtained by high-performance liquid chromatography (HPLC) analysis. The study results demonstrated the increased bioavailability of NRG-SLNs and confirmed the potential clinical applications of NRG-SLNs involving pulmonary delivery.
Materials and methods
Materials
NRG (MW =272.25, purity ≥98%) was purchased from Kang Weisheng Biotech Co., Ltd. (Nanjing, People’s Republic of China), egg-phosphatidylcholine was purchased from Shanghai Taiwei Pharmaceutical Co., Ltd. (Shanghai, People’s Republic of China), and mannitol and glycerol monostearate (GMS) were purchased from Tianjin Bodi Chemical Co., Ltd. (Tianjin, People’s Republic of China). FITC was obtained from Sigma-Aldrich Co. (St Louis, MO, USA), Sephadex gel-50 chromatography was purchased from Blue Quaternary Science & Technology Development Co., Ltd. (Shanghai, People’s Republic of China), Poloxamer188 was purchased from Sigma-Aldrich Co., and Tween-80 was purchased from the Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People’s Republic of China). All the other reagents were of analytical grade, and water was doubly distilled and deionized.
Methods
Selection of lipid materials
A series of lipid materials were selected, including stearic acid, GMS, palmitic acid, and glyceryl behenate (DA). The GCM was used to calculate the differences in the partial solubility parameters between NRG and the lipid matrix.Citation18 The formulation was
Optimization and preparation of NRG-SLNs
On the basis of the outcomes of the monofactor investigation, taking the drug encapsulation efficiency (EE) as an index, four factors including the emulsifying temperature (A), the volume ratio of the oil phase (a mixture of acetone and anhydrous ethanol at 1:1, v/v) to the inner water phase (B, 18 mL water), the mass ratio of drug to lipids (C, 100 mg lipid), and the mass ratio of Poloxamer188 (F68) to Tween-80 (T80; D, with the total concentration of 1.25%) were selected, and a four-factor, three-level orthogonal experimental plan was designed to optimize the preparation of NRG-SLNs. shows the variables and levels of the design.
Table 1 Orthogonal factors and levels of preparation of NRG-SLNs
Optimized NRG-SLNs were prepared by the emulsification and low-temperature solidification method with some slight modification.Citation16,Citation17 Briefly, 10 mg NRG, 100 mg GMS, and 200 mg soya lecithin were mixed and dissolved in a mixture of acetone (3 mL) and anhydrous ethanol (3 mL). The mixture was sonicated and warmed to form an organic phase at 80°C in a water bath. The aqueous phase was prepared by dissolving 125 mg T80 (1.25 w/v%) and 125 mg F68 (1.25 w/v%) in doubly distilled water (18 mL) at 80°C in a water bath. Then, the organic phase was rapidly injected into the water phase through a syringe needle (5 mL) under mechanical stirring (1,500 rpm) to form an emulsion. The resulting suspension was continually stirred at 80°C for ~2 hours to remove ethanol, and the system volume was concentrated to ~10 mL. The resulting emulsion was then quickly solidified in an ice-water bath (10 mL, 0°C–2°C) under mechanical agitation (1,500 rpm) for 1 hour to obtain semitransparent NRG-SLNs. Blank-SLNs were prepared by the same method but without adding NRG. The obtained SLNs suspension was stored in a refrigerator at 4°C.
Characterization of NRG-SLNs
Particle size, polydispersity index, and zeta potential
The particle size, polydispersity index (PDI), and zeta potential of the freshly prepared NRG-SLNs were measured by photon correlation spectroscopy using a zetasizer (Nano ZS™; Malvern Instruments, Malvern, UK) at 25°C. The samples were diluted with double-distilled water, and all measurements were performed in triplicate.
Transmission electron microscopy analysis
The morphology of the NRG-SLNs was observed under transmission electron microscopy (TEM; Tecnai G2F30; FEI Co., Hillsboro, OR, USA) carried out at 200 kV. One drop of the freshly prepared NRG-SLNs suspension was diluted tenfold with distilled water and placed on a copper grid covered with nitrocellulose. Then, each sample was negatively stained with 2% (w/v) phosphotungstic acid and air-dried before observation.
Determination of total drug content
The total drug content of NRG-SLNs was measured by HPLC (L-2400; Hitachi Ltd., Tokyo, Japan) using a ultraviolet-visible detector.Citation7 A Welch C18 column (4.6×200 mm, 5 mm) was used for analysis, and the mobile phase was a mixture of methanol:water (3:1) with 0.5% acetic acid at a flow rate of 1 mL/min. The detection wavelength was 288 nm at 25°C, and the injection volume was 20 µL. NRG-SLNs (0.1 mL) were diluted and demulsified with 5 mL acetone to obtain a clear solution, and then, the total drug content (TDC) was determined by HPLC.
Drug encapsulation efficiency (%EE) determination
The EE was determined by separating the unencapsulated NRG using a Sephadex gel-50 chromatography (1.5×20.0 cm). In brief, 0.5 mL of NRG-SLNs was transferred to a Sephadex gel-50 chromatography with double-distilled water as the eluant. The light blue opalescent eluent that flowed out of the column was collected and diluted to 10.0 mL with acetone. Another 0.5 mL sample of NRG-SLNs was directly diluted with 9.5 mL acetone. Both concentrations of NRG were determined by HPLC at a detection wavelength of 288 nm and the EE is calculated using the following equation: EE (%) = Ws/Wtotal ×100%, where Ws is the amount of NRG loaded in the SLNs and Wtotal the total NRG amount in NRG-SLNs.
In vitro release studies
In vitro NRG release studies were carried out with a freshly prepared NRG-SLN within 24 hours, using the dialysis bag technique. Phosphate-buffered saline (PBS; 0.1 mM, pH 7.4, with 0.5% T80) was used as the simulated blood. The dialysis bags (MW cutoff, 8–12 kDa; USA) were soaked in double-distilled water overnight before use. For the release experiment, 2 mL NRG solution (0.5 mg/mL) and 2 mL NRG-SLNs suspension were transferred to a dialysis bag with the two ends fixed by a clip and then placed in a preheated release medium (50 mL) at 37°C under gentle stirring at 100 rpm. Aliquots of the dissolution medium (0.5 mL) were withdrawn at intervals and passed through a 0.22 µm filter, while the same volume of fresh release medium was added to maintain sink conditions. The concentration of NRG released into the filtrate was analyzed by HPLC as described earlier. All measurements were repeated in triplicate.
Drug-release kinetics
To assess the drug-release kinetics, the release data were plotted using zero-order and first-order kinetics and the Higuchi model, Weibull model, and Ritger–Peppas model. In order to examine the mechanism of drug release in vitro, the regression coefficient (R2) was calculated.Citation19
Preparation of NRG-SLN lyophilized powder
Lyophilization (freeze-drying) was used to increase the stability of SLNs.Citation6 To achieve this, 1 mL mannitol (5% w/v, 8% w/v) and 1 mL lactose (5% w/v, 8% w/v) aqueous solutions were mixed with 2 mL NRG-SLNs suspension at a ratio of 1:2 (v/v) and added to the vial (5 mL). The mixture was then prefrozen at −20°C overnight, and the samples were then transferred quickly to a freeze dryer at −80°C for 24 hours to obtain NRG-SLN lyophilized powder. Finally, 5% mannitol (w/v) was selected as the best cryoprotectant to prepare NRG-SLN lyophilized powder for future structural characterization.
Solid-state characterization
Differential scanning calorimetry
The melting and crystallization behavior of NRG, physical mixture (NRG- and blank-SLNs in a ratio of 1:1), blank-SLNs, and NRG-SLNs were studied by DSC (DSC-60; Shimadzu Corporation, Tokyo, Japan).Citation20,Citation21 For each measurement, accurately weighed samples (5 mg) were sealed in aluminum pans and analyzed over a temperature range of 25°C–280°C under a nitrogen purge (50 mL/min) with a heating rate of 10°C/min.
Powder X-ray diffraction
The samples used for PXRD analysis were the same as those used for DSC analysis. The encapsulation of drug inside the nanoparticles was further confirmed by PXRD (Ultima IV; Rigaku Corporation, Tokyo, Japan). Samples were exposed to Cu·Kα radiation (30 kV, 30 mA) and scanned from 5° (2θ) to 50° (2θ). The step size and step time were 0.02° and 4°/min, respectively.
FT-IR spectroscopy studies
Samples used for FT-IR analysis were the same as those used for DSC analysis. An FT-IR spectrometer (IFS 55; Bruker Corporation, Billerica, MA, USA) was used to record the FT-IR spectra between 400 cm−1 and 4,000 cm−1 using the KBr pellet method.
Short-term stability studies of NRG-SLN lyophilized powder
The lyophilized NRG-SLNs were sealed with in amber-colored glass vials, and stability studies were carried out. Three samples were stored at refrigeration (4°C) and ambient temperatures (25°C) for a period of 3 months. The particle size, zeta potential, PDI, and drug content (%) were determined at intervals (24 hours, 1 month, 2 months, and 3 months). Before determination, the stored samples were redispersed in double-distilled water at 37°C.
Cell evaluation
Cell culture and growth conditions
The cell experiments were approved by the Liaoning Medical University Laboratory Cell Ethics Committee, Jinzhou, People’s Republic of China. Samples of the human lung carcinoma cell line (A549) were obtained from the Nanjing Built Technology Co., Ltd. Cells were grown in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with fetal bovine serum (10% [v/v]) and antibiotics (a mixed solution of penicillin [100 units/mL] and streptomycin [100 µg/mL]) and incubated in a humidified incubator at 37°C with 5% CO2/95% air.
MTT assay
The cell viability was evaluated by MTT assay.Citation22 A549 cells were seeded in 96-well plates at a density of 5.0×104 cells per well in 100 µL solution and incubated for 24 hours to promote adhesion. Then, the cells were added to different formulations of free NRG, NRG-SLNs, and blank-SLNs at a concentration of NRG ranging from 1 µg/mL to 50 µg/mL. The cytotoxicity of blank-SLNs was also evaluated with a lipid concentration equivalent to that of NRG-SLNs. Free NRG was dissolved in dimethylsulfoxide (DMSO) and then diluted in RPMI-1640 medium to obtain a final concentration of DMSO <0.2%. However, the NRG-SLNs and blank-SLNs powder were directly dissolved in and diluted with RPMI-1640 medium. Subsequently, a longer incubation of 72 hours (instead of 24 hours or 48 hours) was used to ensure a high degree of killing, and no cell treatment was taken as the control. MTT solution (5 mg/mL, 20 µL) was added to each well at 37°C, followed by incubation for another 4 hours. Then, the supernatant was removed, and the MTT formazan crystals were dissolved in 100 µL DMSO in each well, and the plates were shaken for 15 minutes while being protected from light. Finally, the absorbance of each well was measured using a microplate reader (Mk3; Thermo Fisher Scientific, Waltham, MA, USA) at 492 nm. The cell viability (%) was calculated using the following equation: %(viability) = (optical density [OD]treat − ODblank)/(ODcontrol − ODblank) ×100% and %cytotoxicity =1− %(viability).
Cellular uptake
For the cellular uptake study, confocal microscopy was used,Citation23,Citation24 and FITC was encapsulated into nanoparticles instead of NRG as a fluorescent probe to examine the cellular uptake of NRG-SLNs. A549 cells were seeded in six-well plates at a seeding density of 5.0×104 cells per well in 1 mL growth medium and incubated for 24 hours to allow attachment and then incubated with FITC-SLNs (at a concentration of 50 µg/mL) at 37°C for 1 hour, 2 hours, and 3 hours. After predetermined intervals, the cells were washed three times with cold PBS to remove noninternalized material and fixed in 2 mL 4% paraformaldehyde for 10 minutes at 4°C. Then washing was carried out three times before adding 1 mL Triton-X100 (1%, v/v, dissolved in PBS) and allowing the samples to stand for 10 minutes at 4°C to lyse the cells. After washing a further three times, the cells were harvested by adding 1 mL of 1% bovine serum albumin at 37°C. Finally, the cells were washed three times with PBS and stained with Hoechst 33342 solution (1 µg/mL) at 37°C for 20 minutes to examine the nuclear morphology. The A549 cells were then washed twice with Hanks’ balanced salt solution, followed by rhodamine-phalloidin in PBS to stain the cell membrane for 30 minutes. Finally, the fixed cells were washed again and then observed under a Radiance 2100 confocal laser scanning microscope (Bio-Rad Laboratories Inc., Hercules, CA, USA).
In vivo pharmacokinetic study
Animals
The animal experiments were approved by the Liaoning Medical University Laboratory Animal Ethics Committee, Jinzhou, People’s Republic of China. Male SD rats (200–250 g) were supplied by the Liaoning Medical University Laboratory Animal Center and were housed in the Animal Care Facility with free access to food for 7 days to allow them to adapt to the new normal conditions. However, the rats were only allowed free access to water before and during the experiment.
Dosing, sampling, and intratracheal instillation technology
Two sample suspensions were prepared (4 mg/mL, for the calculation of NRG): 1) free NRG was dispersed in 1% (w/v) hydroxypropyl methylcellulose aqueous solution and 2) freshly prepared NRG-SLN powders were completely dissolved in double-distilled water. The intratracheal instillation model was selected instead of an inhalation model because the method is an easy and reliable way to control SLNs toxicity and dose.Citation25 According to the maximum tolerated dose and safe dose in rats and in pretests, the amount of NRG for safe pulmonary delivery was determined as 20 mg/kg. The previously described intratracheal instillation method was used with some modification.Citation26,Citation27 Briefly, the animals were anesthetized with ether, cannulated with a laryngoscope to expose the trachea, and two different sample suspensions were instilled into the lungs at an NRG dose of 20 mg/kg. After intratracheal dosing, the animals were held in an upright position for 1 minute to ensure delivery of the dose following the removal of the delivery device.Citation28
Plasma collection
Ten SD rats were divided randomly into two groups. After dosing, 600 µL blood samples were collected by retroorbital venous plexus puncture under mild ether anesthesia at the following times: 0.5 hour, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, and 24 hours. The samples were then quickly transferred to heparinized tubes, and plasma was immediately separated by centrifugation at 8,000 rpm for 10 minutes. The plasma samples were stored at −20°C for further analysis.
Plasma sample preparation and HPLC determination
The plasma samples containing NRG were further processed and assayed according to a previously described method with slight modification.Citation5 In brief, 200 µL aliquots of plasma were transferred to clean Eppendorf (1.5 mL) tubes, followed by the addition of 2% formic acid (50 µL) as a modifier and vortexed for 2 minutes. The plasma samples were extracted with 1.2 mL ethylacetate following vortex agitation for 2 minutes, then centrifuged at 12,000 rpm for 10 minutes, and the upper organic layers were transferred to new Eppendorf tubes and evaporated to dryness with nitrogen at 40°C and 15 psi pressure. Each dried residue was reconstituted with 150 µL mobile phase (a mixture of methanol and water [7:3, v/v]) and then centrifuged at 12,000 rpm for 10 minutes. Subsequently, 20 µL aliquots of each supernatant were analyzed by HPLC using a ultraviolet-visible detector (L-2400; Hitachi Ltd.) at a λmax of 289 nm with a flow rate of 1 mL/min.
Statistical analysis
All data were examined using SPSS statistics 17.0 software and expressed as mean ± standard deviation. Student’s t-test was statistically significant at P-values <0.05.
Results and discussion
Selection of lipid
Although no systematic and standard approach to choosing a suitable solid lipid has been published yet, some criteria have been defined in the published literature, such as qualitative solubility, partitioning behavior, and GCM.Citation29 GCM was used to predict the differences in the partial solubility by comparing the structure and groups of the molecules and calculating their contribution to the partial solubility parameters V, Fd, Fp, and Eh.Citation18 According to GCM, the solubility parameter is used to reflect the square root of the cohesive energy density of the components that hold substances together.Citation30 In addition, the standard to classify the values of Δδ, when Δδ <7.0 MPa1/2, shows that the drug and lipid material are compatible, namely, the lipid material is suitable to incorporate the drug as a carrier; Δδ >10.0 MPa1/2 means the drug and lipid material are incompatible; and a Δδ between 7.0 MPa1/2 and 10.0 MPa1/2 means further study is required to identify the presence of a relatively strong polar or hydrogen bonding effect between the two groups. The values of δd, δp, and δh of each lipid and the differences in partial solubility (Δδp) are listed in . The miscibility of the drug with lipid is one of the most important factors that determine the loading capacity of the drug in the lipid. The values of Δδp indicated that NRG and GMS had the highest compatibility, and the solubility of the drug in GMS was theoretically the highest. Thus, GMS was selected to prepare NRG-SLNs for further study.
Table 2 Partial solubility parameters and the differences between NRG and lipid materials
Formulation optimization and preparation of NRG-SLNs
The results of the L9(3Citation4) orthogonal design are shown in . It was found that the drug EEs (regarded as an index) of all the experiments were within 40%–80%. The range, revealing the relationship between the index and each factor, was used to choose the optimum ingredient formation that showed the degree that various factors affected the index. In this experiment, the ranking of the four factors of the R-value was C>A>D>B, and each level within individual factors was in the order: A: 3>2>1; B: 1>2>3; C: 3>2>1; D: 3>2>1. The Orthogonality Experiment Assistant Version 3.1 software (Sharetop software studio)Citation31 was used to analyze the results, and the optimized formulation was found to be C3A1D3B1 in which the amounts of NRG, GMS, egg-phosphatidylcholine, T80, and F68 were 10 mg, 100 mg, 200 mg, 125 mg, and 125 mg, respectively.
Table 3 EE results and analysis of orthogonal experiment of the preparation of NRG-SLNs
Emulsification and low-temperature solidification is a dependable, standard, and reproducible method for preparing SLNs. There were three different models for drug incorporation into SLNs, namely, 1) drug-enriched core, 2) drug-enriched shell, and 3) solid solution. The drug distribution in SLNs was determined by the preparation method.Citation32,Citation33 In this experiment, T80 as a surfactant can reduce the size and improve the stability of SLNs.Citation11 The melting point of GMS is ~75°C, and, in order to maintain its molten state during the preparation process, the temperature should be >75°C and, finally, the optimized emulsifying temperature was selected to be 80°C. As the relative solubility of NRG was higher in acetone and that of soya lecithin was higher in anhydrous ethanol, we selected the solvent system of acetone/anhydrous ethanol (1:1) as optimal with a volume of 3 mL. The stirring speed is another factor that affects the nanoparticle size.Citation34 If the speed is too low, the particle size will be increased and its stability will also be adversely affected; if the speed is too high, a large amount of foam will be produced, which will adversely affect the emulsifying effect of the surfactant. Lastly, the optimum stirring speed was confirmed to be 1,500 rpm. When solidifying in the cold aqueous phase at 0°C, the liquid lipid is converted to a solid and some drug is dispersed inside to form the core of the particles. Then, the surfactants and soya lecithin containing the drug could be adsorbed on the surface of the core particles. Therefore, we decided that the drug incorporation of NRG-SLNs should be the core–shell model with a drug-enriched shell.
Characterization of NRG-SLNs
Particle size, PDI, and zeta potential
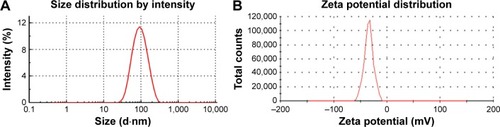
As is well known, the particle size distribution is one of the most important properties used to evaluate the stability of colloidal systems.Citation35 The mean particle diameter of the optimized NRG-SLNs suspension was determined immediately after production to be 98±0.61 nm with a PDI of 0.258±0.058 (). The PDI is an indicator of the homogeneity of the size distribution, and the narrow size distribution and uniform size confirmed the homogeneous nature of the formulation. According to a previous report, particles can be dispersed stably when the absolute value of the zeta potential is >30 mV.Citation36 As shown in , the zeta potential of the optimized NRG-SLNs was approximately −31.4±0.98 mV, which demonstrated that the colloidal dispersion was a physically stable system.
Figure 2 Size distribution (A) and zeta potential (B) of NRG-SLNs.
Morphology of NRG-SLNs
Compared with free NRG dispersed in water (), NRG-SLNs suspension was homogeneous without any apparent drug crystals (). The TEM image () of the optimized NRG-SLNs suspension had a regular spherical shape with a clear and smooth surface and no adhesion. Most of the particles were observed to range from 60 nm to 80 nm. It was reasonable to assume that the diameter measured by TEM was lower than that measured by dynamic light scattering for the same NRG-SLNs suspension, and the dynamic light scattering figures show the wet and extended hydrodynamic radius of NRG-SLNs suspension, while TEM shows the dry and shrunk configuration of NRG-SLNs suspension.Citation17
Figure 3 Images of free NRG in water (A), NRG-SLNs suspension (B), and NRG-SLNs suspension via TEM (C).

TDC and entrapment efficiency
The EE of the optimized NRG-SLNs suspension was found to be 79.11%±0.56%, and an average TDC was estimated to be 9.76±0.43 mg, which was ~97.6% (approaching 100%) in NRG-SLNs, and there was no significant loss of drug(s) during the formulation process. The high values of TDC and EE further confirm the efficiency of the method for the preparation of NRG-SLNs and analysis by HPLC.
In vitro drug-release behavior
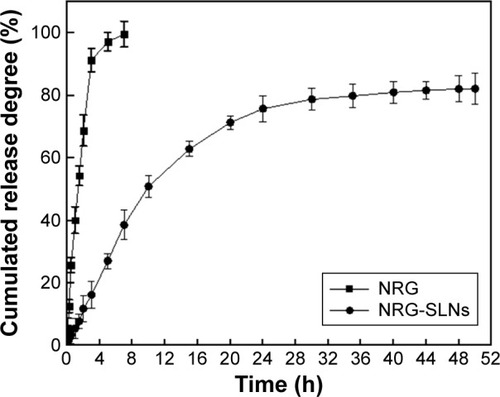
In this study, the dynamic dialysis method was used to measure the drug-release behavior in vitro of free NRG from NRG-SLNs. As the solubility of NRG in aqueous solution is only 41.76±0.51 µg/mL, PBS (0.5% T80 in PBS, pH 7.4) was chosen as a receptor medium to provide sink conditions.Citation6,Citation37 shows the release profile of NRG from NRG-SLNs and NRG solution at predetermined time intervals. It can be seen that the cumulative release of NRG solution was rapid with ~100% being released from the dialysis bag within 5 hours, while NRG-SLNs showed an initial burst release of ~33% in 5 hours and nearly 80% in a sustained manner within 48 hours. This confirmed the prolonged release characteristics of NRG-SLNs. The release of NRG solution fitted a first-order kinetic model (R2=0.9857); however, a Higuchi kinetic model (R2=0.9989) was the best fit for NRG-SLNs. The drug release mechanism of NRG-SLNs could involve drug diffusion, polymer matrix swelling, and the polymer erosion or degradation,Citation17 which corresponded to the view above that NRG-SLNs fitted the core–shell model with a drug-enriched shell. These in vitro release data were similar to those in many previous studies reporting that drug-loaded SLNs provided a controlled release pattern.Citation16,Citation17
Figure 4 In vitro release profile of NRG from NRG-SLNs and free NRG solution by a dialysis method in phosphate-buffered saline (0.5% of Tween-80 in PBS, pH 7.4) at 37°C.
Abbreviations: NRG, naringenin; SLNs, solid lipid nanoparticles; h, hours; PBS, phosphate buffered saline.
Preparation of NRG-SLN lyophilized powder
The size stability profiles of the formulations were not affected markedly with time, while lipid nanocarriers have been shown to exhibit instability over time, although they can be stabilized by lyophilization or adsorption onto a solid matrix.Citation38 The main reason for instability was that the membrane materials were oxidized easily. However, liposomes with SLNs could improve the antioxidation effect of SLNs preparation. In addition, freeze-drying can be used to improve the liposome stability. On one hand, a cryoprotectant could maintain the stability of liposome membranes and significantly reduce the speed of oxidation and hydrolysis of drug and liposome. On the other hand, a cryoprotectant could form a eutectic or glassy substance with water, which would inhibit the growth of ice crystals and reduce extrusion and mechanical damage to the nanoparticles.Citation31 The process of screening an optimal cryoprotectant is described in . When the cryoprotectant was 5% mannitol (w/v), the prepared NRG-SLN lyophilized powder exhibited the smallest changes in particle size, a shorter dissolution time, and a stable solution system.
Table 4 The appearance, dissolution time, solution color, and mean particle size of the lyophilization powder of NRG-SLNs
Characterization of freeze-dried powders
Differential scanning calorimetry
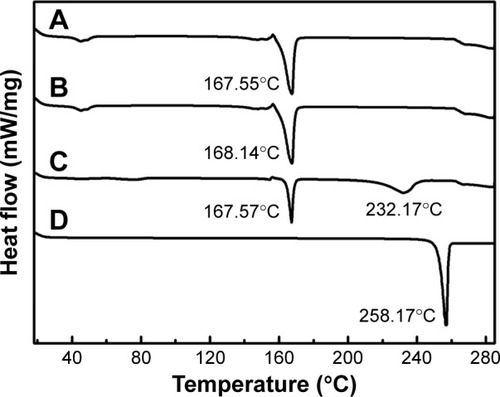
DSC analysis is an important approach to determine the state of incorporated drug.Citation5 The DSC curves of NRG-SLNs, blank-SLNs, NRG, and a physical mixture of blank-SLNs and NRG are shown in . The blank-SLNs only had melting peaks around 168°C for the solid lipid core as well as the lyoprotectant used in freeze-drying. Thermograms for both NRG-SLNs and physical mixture were similar due to the fact that the melting properties of the SLNs were mainly determined by the solid lipid core material.Citation39 Free NRG exhibited a characteristic melting endothermic peak at 258.17°C corresponding to its melting point, which confirmed the crystalline nature of the drug. The physical mixture exhibited an endothermic melting peak at 232.17°C, which can be attributed to the presence of free NRG. Compared with the thermogram of NRG, the endothermic melting of physical mixture shifted to a lower melting temperature. The reason for this may be due to some interactions between NRG and components of the blank-SLNs, especially the lyoprotectant, upon mixing. This would result in a partial conversion of crystalline NRG to its amorphous form and the more amorphous the mixture, the lower the melting enthalpy. A similar observation has been reported elsewhere.Citation40 However, NRG-SLNs did not show any distinct melting peak indicating the absence of crystallinity, namely, the drug was present in an amorphous or molecular state in SLNs.
Figure 5 DSC analysis of NRG-SLNs (A), blank-SLNs (B), physical mixture of NRG and blank-SLNs (C), and NRG (D).
Powder X-ray diffraction
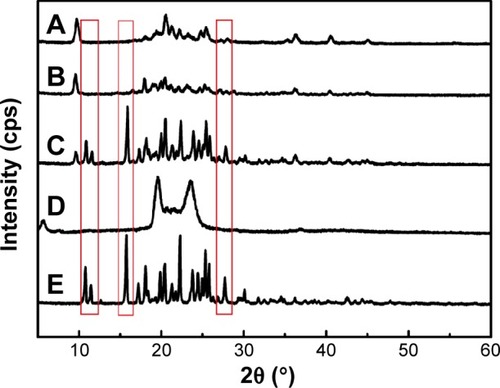
The PXRD study was further used to confirm the nature of the pure NRG after encapsulation into SLNs. The diffraction pattern of the NRG, blank-SLNs, NRG-SLNs, physical mixture of NRG and blank-SLNs and GMS was analyzed (). Characteristic peaks were present in the NRG and physical mixture, which confirmed the crystallinity of the NRG components (, red rectangles). However, the crystalline peaks were absent from the diffraction pattern of blank-SLNs and NRG-SLNs, which indicated that NRG was in an amorphous or disordered crystalline form in SLNs according to the DSC measurements. This disordered crystalline form of NRG inside the SLNs contributes to its sustained release from the NRG-SLNs. The presence of drug in crystalline form inside SLNs hampers its release as such large molecules cannot diffuse easily through the small pores. However, the amorphous or disordered crystalline phase of the drug can allow its easy diffusion through the SLNs, leading to a sustained release of the encapsulated drug.Citation22
Figure 6 X-ray diffraction patterns of NRG-SLNs (A), blank-SLNs (B), physical mixture of NRG and blank-SLNs (C), GMS (D), and NRG (E).
Abbreviations: NRG, naringenin; SLNs, solid lipid nanoparticles; GMS, glycerol monostearate.
FT-IR spectroscopy
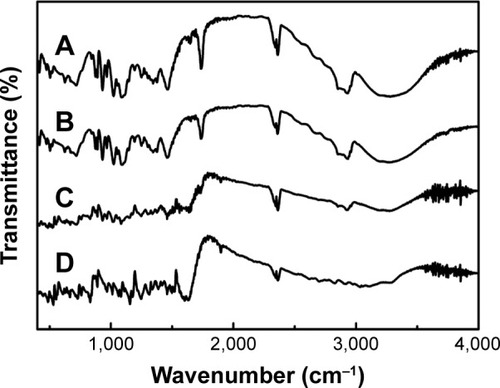
FT-IR spectroscopy is one of the best techniques to evaluate the chemical stability of the encapsulated drug inside the SLNs.Citation41 The FT-IR spectra of NRG-SLNs, blank-SLNs, NRG, and a physical mixture of blank-SLNs and NRG are presented in . The structure of NRG contains 5-OH and 4–C=O with a potential to form intramolecular hydrogen bonds. In the case of NRG alone, a characteristic absorption band appearing around 3,290 cm−1 and 3,117 cm−1 corresponds to O–H stretching, while absorption bands around 1,626 cm−1 and 1,464 cm−1 could be attributed to C=O stretching. These distinctive bands of NRG were also identified in NRG-SLNs, which suggest that NRG is well incorporated into SLNs and supports the chemical stability of NRG in SLNs.
Figure 7 FT-IR spectra of NRG-SLNs (A), blank-SLNs (B), physical mixture of NRG and blank-SLNs (C), and NRG (D).
Stability studies of NRG-SLN lyophilized powder
These studies were designed to investigate the storage stability of lyophilized NRG-SLNs over a prolonged period of time. As previously indicated from zeta potential measurements, all the formulations were expected to be physically stable, at least in the short term. In order to assess the effect of the storage temperature on the stability, NRG-SLN lyophilized powders were monitored with regard to their physical and chemical stability for 3 months of storage in the absence of sunlight at refrigeration (4°C) and ambient temperature (25°C). The results of the stability studies of NRG-SLN lyophilized powder for 3 months are shown in . In our experiment, we found that the color, particle size, zeta potential, and TDC had no significant effect on storage up to 3 months under both conditions, which means that our method is suitable for the production of stable NRG-SLN lyophilized powders. The good stability may be due to the slow transition of lipid in SLNs, the low particle size, and steric effect of T80.Citation42
Table 5 Stability studies of NRG-SLN lyophilized powder for 3 months
Cell evaluation
Cytotoxicity of NRG-SLNs
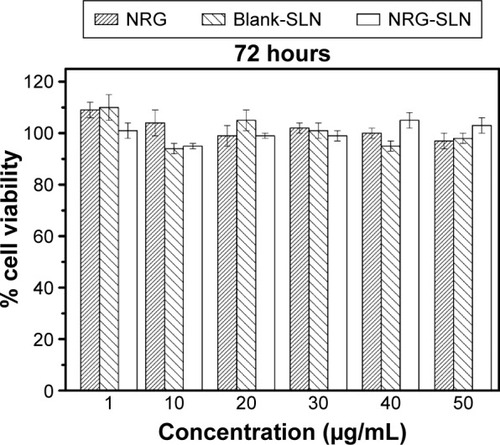
The cytotoxicity of NRG, blank-SLNs, and NRG-SLNs was investigated using the A549 cell line. The results of the cytotoxicity study are shown in . No significant toxicity was observed for all the formulations over a range of concentrations (the NRG concentration of 1–50 µg/mL was equivalent to the lipid concentration of 0.03–1.5 mg/mL) since the percentage cell viability was >94%, and so, the cytotoxicity of the lipid carrier was negligible and was not concentration dependent. The results of the MTT cell viability assay suggest that the SLNs used in this study were nontoxic (even at a high concentration of 50 µg/mL), which is in good agreement with the results described in the literature for similar SLNs.Citation17,Citation19,Citation43 The results also indicated that NRG may have no inhibition in A549 cells corresponding to a previous literature report.Citation41 In summary, all three formations showed no significant toxicity in A549 cells, which further confirmed their safety in normal cells.
Figure 8 Cytotoxicity of NRG, blank-SLNs, and NRG-SLNs (n=3).
Cellular uptake
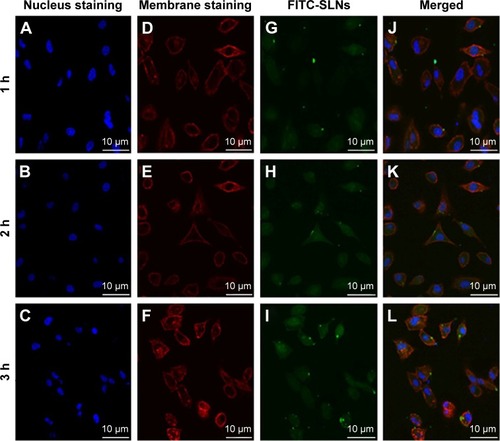
To evaluate whether the prepared NRG-SLNs could efficiently deliver drug into cells, we used fluorescent FITC-labeled SLNs to examine the internalization. Confocal laser scanning microscopy was used to investigate the cellular localization of FITC-SLNs. Cell nuclei were stained with Hoechst 33342, and cell membranes were stained with rhodamine-phalloidin. As shown in , A549 cells were incubated with 50 µg/mL FITC-SLNs in serum-free Dulbecco’s modified eagle medium for a range of incubation times. The 2-hour internalization of FITC-SLNs was significantly greater than that for 1 hour. FITC-SLNs formed nonuniform green fluorescent aggregates and mostly accumulated in the cell membrane region but did not penetrate the nucleus. However, the 3-hour internalization was greater than that at 2 hours. These results show that the cellular uptake or accumulation of FITC-SLNs in A549 cells is highly time-dependent within 3 hours and similar to that in a previous report.Citation44 These results also indicate that our nanocarriers are of a suitable size to be taken up efficiently by cells for drug delivery and nontoxic because they produce no change in the morphology of the cell nuclei.
Figure 9 Confocal microscopy images of A549 cells after 1 h, 2 h, and 3 h incubation at 37°C with FITC-SLNs.
Abbreviations: FITC, fluorescein isothiocyanate; SLNs, solid lipid nanoparticles; h, hours.
Pharmacokinetic studies
HPLC method validation
Before the pharmacokinetic study, an HPLC method was established and validated for quantitative determination of the plasma concentrations of NRG in vivo to assess its pharmacokinetic behavior.Citation4 The retention time of NRG was ~4.5 minutes with a total run time of 15 minutes. The assay was linear over the range 5–1,000 ng/mL with a detection limit of 1 ng/mL, and the standard regression equation of the peak area ratios of NRG (Y) to NRG concentrations (X) was as follows: Y=−20,116X +94,430 (correlation coefficients R2=0.9995). The relative standard deviation of intra- and interday precision at three concentrations (10, 500, and 1,000 ng/mL) was <4.79%, and the accuracy ranged from 97.14% to 104.78%. These results indicate that the developed HPLC method is precise, with good sensitivity and reproducibility for the determination of NRG in rat plasma and meets the requirements for pharmacokinetic studies.
Pharmacokinetics
An in vivo study was performed to quantify NRG after pulmonary administration of NRG formulations. Results of pharmacokinetics studies showed that the pharmacokinetics processes after a single dose of NRG and NRG-SLNs suspension were all fitted noncompartment model. Pharmacokinetic parameters were calculated using DAS 2.0 software, according to the principle of determining the best correlation between the observed and computed concentrations. The statistical comparison of data was performed using the Student’s unpaired t-test at a significance level of 0.05. Peak concentration (Cmax) and time of peak concentration (Tmax) were acquired diametrically from the individual plasma concentration–time profiles. The relative bioavailability (Fr) was calculated using the following formula:
The pulmonary pharmacokinetic parameters of the NRG-SLNs and NRG suspension are listed in . The mean plasma concentration–time profile in rats is shown in . The NRG plasma concentrations were significantly higher for rats treated with NRG-SLNs suspension than for those treated with NRG suspension. The AUC0→24h, AUC0→∞, Cmax, t1/2, mean residence time, and Fr of NRG-SLN formulation after pulmonary administration were approximately 2.53-fold, 3.66-fold (P<0.05), 1.61-fold, 3.64-fold (P<0.05), 3.33-fold (P<0.05), and 2.53-fold higher than those of NRG suspension, respectively. However, the mean clearance rate value for NRG-SLNs was reduced ~3.72-fold compared with the NRG suspension (P<0.05).
Table 6 Pharmacokinetic parameters of NRG after intratracheal administration of NRG suspension and NRG-SLNs suspension (n=5)
Figure 10 The mean plasma concentration–time curve of NRG in rats after a single intratracheal dose (20 mg/kg) of NRG suspension and NRG-SLNs.
Abbreviations: NRG, naringenin; SLNs, solid lipid nanoparticles; h, hours.
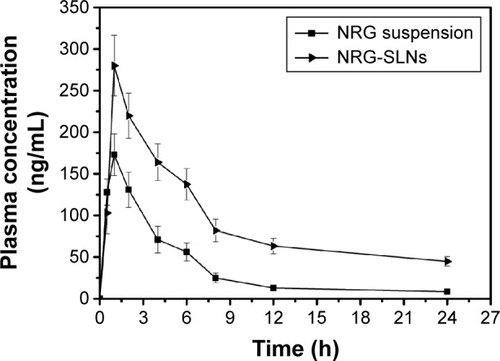
These results showed the NRG-SLNs resulted in an increased absorption of NRG after pulmonary administration. It might be partly attributed to the smaller particle size and the F68 improvement in the absorption of NRG with a moderately inhibited P-glycoprotein efflux system.Citation45 The significantly prolonged t1/2 and mean residence time of the NRG-SLNs suspension compared with free NRG suspension shown in this study may be because 1) the drug was embedded into the solid lipid matrix to protect it from enzymatic degradation, and 2) NRG exhibited sustained release from the SLNs and a prolonged circulation time in blood corresponding to the results of the in vitro release test earlier.Citation46 The current findings show the high clinical treatment potential of SLNs as an effective nanocarrier tool for the pulmonary delivery of NRG.
Conclusion
In our study, a poorly water-soluble drug, NRG, was first successfully incorporated into SLNs – lipidic nanocolloidal system using an emulsification and low-temperature solidification method and optimized using L9(3Citation4) orthogonal design. The formulated NRG-SLNs exhibited nanometer range uniform spherical-shaped particles with a narrow size distribution, high drug content, good entrapment efficacy, and sustained-release profile in vitro with burst release during the initial phase as well as exhibiting subsequent Higuchi release kinetics. Optimized NRG-SLN lyophilized powders were obtained successfully by freeze-drying. FT-IR, DSC, and PXRD studies were used to further confirm that the drug was encapsulated into SLNs in an amorphous form. The NRG-SLN lyophilized powders were stable at 4°C and room temperature for 3 months without any obvious changes. The MTT cell viability assay using a human lung epithelial cancer cell line (A549) demonstrated that the SLNs used in this study were nontoxic. FITC-labeled SLNs were used to examine NRG-SLNs internalization, and the cellular uptake of FITC-SLNs in A549 cells was found to be highly time dependent within 3 hours. The HPLC method was used to detect NRG in plasma. A pharmacokinetic study conducted in male SD rats showed that NRG-SLNs produced a significant improvement in the relative bioavailability compared with the NRG suspension after administration via pulmonary instillation. This work demonstrates that SLNs offer a promising pulmonary delivery system for increasing the bioavailability of poorly water-soluble drugs such as NRG.
Acknowledgments
The authors thank the National Natural Science Foundation of China (no 81302707) and Natural Science Foundation of Liaoning Province (no 2013022052) for their financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
- YenFLWuTHLinLTChamTMLinCCNaringenin-loaded nanoparticles improve the physicochemical properties and the hepatoprotective effects of naringenin in orally-administered rats with CCl(4)-induced acute liver failurePharm Res200926489390219034626
- YangLJMaSXZhouSYPreparation and characterization of inclusion complexes of naringenin with β-cyclodextrin or its derivativeCarbohydr Polym201398186186923987422
- KhanAWKottaSAnsariSHSharmaRKAliJEnhanced dissolution and bioavailability of grapefruit flavonoid naringenin by solid dispersion utilizing fourth generation carrierDrug Dev Ind Pharm201541577277924669978
- KhanAWKottaSAnsariSHSharmaRKAliJSelf-nanoemulsifying drug delivery system (SNEDDS) of the poorly water-soluble grapefruit flavonoid naringenin: design, characterization, in vitro and in vivo evaluationDrug Deliv201522455256124512268
- TsaiMJHuangYBFangJWFuYSWuPCPreparation and evaluation of submicron-carriers for naringenin topical applicationInt J Pharm20154811–2849025615985
- KeWTingtingLRongLPreparation and in vitro release of buccal tablets of naringenin-loaded MPEG-PCL nanoparticlesRSC Adv2014473367233679
- KumarSRandhawaJKPreparation and characterization of paliperidone loaded solid lipid nanoparticlesColloids Surf B Biointerfaces201310256256823104026
- VaghasiyaHKumarASawantKDevelopment of solid lipid nanoparticles based controlled release system for topical delivery of terbinafine hydrochlorideEur J Pharm Sci201349231132223557842
- OmwoyoWNOgutuBOlooFPreparation, characterization, and optimization of primaquine-loaded solid lipid nanoparticlesInt J Nanomedicine2014983865387425143734
- MüllerRHMäderKGohlaSSolid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the artEur J Pharm Biopharm200050116117710840199
- Ezzati Nazhad DolatabadiJHamishehkarHEskandaniMValizadehHFormulation, characterization and cytotoxicity studies of alendronate sodium-loaded solid lipid nanoparticlesColloids Surf B Biointerfaces20141175212824607519
- ShahRMMalherbeFEldridgeDPalomboEAHardingIHPhysicochemical characterization of solid lipid nanoparticles (SLNs) prepared by a novel microemulsion techniqueJ Colloid Interface Sci2014428828629424910064
- YangXLiuYLiuCZhangNBiodegradable solid lipid nanoparticle flocculates for pulmonary delivery of insulinJ Biomed Nanotechnol20128583484222888755
- BiRShaoWWangQZhangNSolid lipid nanoparticles as insulin inhalation carriers for enhanced pulmonary deliveryJ Biomed Nanotechnol200951849220055110
- VarshosazJGhaffariSMirshojaeiSFBiodistribution of amikacin solid lipid nanoparticles after pulmonary deliveryBiomed Res Int2013201313685923984315
- LiHZhaoXMaYZhaiGLiLLouHEnhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticlesJ Control Release2009133323824418951932
- WangJZhuRSunXZhuYLiuHWangSLIntracellular uptake of etoposide-loaded solid lipid nanoparticles induces an enhancing inhibitory effect on gastric cancer through mitochondria-mediated apoptosis pathwayInt J Nanomedicine2014983987399825187702
- JustSSievertFThommesMBreitkreutzJImproved group contribution parameter set for the application of solubility parameters to melt extrusionEur J Pharm Biopharm20138531191119923628829
- JoseSAnjuSSCinuTAAleykuttyNAThomasSSoutoEBIn vivo pharmacokinetics and biodistribution of resveratrol-loaded solid lipid nanoparticles for brain deliveryInt J Pharm20144741–261325102112
- DudhipalaNVeerabrahmaKPharmacokinetic and pharmacodynamic studies of nisoldipine-loaded solid lipid nanoparticles developed by central composite designDrug Dev Ind Pharm201541121968197725830370
- KumarMKakkarVMishraAKChuttaniKKaurIPIntranasal delivery of streptomycin sulfate (STRS) loaded solid lipid nanoparticles to brain and bloodInt J Pharm20144611–222323324286922
- NassimiMSchlehCLauensteinHDA toxicological evaluation of inhaled solid lipid nanoparticles used as a potential drug delivery system for the lungEur J Pharm Biopharm201075210711620206256
- VanditaKShashiBSantoshKGPalKIEnhanced apoptotic effect of curcumin loaded solid lipid nanoparticlesMol Pharm20129123411342123127155
- XuCFLiuYShenSZhuYHWangJTargeting glucose uptake with siRNA-based nanomedicine for cancer therapyBiomaterials20155111125770992
- DriscollKECostaDLHatchGIntratracheal instillation as an exposure technique for the evaluation of respiratory tract toxicity: uses and limitationsToxicol Sci2000551243510788556
- JeongJHanYPolandCAChoWSResponse-metrics for acute lung inflammation pattern by cobalt-based nanoparticlesPart Fibre Toxicol2015125131725967046
- HuoLChenRZhaoLSilver nanoparticles activate endoplasmic reticulum stress signaling pathway in cell and mouse models: the role in toxicity evaluationBiomaterials201561830731526024651
- HuXYangFFQuanLHPulmonary delivered polymeric micelles-pharmacokinetic evaluation and biodistribution studiesEur J Pharm Biopharm20148831064107525460153
- AssiniJMMulvihillEESutherlandBGNaringenin prevents cholesterol-induced systemic inflammation, metabolic dysregulation, and atherosclerosis in Ldlr−/− miceJ Lipid Res201354371172423269394
- ZhangCPengFLiuWNanostructured lipid carriers as a novel oral delivery system for triptolide: induced changes in pharmacokinetics profile associated with reduced toxicity in male ratsInt J Nanomedicine2014921049106324591827
- ZhangHZhangFMYanSJPreparation, in vitro release, and pharmacokinetics in rabbits of lyophilized injection of sorafenib solid lipid nanoparticlesInt J Nanomedicine2012762901291022787390
- ShahMAgrawalYHigh throughput screening: an in silico solubility parameter approach for lipids and solvents in SLN preparationsPharm Dev Technol201318358259022107345
- de JesusMBZuhornISSolid lipid nanoparticles as nucleic acid delivery system: properties and molecular mechanismsJ Control Release2015201311325578828
- NatarajanJVNugrahaCNgXWVenkatramanSSustained-release from nanocarriers: a reviewJ Control Release20141931112213824862321
- JiaYJiJWangFShiLYuJWangDFormulation, characterization, and in vitro/vivo studies of aclacinomycin A-loaded solid lipid nanoparticlesDrug Deliv201514719
- HaoJFangXZhouYDevelopment and optimization of solid lipid nanoparticle formulation for ophthalmic delivery of chloramphenicol using a Box-Behnken designInt J Nanomedicine20116468369221556343
- HuangXChenYJPengDYSolid lipid nanoparticles as delivery systems for gambogenic acidColloids Surf B Biointerfaces2013102239139723010123
- AndeyTSudhakarGMarepallySPatelABanerjeeRSinghMLipid nanocarriers of a lipid-conjugated estrogenic derivative inhibit tumor growth and enhance cisplatin activity against triple-negative breast cancer: pharmacokinetic and efficacy evaluationMol Pharm20151241105112025661724
- NaguibYWRodriguezBLLiXHurstingSDWilliamsRO3rdCuiZSolid lipid nanoparticle formulations of docetaxel prepared with high melting point triglycerides: in vitro and in vivo evaluationMol Pharm20141141239124924621456
- LiJGuoXLiuZPreparation and evaluation of charged solid lipid nanoparticles of tetrandrine for ocular drug delivery system: pharmacokinetics, cytotoxicity and cellular uptake studiesDrug Dev Ind Pharm201440798098723662696
- DasPJPaulPMukherjeeBPulmonary delivery of voriconazole loaded nanoparticles providing a prolonged drug level in lungs: a promise for treating fungal infectionMol Pharm20151282651266425941882
- AggarwalNGoindiSPreparation and in vivo evaluation of solid lipid nanoparticles of griseofulvin for dermal useJ Biomed Nanotechnol20139456457623621015
- WangSChenTChenRHuYChenMWangYEmodin loaded solid lipid nanoparticles: preparation, characterization and antitumor activity studiesInt J Pharm20124301–223824622465546
- SunJBiCChanHMSunSZhangQZhengYCurcumin-loaded solid lipid nanoparticles have prolonged in vitro antitumour activity, cellular uptake and improved in vivo bioavailabilityColloids Surf B Biointerfaces20131111136737523856543
- FanTChenCGuoHDesign and evaluation of solid lipid nanoparticles modified with peptide ligand for oral delivery of protein drugsEur J Pharm Biopharm201488251852824968819
- RamalingamPKoYTEnhanced oral delivery of curcumin from N-trimethyl chitosan surface-modified solid lipid nanoparticles: pharmacokinetic and brain distribution evaluationsPharm Res201532238940225082210