
Abstract
Purpose
The crosstalk between hepatocellular carcinoma (HCC) cells and hepatic stellate cells (HSCs) is one of the important mechanisms of liver cancer metastasis. The relationship between liver cancer metastasis and glycolysis has been extensively studied recently. However, the role of von Willebrand factor (vWF) mediated glycolysis mechanism in liver cancer metastasis is currently unknown.
Methods
Western blot was used to verify the expression of vWF in HCC cells. PAS staining, glycogen and L-lactate content assays were used to reflect cellular glycolysis levels. The ability of cell migration was explored by Wound-healing and Transwell assays. Besides, the effect of vWF on the progression of HCC in vivo was also studied using subcutaneous xenograft model.
Results
vWF derived from HCC cells promoted tumor migration by mediating glycolysis. Besides, vWF participated in the crosstalk between HCC cells and HSCs. HCC cells activated HSCs through vWF-mediated TGFB1 expression and secretion, and activated HSCs upregulated vWF expression in HCC cells through IL-6 secretion feedback. Further, in vitro and in vivo experiments also confirmed the importance of the JAK1/vWF/TGFB1 axis in regulating HSCs-derived IL-6 mediated HCC migration and growth.
Conclusion
In summary, this article demonstrated that IL-6 released from hepatic stellate cells enhanced glycolysis and migration ability of liver cancer cells by activating JAK1/vWF/TGFB1 axis which may also be a potential target for inhibiting liver cancer metastasis.
Keywords:
Introduction
According to current information, primary liver cancer is the sixth most common cancer worldwide in 2020 and the third-leading cause of cancer-related death.Citation1 As one of the types of primary liver cancer, hepatocellular carcinomas (HCCs) account for 70% to 90% of cases around the world.Citation2 Even though great efforts have been made to develop methods for curing HCC during past decades,Citation3 the high recurrence rate has remained a big problem in achieving better outcomes. This is mainly due to the high possibility of intrahepatic metastasis and postoperative recurrence.Citation4 Therefore, elucidating the mechanism of HCC metastasis and recurrence is of great significance.
Tumor microenvironment (TME) consists of multiple cells and cytokines presenting different functions.Citation5 In the TME of HCC, hepatic stellate cells (HSCs) are the main part of stromal cells.Citation6 During HCC progression, cytokine-producing HSCs are activated into myofibroblastic HSCs.Citation7,Citation8 This dynamic shift in HSC subpopulations during chronic liver disease ultimately leads to an irreversible situation. Previous studies have discovered that HSCs could aggravate HCC progression through crosstalk mechanisms.Citation9 For instance, platelet-derived growth factor B (PDGFB),Citation10 C-X-C motif chemokine ligand 1 (CXCL1)Citation11 and hexokinase 1 (HK1)Citation12 released from HSCs could regulate HCC progression by mediating different pathways. Moreover, Interleukin (IL)-6, a major proinflammatory cytokine, released from HSCs has also been demonstrated contributing to HCC progression through directCitation13 or indirectCitation14 interaction with HCC cells. Given the drug resistance during tumor therapy and the diversity of tumor treatment methods, further research is still needed to find the mechanisms mediating interactions between HCC cells and HSCs.
As a multi-step process, solid tumor metastasis can be approximately divided into five parts: (1) invasion of the basement membrane and cell migration; (2) infiltration within surrounding vasculature or lymphatic system; (3) survival in the circulation; (4) extravasation from vasculature to secondary tissue; (5) colonization at secondary sites.Citation15 In this complex process, the von Willebrand factor (vWF) plays a pivotal role in promoting tumor cell metastasis. As a multimeric procoagulant plasma glycoprotein,Citation16 vWF was proven to originate from cancer cells de novoCitation17,Citation18 which was independent of the source of endothelial cells and megakaryocytes.Citation19 The tumor metastasis and glycolysis-promoting function of vWF were reported in a variety of cancer types such as gastric adenocarcinoma,Citation17 lung adenocarcinoma,Citation18 and osteosarcoma.Citation20 Previous studies have found that vWF enhanced HCC invasion and migration through phosphorylated STAT3-mediated MMP2 and MMP9 expression.Citation21 vWF has been proven to promote glycogen metabolism in lung adenocarcinoma cells through upregulating PHKG1 expression.Citation18 However, the function of vWF in HCC glycolysis remains largely unknown. Besides, previous studies elucidating the mechanisms of the pro-migration function of vWF mainly focused on the hematological metastasis of tumor cells (steps 3 and 4). Thus, little is known about whether vWF can exert its function in the process of invasion into the vasculature (steps 1 and 2) and the underlying mechanism remains to be clarified.
In this study, we found that vWF can be expressed by HCC cells and regulated by JAK1 signaling activating by the IL-6 released from HSCs. HCC-derived vWF could exert its role in glycolysis promotion and tumor metastasis aggravation by regulating TGFB1-mediated crosstalk between HCC cells and HSCs. Our findings provided new insight into vWF function in the initial step of tumor metastasis and implied that vWF may hopefully become a potential target for HCC therapy.
Materials and Methods
Cell Lines and Cell Culture
HepG2 and SMMC-7721 cell lines were obtained from the Type Culture Collection of the Chinese Academy of Sciences. Hep3B and LX-2 cell lines were purchased from EK-Bioscience. All cell lines were cultured in DMEM medium with 10% fetal bovine serum (Yeasen) and 100 U/mL penicillin/streptomycin (Invitrogen Corporation) in a 37 °C incubator with 5% CO2.
Co-Culture Systems
Six-well Transwell chambers with 0.4 μm polyester membranes (14112, Labselect) were used for cell co-culture experiments. In the experiment studying the effect of hepatic stellate cells on liver cancer cells, 5 × 105 HepG2/SMMC-7721 cells were added to the lower chamber one day before co-culturing, and 1.5 × 105 LX-2 cells were added to the upper chamber after tumor cell adhesion. On the contrary, when studying the effect of liver cancer cells on hepatic stellate cells, 3 × 105 LX-2 cells were added to the lower chamber on the day before co-culturing, and 2.5 × 105 HepG2/SMMC-7721 cells were added to the upper chamber after LX-2 cell adhesion. After co-culturing for 48 hours, lower chamber cells or supernatant were collected for subsequent experimental analysis.
Vector Construction and Transfection
To knockdown vWF in HCC cells, vWF shRNA was cloned into pLent-U6-GFP-Puro vector (Vigene, Maryland, USA). The vWF pcDNA (pcDNA3.1-WT-vWF) was purchased from Addgene (catalog no.124794, USA). To knockdown TGFB1 and TGFBR2, TGFB1 and TGFBR2 shRNA were cloned into pLent-U6-GFP-Puro vector (Vigene, Maryland, USA). Vector sequences used in this article were listed in Supplementary Table S1. Transfections were performed using Lipo8000TM (C0533, Beyotime, Shanghai, China). For stable vWF overexpression, TGFB1 knockdown HepG2 cells and TGFBR2 knockdown LX-2 cells, the plasmids and lentivirus vectors were transfected into cells, and positive cells were selected by puromycin (MS0011, Maokang Bio, Shanghai, China).
PAS Staining Assay
According to the protocol of the PAS staining kit (G1360, Solarbio), cells were fixed with 70% ethanol for 20 minutes, then discarded the ethanol and air dried them, placed cells in oxidant solution and reacted in the dark for 10 minutes. Cells were washed with ultrapure water, then added Schiff staining solution and reacted in a dark place for 10 minutes. After washing and drying, re-dyed with hematoxylin for 1 minute. Finally, cells were washed with ultrapure water, and images were recorded under a 10 × microscope.
Wound-Healing Assay
When the cell density in the six-well plates reached 100%, utilized a 200 μL pipette tip to create vertical scratches. After cleaning cells twice with PBS, added the serum-free and antibiotic-free culture medium, and then recorded the images with a 5 × microscope at 0 h and 24 h, respectively. The cell migration rate was calculated by Image J software.
Transwell Assay
The pre-treated cells were digested with trypsin and resuspended in a serum-free culture medium. Added 200 μL of cell suspension containing 2 × 104 cells into the upper Transwell chambers (354234, Corning) and added 500 μL culture medium with 10% FBS to the lower chambers. Incubated cells in a 37 °C incubator with 5% CO2 for 48 h, and then removed the chambers and fixed cells with methanol for 20 minutes. After cleaning twice with PBS, stained cells with 5% crystal violet for 5 minutes. After washing off the floating color with PBS, damp cotton swabs were utilized to wipe off the cells on the inner side of the chambers. Observed and recorded the images under a 10 × microscope. Afterward, methanol was used to dissolve the crystal violet of the cells on the lower side of the chambers, and the absorbance was measured at the wavelength of 570 nm for quantification.
Glycogen Content Assay
Collected 5 × 106 cells and follow the protocol of commercial glycogen assay kit (BC0340-50, Solarbio) to quantify the glycogen content in the cells.
L-Lactate Assay
Collected 5 × 106 cells and follow the protocol of commercial L-lactate assay kit (D799851-0050, Sangon Biotech) to quantify the L-lactate content in the cells.
ELISA Assay
Collected the cell supernatant and centrifuged it for 20 minutes under conditions of 4 °C and 1000 g, then took the supernatant for testing. Followed the manufacturer instructions of IL-6 (E-EL-H6156, Elabscience) and TGFB1 (JN22165, Jining Shiye) ELISA kits to quantify protein content in cell medium.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
The total RNA of cells was extracted using Trizol reagent (Invitrogen). cDNA was synthesized from 500 ng total RNA using Hifair® II 1st Strand cDNA Synthesis Kit (11119ES60, Yeasen). qPCR was performed using Hieff UNICON® qPCR SYBR Green Master Mix (11198ES08, Yeasen). All the procedures followed the manufacturer instructions. Total target gene levels were normalized with glycerol triphosphate dehydrogenase (GAPDH), and all results were calculated using the 2−ΔΔCT method. The sequences of former and reverse primers were listed in Supplementary Table S2.
Western Blot
Pre-cold RIPA buffer and protein inhibitors were added to the cell lysate (5 × 106 cells). After putting it at 4 °C for 20 minutes, cell lysate was centrifuged at 12000 rpm for 20 minutes. Then, the supernatant was collected, and protein concentration was determined by the Bradford method. Twenty microgram of protein was loaded and separated on SDS-PAGE and electrotransferred it to a polyvinylidene difluoride membrane. Five percent BSA was used for 2 hours to block membranes before they were blotted overnight with primary antibodies at 4 °C. After incubating with HRP-conjugated antibody at room temperature for 60 minutes. The binding images of target proteins and antibodies were recorded by Tanon Image System (5200S). The antibodies used in this article were listed in Supplementary Table S3.
Immunohistochemical Staining
Tumor tissue sections were deparaffinized in xylene and hydrated in ethanol. Three percent H2O2 was added to eliminate endogenous peroxidase activity. Immersed the slices in 10 mM citrate buffer (pH 6.0) and boiled it for 20 min. After cooling, the sections were blocked with 3% BSA for 1 h, and then slices were incubated with primary antibodies at 4 °C overnight. After washing three times in PBS, slices were incubated with biotin-labeled goat anti-rabbit IgG (Biological Technology, California, USA) for 1 h at room temperature. Added an appropriate amount of streptavidin-peroxidase complex (SABC, Biological Technology) and incubated at 37 °C for 30 min. After washing with PBS, the freshly prepared 3.3′-diaminobenzidine tetrahydrochloride (DAB, Biological Technology) solution was added and then added hematoxylin for about 30s. After the slices were dehydrated, photographs were recorded under a brightfield microscope.
Animal Experiments
Balb/c nude mice were obtained from JSJ laboratories. Five 6-week-old Balb/c nude mice were randomly assigned to each group. 1×107 HepG2 and 1×106 LX-2 cells with different treatments were subcutaneously co-injected into the left flanks of nude mice. Animals were euthanized by CO2 asphyxiation after 4 weeks, and tumors were saved at −80 °C for subsequent assays. Animal experiments were conducted in mice using protocols approved by the IACUC of East China University of Science and Technology [grant number ECUST-2021-06002]. Animal husbandry protocols followed the Declaration of Helsinki.
Statistical Analysis
Statistical analyses were carried out by GraphPad Prism 9.4.0 software. All the experiments were repeated at least three times and each value represented the mean ± SD. Student’s t-test (two-tailed) was used to evaluate the statistical significance between the two groups. P < 0.05 was considered statistically significance.
Results
vWF Derived from HCC Cells Promotes Tumor Migration by Inducing Glycolysis in vitro
Our previous research has demonstrated that vWF derived from lung adenocarcinoma (LUAD) cells de novo could mediate glycolysis in LUAD cells.Citation17,Citation18 Here, we collected hepatocellular carcinoma gene expression data from the GEPIA database (http://gepia.cancer-pku.cn/) and found that vWF was highly expressed in the hepatocellular carcinoma tissues (Supplementary Figure S1A). Western blotting data also confirmed that vWF was expressed in HepG2, SMMC-7721 and Hep3B cell lines ().
Figure 1 vWF derived from HCC cells promotes tumor migration through inducing glycolysis in vitro. (A) The expression levels of vWF in L-02, HUVEC and HCC cell lines. (B–D) PAS staining, glycogen and L-lactate content of vWF overexpression and knockdown HCC cells. ***P<0.001, compared with vWF psilence group; #P<0.05, ###P<0.001, compared with vWF pcDNA group. (E and F) Wound-healing and Transwell assays revealed the migration ability of HCC cells under the treatment of vWF OE and 0.5 μM CP-91149.
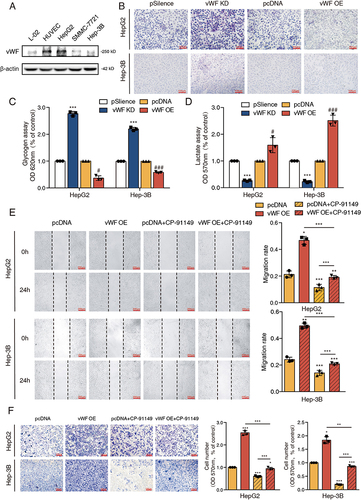
To explore whether vWF could mediate tumor cell migration through inducing glycolysis, we first constructed vWF overexpression (OE) and knockdown (KD) plasmids and confirmed their efficiency in HepG2 and SMMC-7721 cell lines by Western blot (Supplementary Figure S1B and S1C). The PAS staining and glycogen assay Results showed the accumulation of glycogen in HCC cells after vWF knocking down while the consumption of glycogen when vWF was overexpressed ( and ). Besides, the opposite results of L-lactate content in HCC cells after transfecting vWF KD and OE plasmids further confirmed that vWF could regulate glycolysis in HCC cells (). Then, we combined glycogen phosphorylase inhibitor CP-91149 (HY-13525, MCE, Shanghai, China) with vWF OE to explore the relationship between migration and glycolysis induced by vWF. Wound-healing and Transwell assays revealed that the attenuated migration ability of HCC cells treated with 0.5 μM CP-91149 was restored after transfecting the vWF OE plasmid ( and ). These data suggested that vWF derived from HCC cells could aggravate tumor cell migration by regulating glycolysis in vitro.
vWF Mediates the Crosstalk Between HCC Cells and HSCs
Hepatic stellate cells are the main part of stromal cells in the HCC tumor microenvironment.Citation8 Interestingly, in GEPIA database, we found that the expression level of ACTA2 (also called α-SMA), a classic biomarker for activation of hepatic stellate cells, was correlated with vWF expression in HCC tumor tissues (). Besides, we constructed nude mice subcutaneous xenograft model by co-injecting 1 × 107 HepG2 cells and 1 × 106 LX-2 cells subcutaneously into nude mice. The results of solid tumor immunohistochemistry indicated that α-SMA was highly expressed and distributed around the tumor area. Simultaneously, vWF was highly expressed in tumor regions ().
Figure 2 vWF mediates the crosstalk between HCC cells and HSCs. (A) The expression correlation of vWF and ACTA2 in HCC tissues. (B) The distribution area of α-SMA and vWF in tumor region. Arrows point to feature areas. (C) vWF expression levels of HCC cells with or without LX-2 co-culturing for 48 h. (D) The influence of co-culturing with HCC cells expressing different vWF levels to the activation of LX-2 cells. (E and F) Migration ability of HCC cells expressing different vWF levels with LX-2 co-culturing for 48 h reflected by Wound-healing and Transwell assays.
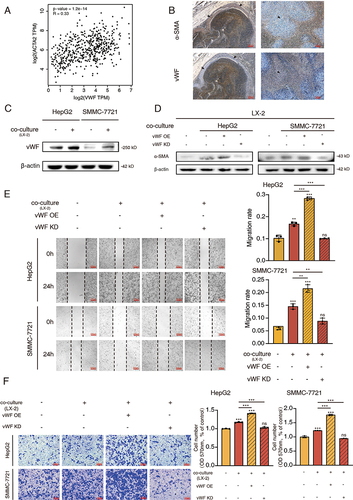
To investigate whether vWF could participate in the interactions between HCC cells and HSCs, co-culture systems were utilized for further research. As shown in , vWF was significantly upregulated in HCC cells co-culturing with LX-2 cells. Similarly, the α-SMA level of LX-2 cells was upregulated while co-culturing with HCC cells and enhanced or attenuated expression of α-SMA was observed, respectively, when vWF was overexpressed or knocked down in the co-culturing HCC cells (). Besides, Wound-healing and Transwell assays also demonstrated that co-culture with LX-2 cells could enhance the migration ability of HCC cells. This enhanced migration ability can be further improved or reduced when the co-culturing HCC cells were transfected with vWF OE or KD plasmids ( and ). All the results indicated that vWF plays a crucial role in the communications of HCC cells and HSCs.
IL-6/JAK1 Signaling Pathway Activated by Co-Culture System Regulates vWF-Mediated Glycolysis and Migration of HCC Cells
As proven in the former part, vWF could participate in the crosstalk of HCC cells and HSCs, thus mediating their bioprocess. Therefore, we then wondered how HSCs regulated migration and glycolysis of HCC cells through mediating vWF expression. Firstly, we conducted heatmap and GSEA analysis by recruiting the GSE62455 dataset containing the mRNA expression data of HCC cells with or without HSCs co-culturing. The results of the heatmap showed that many proteins correlating with the activation of JAK/STAT signaling pathway were significantly upregulated after HSCs co-culturing. The GSEA enrichment score of JAK/STAT signaling pathway further demonstrated this finding ().
Figure 3 IL-6/JAK1 signaling pathway activated by co-culture system regulates vWF mediated glycolysis and migration of HCC cells. (A) Heatmap results (left) of the top 100 differentially expressed genes in GSE62455 dataset and GSEA results (right) of JAK/STAT signaling pathway. (B) IL-6 expression levels of LX-2 cells with or without HCC cells co-culturing for 48 h. (C) vWF expression levels in HepG2 cells under the treatment of gradient concentrations of exogenous IL-6 for 48 h. (D) vWF expression levels in HCC cells with or without 3 μM pyridone 6 treatment for 48 h. (E) The phosphorylation levels of JAK1 in HCC cells with or without LX-2 cells co-culturing for 48 h. (F) Glycogen content of HepG2 cells treating with vWF OE and pyridone 6 detected by PAS staining. (G and H) Migration ability of HepG2 cells treating with vWF OE and 3 μM pyridone 6 reflected by Wound-healing and Transwell assays.
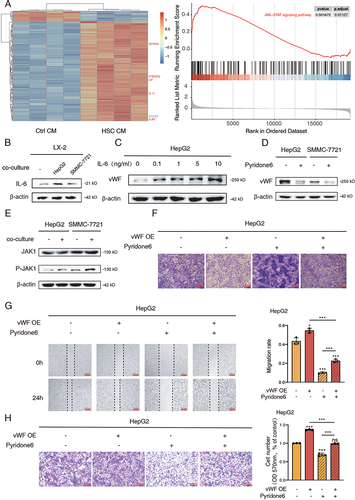
Owing to that IL-6 is a classic upstream protein of JAK/STAT signaling pathway,Citation22 we next tested the secretion and expression of IL-6 in cell medium and LX-2 cells. Interestingly, we found that the secretion and expression of IL-6 by LX-2 cells was substantially increased after co-culturing with HCC cells through ELISA assays and Western blot (Supplementary Figure S2A and 3B). Subsequently, we utilized gradient concentrations of exogenous IL-6 protein treating HCC cells and found that vWF expression could be markedly upregulated even under 0.1 ng/mL of IL-6 ( and Supplementary S2B). These data indicated that HCC cells could stimulate IL-6 synthesis and secretion by LX-2 cells which could influence vWF expression in HCC cells in turn. Besides, JAK signaling pathway inhibitor pyridone 6 (SD4758, Beyotime, Shanghai, China) was adopted for further experiments. As shown in , the vWF expression of HCC cells was significantly inhibited by 3 μM pyridone 6. To clarify which isoform of JAK proteins exhausted its function after co-culturing with LX-2 cells, Western blot was adopted and proved that the JAK1 isoform was highly phosphorylated, while the JAK2 isoform sustained ( and Supplementary Figure S2C). Therefore, these data implied that vWF expression in HCC cells can be mediated by LX-2 cells through IL-6/JAK1 signaling.
Finally, the function of JAK1 signaling in regulating vWF-mediated glycolysis and cell migration was explored. The effect of pyridone 6 on JAK1 phosphorylation under normal or co-culturing conditions was confirmed by Western blot (Supplementary Figure S2D). The results of PAS staining and glycogen assays showed that pyridone 6 promoted the accumulation of glycogen, while vWF OE consumed accumulated glycogen again (, Supplementary Figure S2E, S2F and S2G). The opposite cell L-lactate content further confirmed the regulatory effect of JAK1 signaling on vWF-mediated glucose metabolism (Supplementary Figure S2H and S2I). Similarly, Wound-healing and Transwell assays also manifested that the cell migration inhibitory function by pyridone 6 can be rescued through transfecting vWF OE plasmids into HCC cells (, Supplementary Figure S2J and S2K). Taken together, all the data indicated that HCC cells promoted the secretion of IL-6 from HSCs which in turn activated JAK1 signaling and aggravated HCC glycolysis and migration through mediating vWF expression.
vWF-Mediated TGFB1 Secretion from HCC Cells Activates LX-2 Cells Which Enhance HCC Migration and TGFB1 Expression in Turn
To further explored how vWF mediated the activation of LX-2 cells, several cytokines related to HSCs activation were sortedCitation23,Citation24 and screened by qPCR. The results showed that the expression of TGFB1 and PDGFB in HepG2 cells was significantly upregulated after co-culturing with LX-2 cells (). We then tested the relationship between these two cytokines and vWF mRNA by qPCR and found that the relative mRNA level of TGFB1 was significantly higher than that of PDGFB after transfection with vWF OE plasmids into HCC cells ( and Supplementary Figure S3A). Heatmap results in the former part also showed that TGFB1 was upregulated in the HSC conditional medium-treated HCC cells (). ELISA assays identified TGFB1 concentration in the medium was increased with vWF OE (). Western blot results confirmed that the expression level of α-SMA in LX-2 cells was positively related to TGFB1 in a dose-dependent manner and even 0.1 ng/mL TGFB1 could highly activate LX-2 cells (). To further confirm whether pyridone 6 could regulate TGFB1 expression through mediating vWF, western blot was conducted and showed that vWF markedly upregulated TGFB1 expression, while pyridone 6 could reverse such promoting effect ().
Figure 4 LX-2 enhances vWF mediated TGFB1 expression and secretion by activating the JAK signaling pathway. (A) qPCR results of cytokines related to hepatic stellate cell activation in HCC cells with or without LX-2 co-culturing for 48 h. (B) Relative mRNA expression data of TGFB1 in HCC cells under pcDNA and vWF OE conditions. ***P<0.001, compared with HepG2 vWF pcDNA group; ###P<0.001, compared with SMMC-7721 vWF pcDNA group. (C) The concentrations of TGFB1 in cell medium was detected by ELISA.**P<0.01, compared with HepG2 vWF pcDNA group; ##P<0.01, compared with SMMC-7721 vWF pcDNA group. (D) Western blot results of LX-2 activation levels with the treatment of gradient concentrations of exogenous TGFB1 for 48 h. (E) TGFB1 expression in HCC cells with the treatment of vWF OE and 3 μM pyridone 6 reflected by Western blotting. (F and G) Functions of TGFB1 and TGFB1R2 in mediating the migration ability of HepG2 cells by Wound-healing. (H and I) Functions of TGFB1 and TGFB1R2 in mediating the migration ability of HepG2 cells by Transwell assays.
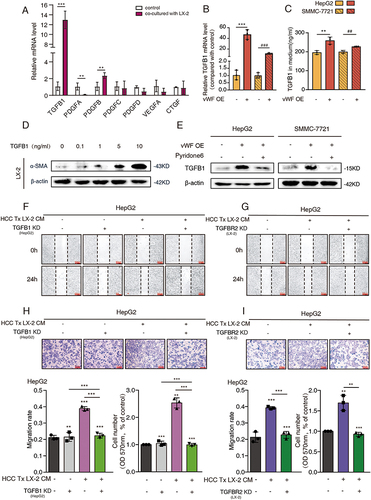
In addition, the effect of TGFB1 and its receptor TGFBR2 on regulating the migration ability of HCC cells was further explored. The transfected efficiency of TGFB1 and TGFBR2 shRNA was confirmed by Western blot (Supplementary Figure S3B and S3C). The results of Wound-healing assays showed that the migration ability of HCC cells was increased obviously with the treatment of HSC conditional medium. On this base, the attenuated migration ability was observed when the TGFB1 shRNA was transfected into HCC cells or TGFBR2 shRNA was transfected into LX-2 cells (, S3D and S3E). Furthermore, Transwell assays also demonstrated this conclusion (, S3F and S3G). Therefore, we found that activating HSCs promoted vWF-mediated TGFB1 expression and secretion through activating JAK1 signaling. Thus, the upregulated TGFB1 in the medium could further activate HSCs and aggravate HCC migration by interacting with the TGFBR2 receptor on the LX-2 cells.
Activated HSCs Enhance HCC Cells’ Glycolysis and Migration Through JAK1/vWF/TGFB1 Axis in vitro
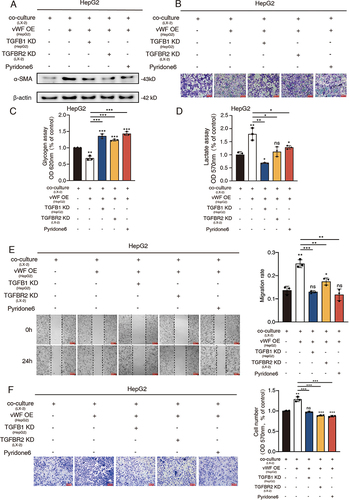
To better understand the integration of the mechanisms we have found in the former parts, various experimental groups were designed for the exploration afterward. At first, α-SMA levels of LX-2 cells were detected by Western blot and the results revealed that vWF overexpressing HCC cells highly activated LX-2 cells, while the LX-2 activation levels of TGFB1 KD, TGFBR2 KD and pyridone 6 treating groups were relatively lower ( and S4A). Besides, glycolysis reflected by PAS staining and glycogen content confirmed the glucose consumption-promoting ability of vWF while interfering with the expression of TGFB1 and TGFBR2 or inhibiting the JAK1 signaling could promote the accumulation of glucose (, S4B and S4C). Additionally, a similar conclusion was verified by the relative L-lactate content reverse with glucose ( and S4D). Moreover, migration ability displayed by Wound-healing and Transwell assays also indicated the pro-migration function of the vWF OE group while the suppression function of the other three treatment groups (, S4E and S4F). Altogether, all the results revealed the mechanism that vWF derived from HCC cells facilitated cancer cell migration and glycolysis ability by regulating TGFB1 mediated LX-2 activation which upregulated vWF expression of HCC cells through activating JAK1 signaling in turn.
Figure 5 TGFB1 derived from HCC cells promotes cancer cell migration and glycolysis through activating HSCs. HepG2 cells transfected with vWF OE and TGFB1 KD or treated by pyridone 6 co-cultured with LX-2 cells transfected with TGFBR2 KD were used for (A) Western blot analysis of α-SMA levels of LX-2 cells; (B and C) Glycogen content reflected by PAS staining and glycogen assays; (D) L-lactate content tested by L-lactate assays; (E and F) Migration ability was examined by Wound-healing and Transwell assays.
HSCs Enhance Tumor Growth Through JAK1/vWF/TGFB1 Axis in vivo
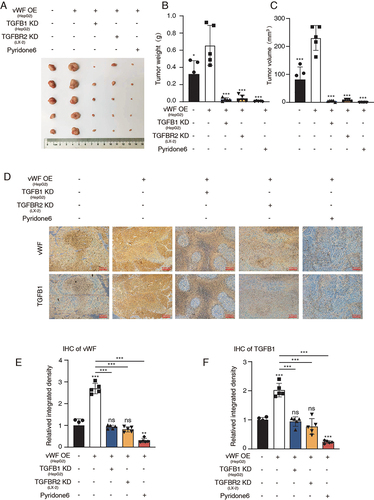
To further explore the JAK1/vWF/TGFB1 axis in mediating tumor growth in vivo. A subcutaneous xenograft model was constructed by co-injecting 1 × 107 HepG2 cells and 1 × 106 LX-2 cells subcutaneously into nude mice to study vWF-mediated tumor growth. Tumor volume and weight data showed that vWF OE enhanced the proliferation of HCC cells, while further suppressing the expression of TGFB1 and TGFBR2 or inhibiting JAK1 signaling decreasing proliferation ability (). Moreover, the immunohistochemistry results of vWF and TGFB1 distribution in the solid tumor verified the positive correlation of these two proteins (). The relative protein intensity of different treatment groups further demonstrated the importance of JAK1/vWF/TGFB1 axis in regulating HCC growth ( and ). In brief, these findings showed that HSCs could promote tumor growth by mediating JAK1/vWF/TGFB1 axis in vivo.
Figure 6 HSCs enhance tumor growth through JAK1/vWF/TGFB1 axis in vivo. Cells with different treatment were injected subcutaneously into nude mice and solid tumors were collected for the further analysis. (A) Tumor images; (B and C) Tumor weight and volume; (D) IHC staining results of vWF and TGFB1 in different groups; (E and F) IHC score results of vWF and TGFB1 in different groups.
Discussion
Tumor metastasis is the main leading cause of HCC-related death worldwide.Citation4 Despite significant efforts to reduce patient metastasis and recurrence rates that have been made over the past decades, the five-year survival rate of patients after surgery was still not optimistic because of potential metastatic lesions.Citation25 Thus, treatment methods are necessary to be continuously innovated. Recent studies have shown that hepatic stellate cells play an important role in the progression of hepatocellular carcinoma metastasis.Citation26 In addition, more and more research focused on the source of tumor metastasis motivation, especially the area related to glucose metabolism.Citation27–29 Interestingly, our previous work has demonstrated the correlation of glycolysis and vWF in lung adenocarcinoma. However, whether similar conclusions can be found in HCC remains unclear, and how vWF participates in the crosstalk of HCC cells and HSCs still need to be uncovered. Given that current research on vWF mainly focused on coagulation functionCitation30,Citation31 and tumor hematogenous metastasis,Citation32,Citation33 there was little research about vWF function on the initial steps of tumor metastasis. Therefore, in this article, we investigated the role of vWF in mediating crosstalk between hepatic stellate cells and hepatocellular carcinoma cells and delved into potential regulatory mechanisms.
We first confirmed the expression of vWF in liver cancer cell lines and then tested the glycolysis level of HCC by constructing vWF shRNA and overexpression plasmids. The results showed that vWF can promote the glycogen consumption-ability of liver cancer cells. In addition, we found that the enhancement of tumor metastasis induced by vWF was significantly related to glycolysis by combining with glycogen phosphorylase inhibitor CP-91149. To investigate whether vWF can participate in the crosstalk between hepatic stellate cells and liver cancer cells, we adopted two co-culture systems to detect the interaction between the two cells. The results indicated that co-culture can significantly upregulate the expression of vWF in liver cancer cells, and overexpressing or knocking down vWF in liver cancer cells can promote or inhibit the activation of hepatic stellate cells. Wound-healing and Transwell experiments also showed that overexpression of vWF in hepatoma cells can further promote the enhanced tumor cell migration ability of co-culturing ( and ).
On this basis, we further explored the molecular mechanism by which hepatic stellate cells regulate vWF-mediated glycolysis and tumor metastasis in liver cancer cells. We first conducted differential genes heatmap and GSEA analysis by utilizing the GEO dataset GSE62455 and found that the JAK/STAT pathway was highly activated under co-culture conditions. Afterward, we analyzed the expression and secretion of IL-6, the classic upstream regulatory protein of the JAK/STAT pathway in hepatic stellate cells. The results indicated that co-culture can significantly promote the expression and secretion of IL-6 in hepatic stellate cells, and the secretion of IL-6 was sufficient to significantly increase the expression of vWF in liver cancer cells. Interestingly, a previous study also discovered that IL-6 released from hepatic stellate cells expanded the population of myeloid-derived suppressor cells which in turn promoted HCC progression.Citation14 The direct and indirect impact of HSCs-derived IL-6 on HCC progression implied its role as a regulator in HCC TME. In the study of JAK protein subtypes that mediated vWF expression, we found that JAK1 was significantly phosphorylated under co-culture conditions, while the phosphorylation level of JAK2 remained unchanged. Later, in the study on the glycolysis and migration levels of liver cancer cells using JAK inhibitor pyridone 6, it was found that the JAK signaling pathway can affect the glycolysis and migration levels of tumor cells through vWF.
To investigate the mechanism of vWF expression in liver cancer cells mediating the activation of hepatic stellate cells, we first screened seven cytokines related to hepatic stellate cell activationCitation23,Citation24 through qPCR. The results showed that the relative mRNA expression levels of TGFB1 and PDGFB significantly increased after co-culturing, and the relative mRNA expression level of TGFB1 was higher than PDGFB after vWF overexpressing in HCC cells. The results of ELISA also showed that overexpression of vWF can significantly promote the secretion of TGFB1 in liver cancer cells, and this promoting effect was significantly reflected in the increased activation of hepatic stellate cells. Our further pathway integrity validation experiments also demonstrated the important role of the JAK1/vWF/TGFB1 axis in hepatic stellate cell-regulated liver cancer metastasis and glycogen metabolism in vitro. The results of the nude mice subcutaneous xenograft model also confirmed the role of JAK1/vWF/TGFB1 axis mediated by hepatic stellate cells in promoting tumor growth in vivo. However, although we have preliminarily validated the role of IL-6 derived from hepatic stellate cells in regulating vWF-mediated HCC glycolysis and migration in vitro experiments, in vivo studies with targeted hepatic stellate cells’ IL-6 depletion tools will further refine the mechanism of hepatic stellate cells in mediating HCC progression.
Conclusion
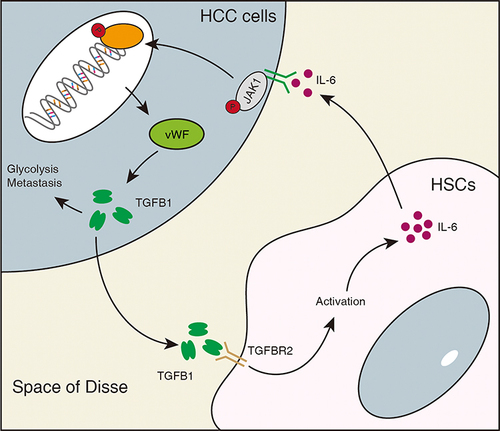
In summary, our study suggests that vWF can be expressed in liver cancer cells and can enhance the activation of hepatic stellate cells by promoting the secretion of TGFB1 from liver cancer cells. In addition, activated hepatic stellate cells can also stimulate the JAK1 signaling pathway in liver cancer cells by secreting IL-6, thereby increasing the expression of vWF and enhancing the glycolysis and migration ability of liver cancer cells (). Our findings enrich the molecular mechanisms by which hepatic stellate cells regulate HCC progression, and IL-6 derived from hepatic stellate cells regulates the JAK1/vWF/TGFB1 axis in liver cancer cells, which is also a potential therapeutic target for liver cancer metastasis.
Figure 7 Schematic diagram illustrating the mechanism of HSC-derived IL-6 in promoting vWF mediated HCC glycolysis and migration. IL-6 from hepatic stellate cells promotes glycolysis and migration of HCC through the JAK1/vWF/TGFB1 axis. In addition, TGFB1 secreted by HCC cells further promotes the activation of hepatic stellate cells.
Data Sharing Statement
Data and materials are available.
Ethics Statement
Animal experiments conducted in mice were using protocols approved by the IACUC of East China University of Science and Technology [grant number ECUST-2021-06002]. Animal husbandry protocols followed the Declaration of Helsinki.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
We acknowledge TCGA and GEO database for providing their meaningful data.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
- Wu J, Yang S, Xu K, et al. Patterns and trends of liver cancer incidence rates in eastern and southeastern asian countries (1983-2007) and predictions to 2030. Gastroenterology. 2018;154(6):1719–1728. doi:10.1053/j.gastro.2018.01.033
- Tumen D, Heumann P, Gulow K, et al. pathogenesis and current treatment strategies of hepatocellular carcinoma. Biomedicines. 2022;10(12):3202–3242. doi:10.3390/biomedicines10123202
- Niu ZS, Wang WH, Niu XJ. Recent progress in molecular mechanisms of postoperative recurrence and metastasis of hepatocellular carcinoma. World J Gastroenterol. 2022;28(46):6433–6477. doi:10.3748/wjg.v28.i46.6433
- Chen C, Wang Z, Ding Y, Qin Y. Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front Immunol. 2023;14:1133308. doi:10.3389/fimmu.2023.1133308
- Liu P, Kong L, Liu Y, et al. A key driver to promote HCC: cellular crosstalk in tumor microenvironment. Front Oncol. 2023;13:1135122. doi:10.3389/fonc.2023.1135122
- Filliol A, Saito Y, Nair A, et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature. 2022;610(7931):356–365. doi:10.1038/s41586-022-05289-6
- Barry AE, Baldeosingh R, Lamm R, et al. Hepatic Stellate Cells and Hepatocarcinogenesis. Front Cell Dev Biol. 2020;8:709. doi:10.3389/fcell.2020.00709
- Akkiz H. Emerging role of cancer-associated fibroblasts in progression and treatment of hepatocellular carcinoma. Int J Mol Sci. 2023;24(4):3941–3962. doi:10.3390/ijms24043941
- Zhang R, Gao X, Zuo J, et al. STMN1 upregulation mediates hepatocellular carcinoma and hepatic stellate cell crosstalk to aggravate cancer by triggering the MET pathway. Cancer Sci. 2020;111(2):406–417. doi:10.1111/cas.14262
- Li S, Hu X, Yu S, et al. Hepatic stellate cell‐released CXCL1 aggravates HCC malignant behaviors through the MIR4435‐2HG / miR‐506‐3p/ TGFB1 axis. Cancer Sci. 2022;114(2):504–520. doi:10.1111/cas.15605
- Chen QT, Zhang ZY, Huang QL, et al. HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat Metab. 2022;4(10):1306–1321. doi:10.1038/s42255-022-00642-5
- Makino Y, Hikita H, Kodama T, et al. CTGF mediates tumor-stroma interactions between hepatoma cells and hepatic stellate cells to accelerate hcc progression. Cancer Res. 2018;78(17):4902–4914. doi:10.1158/0008-5472.CAN-17-3844
- Hsieh CC, Hung CH, Chiang M, Tsai YC, He JT. Hepatic stellate cells enhance liver cancer progression by inducing myeloid-derived suppressor cells through interleukin-6 signaling. Int J Mol Sci. 2019;20(20):5079. doi:10.3390/ijms20205079
- Hapach LA, Mosier JA, Wang W, Reinhart-King CA. Engineered models to parse apart the metastatic cascade. NPJ Precis Oncol. 2019;3(1):20–28. doi:10.1038/s41698-019-0092-3
- Patmore S, Dhami SPS, O’Sullivan JM. Von Willebrand factor and cancer; metastasis and coagulopathies. J Thromb Haemost. 2020;18(10):2444–2456. doi:10.1111/jth.14976
- A-j Y, Wang M, Wang Y, et al. Cancer cell-derived von Willebrand factor enhanced metastasis of gastric adenocarcinoma. Oncogenesis. 2018;7(1):12–25. doi:10.1038/s41389-017-0023-5
- Gu J, Qi Y, Lu Y, et al. Lung adenocarcinoma-derived vWF promotes tumor metastasis by regulating PHKG1-mediated glycogen metabolism. Cancer Sci. 2022;113(4):1362–1376. doi:10.1111/cas.15298
- Colonne CK, Favaloro EJ, Pasalic L. The Intriguing Connections between von Willebrand Factor, ADAMTS13 and Cancer. Healthcare. 2022;10(3):557–575. doi:10.3390/healthcare10030557
- Liu L, Pan J, Wang H, et al. von Willebrand factor rescued by miR-24 inhibition facilitates the proliferation and migration of osteosarcoma cells in vitro. Biosci Rep. 2018;38(6):372–382. doi:10.1042/BSR20180372
- Liu Y, Wang X, Li S, et al. The role of von Willebrand factor as a biomarker of tumor development in hepatitis B virus-associated human hepatocellular carcinoma: a quantitative proteomic based study. J Proteomics. 2014;106:99–112. doi:10.1016/j.jprot.2014.04.021
- Li Y, Zhao J, Yin Y, et al. The Role of IL-6 in fibrotic diseases: molecular and cellular mechanisms. Int J Biol Sci. 2022;18(14):5405–5414. doi:10.7150/ijbs.75876
- Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;8(11):1419–1454. doi:10.3390/cells8111419
- Yan Y, Zeng J, Xing L, Li C. Extra- and intra-cellular mechanisms of hepatic stellate cell activation. Biomedicines. 2021;9(8):1014–1031. doi:10.3390/biomedicines9081014
- Byrd K, Alqahtani S, Yopp AC, Singal AG. Role of multidisciplinary care in the management of hepatocellular carcinoma. Semin Liver Dis. 2021;41(1):1–8. doi:10.1055/s-0040-1719178
- Quiroz Reyes AG, Lozano Sepulveda SA, Martinez-Acuna N, et al. Cancer stem cell and hepatic stellate cells in hepatocellular carcinoma. Technol Cancer Res Treat. 2023;22:1–13. doi:10.1177/15330338231163677
- Feng J, Li J, Wu L, et al. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2020;39(1):126–145. doi:10.1186/s13046-020-01629-4
- Liu C, Xu K, Liu J, et al. LncRNA RP11-620J15.3 promotes HCC cell proliferation and metastasis by targeting miR-326/GPI to enhance glycolysis. Biol Direct. 2023;18(1):15–30. doi:10.1186/s13062-023-00370-0
- Yu Q, Dai W, Ji J, et al. Sodium butyrate inhibits aerobic glycolysis of hepatocellular carcinoma cells via the c-myc/hexokinase 2 pathway. J Cell Mol Med. 2022;26(10):3031–3045. doi:10.1111/jcmm.17322
- Kanaji S, Fahs SA, Shi Q, Haberichter SL, Montgomery RR. Contribution of platelet vs. endothelial VWF to platelet adhesion and hemostasis. J Thromb Haemost. 2012;10(8):1646–1652. doi:10.1111/j.1538-7836.2012.04797.x
- Pablo-Moreno JA, Serrano LJ, Revuelta L, Sanchez MJ, Liras A. The vascular endothelium and coagulation: homeostasis, disease, and treatment, with a focus on the von Willebrand factor and factors VIII and V. Int J Mol Sci. 2022;23(15):8283–8325. doi:10.3390/ijms23158283
- O’Sullivan JM, Preston RJS, Robson T, O’Donnell JS. Emerging Roles for von Willebrand Factor in Cancer Cell Biology. Semin Thromb Hemost. 2018;44(2):159–166. doi:10.1055/s-0037-1607352
- Qi Y, Chen W, Liang X, et al. Novel antibodies against GPIbα inhibit pulmonary metastasis by affecting vWF-GPIbα interaction. J Hematol Oncol. 2018;11(1):117–134. doi:10.1186/s13045-018-0659-4