
Abstract
It is recognized that myeloid differentiation protein 2 (MD-2), a coreceptor of toll-like receptor 4 (TLR4) for innate immunity, plays an essential role in activation of the lipopolysaccharide signaling pathway. MD-2 is known as a neoteric and suitable therapeutical target. Therefore, there is great interest in the development of a potent MD-2 inhibitor for anti-inflammatory therapeutics. Several studies have reported that xanthohumol (XN), an anti-inflammatory natural product from hops and beer, can block the TLR4 signaling by binding to MD-2 directly. However, the interaction between MD-2 and XN remains unknown. Herein, our work aims at characterizing interactions between MD-2 and XN. Using a combination of experimental and theoretical modeling analysis, we found that XN can embed into the hydrophobic pocket of MD-2 and form two stable hydrogen bonds with residues ARG-90 and TYR-102 of MD-2. Moreover, we confirmed that ARG-90 and TYR-102 were two necessary residues during the recognition process of XN binding to MD-2. Results from this study identified the atomic interactions between the MD-2 and XN, which will contribute to future structural design of novel MD-2-targeting molecules for the treatment of inflammatory diseases.
Introduction
Inflammatory responses are triggered by pathogenic bacteria or toxin-mediated injury, which are mediated by pattern receptors expressed by immune cells.Citation1 However, the subsequent production of toxic proinflammatory molecules may result in undesirable outcomes, largely due to collateral tissue damage. Such uncontrolled inflammation plays an important role in the pathogenesis of diseases, such as cardiovascular diseases and sepsis.Citation2–Citation4 For instance, the extensively studied immunostimulatory component of bacteria, lipopolysaccharide (LPS), causes systemic inflammation and sepsis.Citation5–Citation7
LPS-induced inflammation is mediated by the toll-like receptor 4 (TLR4). In this pathway, LPS-binding protein captures LPS, transferring it to CD14 (cluster of differentiation 14)Citation8 for delivery to a coreceptor, myeloid differentiation protein 2 (MD-2).Citation9 TLR4 is a type-I transmembrane protein with a characteristic of horseshoe/solenoid-shaped ectodomain. MD-2, contains 160 amino acid residues, adopts a β-cup fold with two antiparallel β-sheets that forms a deep hydrophobic pocket for LPS recognition.Citation10,Citation11 LPS directly binds to the hydrophobic pocket in MD-2 protein. Upon LPS binding, the TLR4/MD-2 complex undergoes dimerization and recruits its downstream adaptors.Citation9,Citation10 Two major downstream adaptor molecules of MyD88 and TIR-domain-containing adapter-inducing interferon-β are triggered by the dimerization of two TLR4/MD-2 complexes, which then activates the intracellular proinflammatory cascades.Citation12 In general, MD-2 plays an indispensable role in the activation of the LPS-TLR4 signaling pathway and thus has been confirmed as a suitable target for the therapeutic inhibition of TLR4 signaling.Citation13,Citation14
The study of MD-2, as a target for anti-inflammatory therapy, has drawn great attention and a variety of MD-2 inhibitors have been reported, among which lipid IVa is a popular lipid-like antagonist. Lipid IVa, an underacylated lipid A with four fatty acid chains from Escherichia coli, is an intermediate in the biosynthetic pathway of LPS and can inhibit LPS with four acyl chains invading the hydrophobic pocket of MD-2.Citation15 In the meantime, some other natural and synthetic nonlipid compounds have also been tested. For example, JTT-705 and auranofin form covalent bonds with residue Cys-133 of MD-2, while compounds like taxanes and curcumin form noncovalent interactions embedded into the hydrophobic pocket of MD-2.Citation16–Citation18
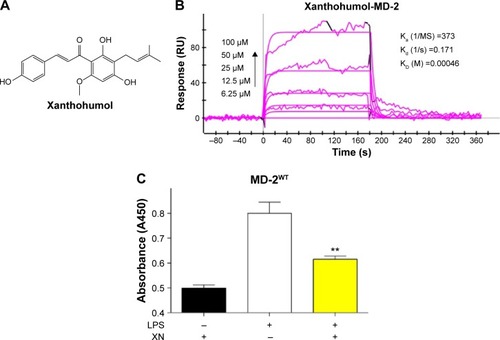
Xanthohumol (XN) () is a prenylated chalcone-type flavonoid of Humulus lupulus L. (Cannabaceae) and it is also used to make commercial flavoring products; XN has a variety of pharmacological functions including anti-inflammation, prevention and treatment of diabetes and antioxidation.Citation19–Citation21 Previous researchers suggested that XN’s anti-inflammatory activities and chemopreventive effects are attributed to blocking LPS binding to MD-2. Peluso et alCitation22 reported that XN can suppress LPS-stimulated inflammatory signaling through direct competitive binding to the hydrophobic cavity of MD-2. However, the binding mechanism of XN in MD-2 pocket are still unclear, preventing effective therapeutic design of XN targeting the MD-2 for the treatment of inflammatory diseases.Citation22 In the present study, we used both computational and experimental methods to explore the interaction between XN and MD-2. Our results indicated that XN can embed into the hydrophobic pocket of MD-2 and form two stable hydrogen bonds with residues ARG-90 and TYR-102.
Figure 1 XN’s binding activity to MD-2 protein.
Abbreviations: ELISA, enzyme-linked immunosorbent assay; LPS, lipopolysaccharide; MD-2, myeloid differentiation protein 2; SEM, standard error of the mean; SPR, surface plasmon resonance; XN, xanthohumol; Ka, association (‘on rate’); Kd, dissociation rates (‘off rate’); KD, equilibrium dissociation constant (‘binding constant’); WT, wild type.
Materials and methods
Reagents
The mouse RAW 264.7 macrophages were purchased from ATCC (Manassas, VA, USA). The recombinant human MD-2 (rhMD-2, or rhMD-2 mutant) proteins were purchased from Biowit Technologies (Shenzhen, People’s Republic of China); XN, LPS (from Salmonella typhosa); and the anti-MD-2 antibody from eBioscience (San Diego, CA, USA). No ethical approval was required for the use of these cell lines.
Surface plasmon resonance analysis
The binding affinity of XN to rhMD-2 and rhMD-2 mutants was determined using a ProteOn XPR36 Protein Interaction Array system (Bio-Rad Laboratories, Hercules, CA, USA) with an HTE sensor chip (ProteOn™, #176-5033). Briefly, MD-2 protein (in acetate acid buffer pH 5.5) was loaded to the sensors that were activated with 10 mM NiSO4. The XN samples (at 100, 50, 25, 12.5, and 6.25 µM) were prepared with running buffer (phosphate buffered saline [PBS], 0.1% sodium dodecyl sulfate, 5% dimethyl sulfoxide). Sensor and sample plates were placed on the instrument. The XN samples were then captured in flow cell 1, leaving the second flow cell as a blank. Five concentrations were injected simultaneously at a flow rate of 30 µM/min for 120 seconds of association phase, followed with 120 seconds of dissociation phase at 25°C. The final graphs were obtained by subtracting blank sensorgrams from the duplex or quadruplex sensorgrams. Data were analyzed with ProteOn manager software. KD (equilibrium dissociation constant [‘binding constant’]) was calculated by global fitting of the kinetic data from various concentrations of XN using 1:1 Langmuir binding model.
Enzyme-linked immunosorbent assay
A 96-well microplate was coated overnight at 4°C with anti-human MD-2 antibody in 10 mM Tris–HCl buffer (pH 7.5). The plate was washed with PBS Tween-20 and blocked with 3% bovine serum albumin in high purity water for 1.5 hours at room temperature. rhMD-2 (4 µg/mL) in 10 mM Tris–HCl buffer (pH 7.5) was added to a precoated plate and incubated for 1.5 hours at room temperature. After washing with PBS Tween-20, biotin-labeled LPS (InvivoGen, San Diego, CA, USA) was incubated for 1 hour at room temperature. Compound XN (0.1 or 1 µM) was added to the biotin-LPS before addition to the wells. After further washing streptavidin-conjugated horseradish peroxidase (Beyotime, Shanghai, People’s Republic of China) was added to the wells for 1 hour at room temperature. The horseradish peroxidase activity was determined using TMB substrate solution (eBioscience, San Diego, CA, USA). The optical density of each well was measured at 450 nm. Data were expressed as mean values (± standard deviation) of three separate experiments, each performed in duplication.
Molecular docking
The binding pose of probe XN in MD-2’s binding site was predicted by the software Glide (Schrödinger, Inc., NY, USA). The crystal structure of MD-2 in complex with lipid IVa was downloaded from the protein data bank (PDB code: 2E59) and prepared using the Protein Preparation Wizard in Maestro (Schrödinger, Inc.).Citation15 The process included removal of all nonbonded heteroatoms and water molecules. Hydrogen bonds were added and optimized to the structure. Other preparation steps involved removal of bad contents, optimization of bond lengths and creation of disulfide bonds. The XN was built by using Maestro and converted to a 3D structure from the 2D structure using LigPrep (Schrödinger, Inc.). The resulting structures were saved in Maestro format.
The crystal structure of MD-2 was used to generate grid with van der Waals radius scaling 1.00 and partial charge cut-off at 0.25. The scoring grid was generated by enclosing the residues 30 Å around lipid IVa in the binding site using Receptor Grid Generation (Schrödinger, Inc.), following the standard procedure. The receptor grid file and the prepared XN were docked using Glide standard precision, while the ligand sampling was set to be flexible. Glide score, an empirical docking scoring functions implemented by the Optimized Potentials for Liquid Simulations 2005 force field, was used to infer the affinity and further analyze the binding mode and molecular dynamics (MD) simulations.
MD simulations
MD simulations of XN/MD-2 complex system and apo MD-2 system were carried out, respectively. The initial XN/MD-2 complex came from the top ranked binding pose in the glide docking results. Before MD simulation, XN was extracted from the XN/MD-2 complex and the charge was prepared by gaussian09 and antechamber.Citation23–Citation25 Molecular mechanics parameters from the ff99SB and GAFF force fields were assigned to the protein and the ligand, respectively, using the LEaP module of AMBER (Assisted Model Building with Energy Refinement) 11 software packages.Citation26–Citation29 The MD-2 structure of apo MD-2 system was prepared by Protein Preparation Wizard of Maestro. All the water molecules and the specified ligand were deleted. The two systems were all solvated in a box of TIP3P water molecules with a hydration shell of 10 Å. In addition, an appropriate number of chloride ions were used to neutralize these two systems.
MD simulation was carried out with AMBER 11 software packages and the two systems were using the same protocol.Citation30,Citation31 Prior to the MD productive simulation, we performed an equilibration protocol consisting of an initial minimization of the water box of 1,000 steps, 500 steps for the steepest descent and 500 steps in the conjugate gradient. Then, the TIP3P water box was heated at constant volume until 300 K using a time constant for the heat bath with a coupling time of 100 ps. Equilibration was at 300 K and constant pressure of 100 ps. Before the production of MD, the whole system was equilibrated at 100 ps at a constant pressure of 1 bar and turned on the Langevin temperature scaling with a collision frequency of 2 ps. The last step used 50 ns of MD simulation without any restraints. Furthermore, nonbonded interactions were cut off at 8.0 Å. Periodic boundary conditions were turned on in every step of the whole process.Citation32 Particle mesh Ewald was also performed to deal with the long-range electrostatic interactions under periodic boundary conditions.Citation33 The SHAKE method was used to constrain hydrogen atoms and the time step was set to 2 fs.Citation34,Citation35 The coordinates were saved every 10 ps for the subsequent analysis.
Results and discussion
Determination of the direct binding of XN to MD-2
Previous studies reported that MD-2 is a potential target for XN but the binding mode of XN to MD-2 is still unknown.Citation22 In this study, we performed biochemical and computational experiments to determine the molecular interactions between MD-2 and XN. At the beginning, surface plasmon resonance (SPR) experiments were carried out to assess the binding affinity of XN to MD-2. As shown in , XN bound to the MD-2 protein in a dose-dependent manner with a relatively high affinity with a KD value of 0.00046 M. Furthermore, using a biotin–streptavidin-based enzyme-linked immunosorbent assay (ELISA) system, we found that biotin-tagged LPS (biotin-LPS) bound rhMD-2 previously coated onto plates, while coincubation with XN significantly reduced binding of biotin-LPS with rhMD-2 (). These results suggest that XN has the same binding site as LPS with a higher binding affinity.
The binding mode of XN to MD-2
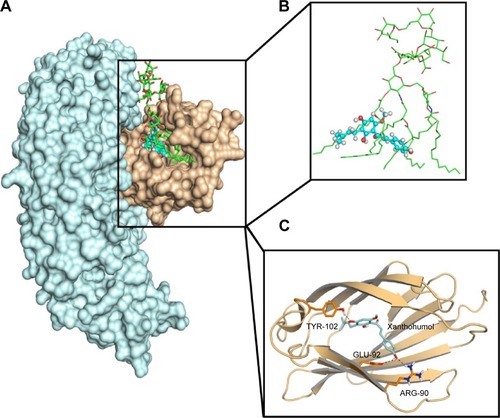
Molecular docking was used to analyze the binding mode of XN to MD-2. Docking results showed that XN could be inserted into the large hydrophobic binding pocket of MD-2 and easily occupy a large portion of the LPS binding site (), which was consistent with our previous experimental results. In addition, other important interactions to MD-2 were also observed such as hydrogen bonding to residues ARG-90, GLU-92, and TYR-102 (). As with previous study, XN and related prenylated flavonoids had the same interactions with the TYR-102 residue, underscoring the role of TYR-102 in XN’s binding to MD-2. Our results were similar with the previously reported study. Additionally, based on the molecular docking results, we selected a docked complex that interacted with TYR-102 with lowest binding energy.
Figure 2 Molecular docking analysis of XN to the activity cavity of MD-2.
Abbreviations: LPS, lipopolysaccharide; MD-2, myeloid differentiation protein 2; TLR4, toll-like receptor 4; XN, xanthohumol.
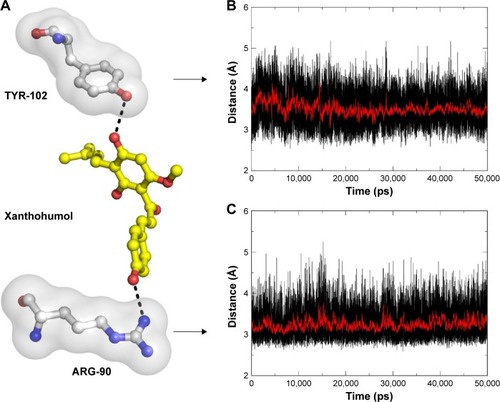
The stability of the docked complex
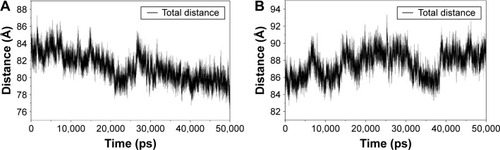
The results from the above docking analysis lead us to explore the dynamic behavior of the XN/MD-2 complex. In order to confirm the stability of docked conformation and to probe the function of XN on MD-2, conventional MD simulations on XN/MD-2 complex and apo MD-2 were conducted. For the initial structure as reference, the root-mean square deviation (RMSD) was detected for all Cα atoms in the proteins and all heavy atoms of XN. From our study, the trajectories that we established were more stable after 10 ns, therefore, only the latter part of the trajectories were considered for further analysis. As shown in , after 10 ns of MD simulation, the RMSD values of Cα atoms for MD-2 and XN gradually stabilized to 3.0 and 0.4 Å, respectively. However, the apo MD-2 converged the RMSD values in the system at 3.5 Å after 10 ns simulation (). Therefore, the above results suggest that two systems became stable in the end. Then, we extracted the initial and last snapshots from MD trajectories of two systems to verify the conformation change of the two systems. As depicted in , the XN/MD-2 complex should be stable; the docked XN/MD-2 complex in the end snapshot of the MD simulation structure and the initial structure were examined to show that they overlapped with slight positional difference. But the apo MD-2 showed a significant deviation until the end of the simulation, resulting in a backbone RMSD of about 3.5 Å that revealed a greatly changed conformation. What’s more, to quantify the “openness” of the hydrophobic cavity of MD-2, we cited a collective distance (Dtotal) defined as the total distance between the center of mass of MD-2 and to each center of mass of β strand.Citation36 In , the time evolution of Dtotal for apo MD-2 as well as the values of Dtotal became smaller about 5 Å than from the beginning (). On the contrary, in , values of Dtotal of XN/MD-2 complex increased slightly, which indicate that XN can stabilize the XN/MD-2 complex (). Garate and OostenbrinkCitation36 also had previously reported the same result with the simulated apo MD-2 and showed that the cavity of MD-2 closes rapidly in the absence of any ligand, indicating a strong hydrophobic nature and the instability of apo MD-2. Therefore XN not only directly binds to MD-2, but also plays a crucial role in the hydrophobic cavity of the MD-2 conformation.
Figure 3 Backbone RMSDs are shown as a function of time for apo MD-2 and XN/MD-2 complex structures at 50 ns.
Abbreviations: MD-2, myeloid differentiation protein 2; RMSD, root-mean square deviation; XN, xanthohumol.
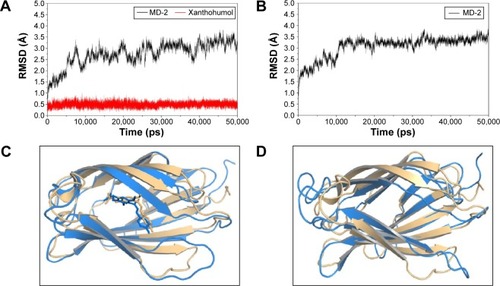
Figure 4 The “openness” of apo MD-2 and XN/MD-2 complex.
Abbreviations: MD-2, myeloid differentiation protein 2; XN, xanthohumol.
The main driving force for XN/MD-2 binding is a combination of several strong hydrogen bonds, which plays an important role in the protein-ligand recognition process. An analysis of the hydrogen bonds was then carried out to explore the interactions between MD-2 and XN where the most frequent hydrogen bonding partner was considered. As shown in , the distance between atoms in the docked complex, which form hydrogen bonds, was monitored during the whole MD simulation process and plotted. As a result, vast profiles of hydrogen bonds were observed, which fluctuated from 2.5 to 4.5 Å with an average of 3.0–3.5 Å hydrogen bonds in the XN/MD-2 complex. Regarding the XN/MD-2 complex, amidogens of ARG-90 and the hydroxyl of TYR-120 formed hydrogen bonds with hydroxyl and methoxy groups of XN, respectively, but ARG-90 was more stable than TYR-120 (). Moreover, although the distance between the two mentioned hydrogen bonds fluctuated slightly, they stabilized slightly with a distance ~3–3.5 Å in 50 ns of MD simulation. The detailed distances of evolutions in the two hydrogen bonds are illustrated in . Lastly, it is unknown as to why the hydrogen bond between GLU-92 and XN disappeared rapidly compared to the docking complex. This may be related to an inadequate amount of attraction.
Figure 5 Key hydrogen interactions between XN and residues ARG-90 and TYR-102 of MD-2.
Abbreviations: MD-2, myeloid differentiation protein 2; XN, xanthohumol.
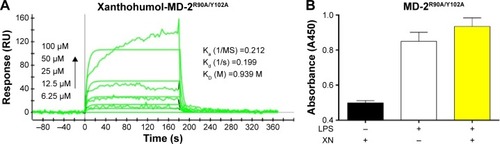
The weak interaction between XN and MD-2R90A/Y102A mutant
Based on the computational results in previous studies, we hypothesized that if residues ARG-90 and TYR-102 transform into alanine, the binding affinity may decline. To test our hypothesis, we analyzed the double-mutant protein MD-2R90A/Y102A using SPR to determine whether these two residues would affect the binding affinity of XN to MD-2 (). We found that the KD of XN bound to MD-2R90A/Y102A was 0.939 M, which was >2,000 times higher than that of the wild-type MD-2. In addition, ELISA assay showed that XN was not able to prevent the binding of biotin-LPS with the MD-2R90A/Y102A mutant (). Furthermore, the side chains in the mutation of MD-2 R90A/Y102A were unable to attract hydrogen bonds to XN, indicating that ARG-90 and TYR-102 were the key amino acids on MD-2 that bind XN. These two results further support the notion that the residues ARG-90 and TYR-102 are essential to the recognition process of XN binding to MD-2 proteins.
Figure 6 XN shows low binding affinity to the MD-2R90A/Y102A mutant.
Abbreviations: ELISA, enzyme-linked immunosorbent assay; LPS, lipopolysaccharide; MD-2, myeloid differentiation protein 2; SPR, surface plasmon resonance; XN, xanthohumol; Ka, association (‘on rate’); Kd, dissociation rates (‘off rate’); KD, equilibrium dissociation constant (‘binding constant’).
Conclusion
In this report we described the interaction mode between XN and MD-2 protein. SPR and ELISA experiments were performed to assess the binding affinity and the binding mode of XN to the MD-2 wild-type protein. We applied molecular docking and molecular dynamic simulations to model the dynamic behavior of molecules and macromolecular assemblies at an atomic level. Moreover, the MD-2 site-directed mutants were used to verify the residues of ARG-90 and TYR-102, both of which are crucial for XN to compete with LPS. We found that XN formed two stable hydrogen bonds to ARG-90 and TYR-102 of MD-2, stabilizing the complex. We put forward a strategy to increase hydrophobic property and add hydrogen bond donors or hydrogen bond acceptors for the purpose of forming hydrogen bonds with GLU-92, which was mentioned in the docking study. What’s more, we can retain some pharmacophores and employ scaffold hopping method, known as lead hopping, to discover novel potent MD-2 inhibitors.Citation37,Citation38 In conclusion, by combining theoretical and experimental approaches, we characterized a structure-based drug design of a potential anti-inflammatory inhibitor by targeting the MD-2 protein in the TLR4 signaling pathway.
Acknowledgments
Financial support was provided by the National Natural Science Funding of China (21272179, 81473242, and 81570027), and Zhejiang Provincial Natural Science Foundation (LY14C050004).
Supplementary materials
Video S1 50 ns of apo MD-2 protein molecular dynamics simulation.
Abbreviation: MD-2, myeloid differentiation protein 2.
https://www.youtube.com/watch?v=xiehyaDcvYw
Video S2 50 ns of xanthohumol/MD-2 complex molecular dynamics simulation.
Abbreviation: MD-2, myeloid differentiation protein 2.
Disclosure
The authors report no conflicts of interest in this work.
References
- HechtGInnate mechanisms of epithelial host defense: spotlight on intestineAm J Physiol19992773 Pt 1C351C35810484321
- RossRAtherosclerosis – an inflammatory diseaseN Engl J Med199934021151269887164
- SacksGPStudenaKSargentKRedmanCWNormal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsisAm J Obstet Gynecol1998179180869704769
- BergAHSchererPEAdipose tissue, inflammation, and cardiovascular diseaseCirc Res200596993994915890981
- MartinezFOSicaAMantovaniALocatiMMacrophage activation and polarizationFront Biosci20081345346117981560
- ReiterRJMelchiorriDSewerynekEA review of the evidence supporting melatonin’s role as an antioxidantJ Pineal Res19951811117776173
- RaetzCRHWhitfieldCLipopolysaccharide endotoxinsAnnu Rev Biochem20027163570012045108
- WrightSDRamosRATobiasPSUlevitchRJMathisonJCCD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding proteinScience19902494975143114331698311
- ParkSHKimNDJungJKLeeCKHanSBKimYMyeloid differentiation 2 as a therapeutic target of inflammatory disordersPharmacol Ther2012133329129822119168
- ParkBSSongDHKimHMChoiBSLeeHLeeJOThe structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complexNature200945872421191119519252480
- ShimazuRAkashiSOgataHMD-2, a molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4J Exp Med1999189111777178210359581
- ZhangHTayPNCaoWLiWLuJIntegrin-nucleated toll-like receptor (TLR) dimerization reveals subcellular targeting of TLRs and distinct mechanisms of TLR4 activation and signalingFEBS Lett20025321–217117612459484
- ChristiansenDBrekkeOLStenvikJLambrisJDEspevikTMollnesTEDifferential effect of inhibiting MD-2 and CD14 on LPS-versus whole E. coli bacteria-induced cytokine responses in human bloodAdv Exp Med Biol201294623725121948372
- RohELeeHSKwakJAMD-2 as the target of nonlipid chalcone in the inhibition of endotoxin LPS-Induced TLR4 activityJ Infect Dis201120371012102021402551
- OhtoUFukaseKMiyakeKSatowYCrystal structures of human MD-2 and its complex with antiendotoxic lipid IVaScience200731658311632163417569869
- Mancek-KeberMGradisarHInigo PestanaMMartinez de TejadaGJeralaRFree thiol group of MD-2 as the target for inhibition of the lipopolysaccharide-induced cell activationJ Biol Chem200928429194931950019473973
- ResmanNGradisarHVaslJKeberMMPristovsekPJeralaRTaxanes inhibit human TLR4 signaling by binding to MD-2FEBS Lett2008582283929393418977229
- GradisarHKeberMMPristovsekPJeralaRMD-2 as the target of curcumin in the inhibition of response to LPSJ Leukoc Biol200782496897417609337
- ZhangXLZhangYDWangTGuoHYLiuQMSuHXEvaluation on antioxidant effect of xanthohumol by different antioxidant capacity analytical methodsJ Chemistry2014201416
- KiyofujiAYuiKTakahashiKOsadaKEffects of xanthohumol-rich hop extract on the differentiation of preadipocytesJ Oleo Sci201463659359724829131
- ChoYCKimHJKimYJDifferential anti-inflammatory pathway by xanthohumol in IFN-gamma and LPS-activated macrophagesInt Immunopharmacol20088456757318328448
- PelusoMRMirandaCLHobbsDJProteauRRStevensJFXanthohumol and related prenylated flavonoids inhibit inflammatory cytokine production in LPS-activated THP-1 monocytes: structure-activity relationships and in silico binding to myeloid differentiation protein-2 (MD-2)Planta Med201076141536154320309792
- BurgerSKLacasseMVerstraelenTDrewryJGunningPAyersPWAutomated parametrization of AMBER force field terms from vibrational analysis with a focus on functionalizing dinuclear zinc(II) scaffoldsJ Chem Theory Comput20128255456226596604
- KhavrutskiiIVLeglerPMFriedlanderAMWallqvistAA reaction path study of the catalysis and inhibition of the Bacillus anthracis CapD gamma-glutamyl transpeptidaseBiochemistry201453446954696725334088
- MuddanaHSSapraNVFenleyATGilsonMKThe SAMPL4 hydration challenge: evaluation of partial charge sets with explicit-water molecular dynamics simulationsJ Comput Aided Mol Des201428327728724477800
- BeauchampKALinYSDasRPandeVSAre protein force fields getting better? a systematic benchmark on 524 diverse NMR measurementsJ Chem Theory Comput2012841409141422754404
- WickstromLOkurASimmerlingCEvaluating the performance of the ff99SB force field based on NMR scalar coupling dataBiophys J200997385385619651043
- HornakVAbelROkurAStrockbineBRoitbergASimmerlingCComparison of multiple amber force fields and development of improved protein backbone parametersProteins200665371272516981200
- WangJWolfRMCaldwellJWKollmanPACaseDADevelopment and testing of a general amber force fieldJ Comput Chem20042591157117415116359
- CaseDACheathamTEDardenTThe Amber biomolecular simulation programsJ Comput Chem200526161668168816200636
- Salomon-FerrerRCaseDAWalkerRCAn overview of the Amber biomolecular simulation packageWiley Interdisciplinary Rev: Comput Mol Sci201332198210
- Munoz-GarciaJCCorzanaFde PazJLAnguloJNietoPMConformations of the iduronate ring in short heparin fragments described by time-averaged distance restrained molecular dynamicsGlycobiology201323111220122923903025
- SaguiCDardenTAMolecular dynamics simulations of biomolecules: long-range electrostatic effectsAnnu Rev Biophys Biomol Struct19992815517910410799
- HessBP-LINCS: a parallel linear constraint solver for molecular simulationJ Chem Theory Comput20084111612226619985
- HessBBekkerHBerendsenHJCFraaijeJLINCS: a linear constraint solver for molecular simulationsJ Comput Chem1997181214631472
- GarateJAOostenbrinkCLipid a from lipopolysaccharide recognition: structure, dynamics and cooperativity by molecular dynamics simulationsProteins201381465867423184816
- MartinYCMuchmoreSBeyond QSAR: lead hopping to different structuresQSAR Combinator Sci2009288797801
- SchneiderGSchneiderPRennerSScaffold-Hopping: how far can you jump?QSAR Combinator Sci2006251211621171