
Abstract
Poly(ADP-ribose) polymerase inhibitors (PARPi) have been in clinical use since 2014 for certain patients with germline BRCA1/2 mutations, but as evidence and approvals for their use in a wider range of patients grow, the question of how best to identify patients who would benefit from PARPi becomes ever more complex. Here, we discuss the development and current state of approved selection testing for PARPi therapy and the ongoing efforts to define a broader range of homologous recombination repair deficiencies that are susceptible to PARP inhibition.
Introduction
Prior to the 1980s, the approach to cancer therapy was defined by the use of non-selective chemotherapeutic agents that destroyed both normal and malignant cells actively duplicating their DNA.Citation1 A multitude of advancements in molecular biology during the 1990s permitted researchers to better recognize DNA structure, mechanisms of DNA damage, and the cellular basis of malignant transformation.Citation2,Citation3 With this understanding, new targets have been identified over the past 20 years that can be modulated or blocked by biotechnological drugs, such as monoclonal antibodies and small molecules,Citation4,Citation5 and have ushered in a new era of cancer therapy leading to national precision medicine initiatives and improved personalized therapeutic treatment options for patients.
It is almost 60 years since the discovery of the [poly(ADP-ribose) polymerase]-1 (PARP-1) nuclear enzyme,Citation6,Citation7 40 years since the identification of the role it plays in DNA damage repair,Citation8 and over a decade since the first clinical trials of the innovative small molecules inhibiting PARP (PARP inhibitors; PARPi) were reported.Citation9 Building further on the premise of precision medicine, this past decade has brought a singularly transformative improvement in diagnostic testing for patients with cancer, including genomic analysis for the identification of inherited and epigenetic tumor mutations, highly sensitive detection of tumor-derived DNA in blood, immune marker changes, RNA and transcriptomic testing; and in response to these technological processes, rapid adaptive changes in clinical trial design. A better understanding of the DNA damage response (DDR) pathway, a network of hundreds of proteins that sense, signal, and repair DNA,Citation10–Citation14 has also led to the mechanistic recognition that loss of one or more of these DDR mechanisms often occurs during the development of cancer, and the pursuit of biomarkers of clinical significance.Citation15
The Role of Homologous Recombination Deficiency in PARPi Response
DNA damage occurs continuously, leading to signal sensing and repairing of this damage via the DDR pathways that form part of various physiological processes involving an estimated 450 genes.Citation10,Citation11 The type of DNA damage that is caused as well as the timing of the same in the cell division cycle dictates the type of repair mechanism utilized;Citation16 some forms of DNA damage can be repaired quickly but more structurally complex repairs require the cell to arrest division to efficiently facilitate this. Repair mechanisms include base excision repair (BER) for single-strand breaks (SSBs), nucleotide excision repair (NER), and mismatch repair of bases (MMR). Double-strand breaks (DSBs) are postulated to be the most genotoxic and fidelitous repair is crucial to cell survival.Citation17 There are at least 3 mechanisms of DSB repair, including homologous recombination (HR) repair, non-homologous end joining (NHEJ), and micro-homology mediated end joining (MMEJ).Citation10 Non-cancerous cells can use the second copy of the DNA helix as a template for the repair of DSBs via HR repair, but cells with a compromised HR repair will use other error-prone mechanisms such as NHEJ which lead to genomic instability. The loss of HR repair capability through mutation, both germline and somatic, or epigenetic changes in one of the HR repair genes creates an associated genomic instability denominated HR deficiency or HRD.
PARP-1 is one of the best described and most abundant enzymes in the super family of PARP proteins, which controls cellular-level repair of SSBs through BER. Normally, once DNA damage is detected, PARP enzymes bind to the DNA lesion and facilitate posttranslational ADP-ribosylation using NAD+ (nicotinamide adenine dinucleotide) as a substrate, which permits uncoiling of the chromatin for repair by engaging H1/H2B histones.Citation18 Additionally, PARP-1 is increasingly recognized as playing a role in DSB detection and repair.Citation19 The ADP-ribosylation (or “PAR-ylation”) process is postulated to be regulated by the helical subdomain of PARP-1, which borders an ADP-ribosyl transferase fold that can selectively permit access of NAD+ after DNA binding, and these modifications recruit other factors that effectively complete DNA repair.Citation20–Citation22 This process is also the target mechanism of action for PARPi: b-nicotinamide adenine dinucleotide (NAD+)-competitive inhibitors.Citation23
Four PARPi have received one or more FDA approvals: olaparib (Lynparza, AstraZeneca), niraparib (Zejula, GlazoSmithKline), rucaparib (Rubraca, Clovis Oncology, Inc.), and talazoparib (Talzenna, Pfizer). At least two others (veliparib [AbbVie] and pamiparib [BeiGene, Ltd.]) are in clinical development. Though cytotoxicity has been recognized to be variable between these inhibitors, more recently, these differences have been postulated to be secondary to the ability of each agent to “trap” PARP enzymes on DNA.Citation24 More specifically, PARPi not only block the enzymatic activity of PARP preventing auto-PARylation, but subsequently impede the dissociation of PARP-1 from the damaged chromatin by stabilizing the PARP-DNA complexes, which directly leads to stalled replication forks and subsequent formation of DSBs ().Citation25,Citation26
Figure 1 Mechanism of PARP inhibitor (PARPi) cytotoxicity. The presence of PARPi prevents repair of single-strand DNA breaks (SSBs). SSBs are ultimately converted to double-strand breaks (DSBs) during replication. In homologous recombination (HR)-deficient tumors, the DSBs cannot be resolved leading to cell death.
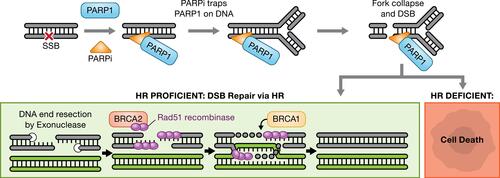
The activity of PARPi was initially demonstrated preclinically in the setting of deleterious germline breast cancer 1 or 2 (BRCA1/2) gene mutations in cell lines and patient-derived xenograft (PDX) models. BRCA1/2 are key HR repair genes and the interplay between PARPi and BRCA1/2 loss results in intolerable levels of genomic instability and cell death, exemplifying the concept of synthetic lethality (ie, the combination of two non-lethal cellular defects causing cell death).Citation27,Citation28 Deleterious mutations in the BRCA genes are a canonical example of HR deficiency; however, HR deficiency can also occur through other mechanisms including germline, somatic, or methylation changes in other HR repair genes.Citation29–Citation31 Multiple assays have been developed to identify defects in HR repair and define an HR deficiency phenotype, with the expectation that the combination of tumors bearing the HR deficiency phenotype and PARP inhibition will result in clinical benefit. The approval and use of PARPi have been heavily dependent on companion diagnostic assays and defining this HR deficiency phenotype.
Since the first approval of olaparib by both the FDA and the EMA in 2014 for the treatment of patients with germline mutations in BRCA1/2 (gBRCAm) and high-grade serous ovarian, fallopian tube and peritoneal carcinoma (HGSC), a clinical “proof-of-concept” confirmation of synthetic lethality, there has been a concerted effort by the research community to explore the occurrence of this vulnerability across tumor types beyond both BRCA deleterious mutations and ovarian cancer. Together with the commercialization of next-generation sequencing testing, validation of HR deficiency as a novel predictive biomarker has become a focus of patient selection strategies for PARPi therapy and a multitude of assays for HR deficiency have been created. Miller et al recently characterized three separate types of HR deficiency testing strategies: 1) HR repair gene-level tests (gene-based assays of germline and somatic BRCA mutations, non-BRCA gene mutations, variants of unknown significance, and HR gene promoter methylation), 2) genomic scars or mutational signatures (such as genomic instability and mRNA expression profiles), and 3) functional assays that can indicate HR deficiency or proficiency (such as Rad51 foci induction).Citation29 Below, we discuss the capabilities of each of these assay types.
Genomic Approaches for Identifying PARPi Sensitivity
Gene-Based Assays
HR deficiency was initially described in the context of germline BRCA1 and BRCA2 pathogenic mutations ().Citation32 Germline mutations in BRCA1/2 are the gold standard for HR deficiency and are found in up to 7% of unselected breast cancer, in up to 15% of triple-negative breast cancers and in 14% of high-grade serous ovarian cancers.Citation33,Citation34 Somatic BRCA1/2 alterations occur in the same tumor types. They manifest as HR-deficient in the same way as germline BRCA1/2 alterations and confer equivalent sensitivity to PARPi and platinum-based regimens.Citation35 There are two FDA-approved companion diagnostic (CDx) tests for BRCA1/2. Myriad Genetics’ BRACAnalysis CDx is indicated for the identification of germline BRCA1/2 mutations from whole blood in breast, pancreatic, ovarian and prostate cancers, and FoundationFocus CDxBRCA from Foundation Medicine is indicated for FFPE tumor tissue to identify deleterious BRCA1/2 alterations in ovarian cancer patients.Citation36
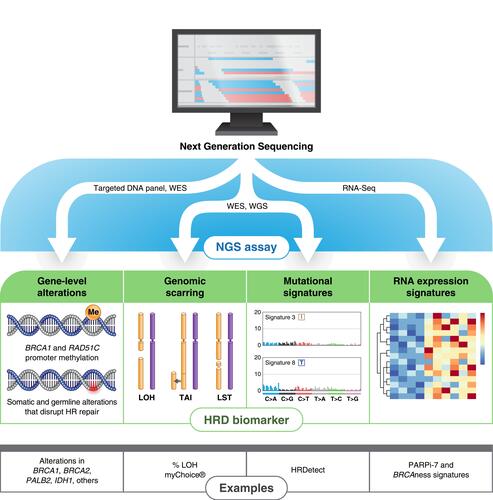
Figure 2 Genomic approaches for assessing homologous recombination (HR) deficiency. Next-generation sequencing (NGS) assays may use targeted DNA panels, whole-exome sequencing (WES), whole-genome sequencing (WGS), or RNA-Seq to identify complex genomic biomarkers of HR deficiency. Targeted gene panels are sufficient to measure a clinically relevant subset of HR-related genes. Genomic scars and mutational signatures are most accurately measured by either WES or WGS, which sample the entire exome or genome, respectively. RNA-Seq has mostly replaced microarray and other platforms for evaluating gene expression signatures such as PARPi-7 and BRCAness.
In addition to pathogenic alterations in BRCA1, methylation of the BRCA1 promoter can also compromise the function of the BRCA1 protein product. Interestingly, BRCA1 promoter methylation (PM) appears to be mutually exclusive with BRCA1 mutation.Citation37 BRCA1 PM is relatively common in triple negative breast and high-grade ovarian cancer (HGOC), where it has been observed in ~30% and 11–17% of patients, respectively.Citation37,Citation38 Swisher et al found that women with BRCA1 PM may benefit from the PARPi rucaparib in the ARIEL2 trial, a study in women with HGOC.Citation37 In this study, patients with high BRCA1 PM levels, as assessed in tumor specimens obtained just prior to enrollment, had an objective response rate (ORR) of 44% compared with an ORR of just 5–10% in patients who were BRCA1 methylation low or unmethylated. The BRCA1 PM ORR was similar to the 39% ORR observed in the 89 evaluable patients enrolled in the ARIEL2 trial with BRCA1 mutations, suggesting that BRCA1 PM patients will derive equivalent benefit to PARPi. Additionally, BRCA1 methylation status was found to potentially change over time, with reversion of methylated back to an unmethylated state potentially conferring resistance to rucaparib. Besides BRCA1/2, there are multiple other gene products that are necessary for successful HR repair. These include genes encoded by MRE11, RAD51, ATM, CDK12, PALB2, CHEK2, RAD51C, RAD51D, BRIP1, and BARD1, as well as other Fanconi anemia genes. Mutations in many of these occur so infrequently that it is not yet clear whether pathogenic or likely pathogenic alterations will confer the same sensitivity to PARPi as alterations in BRCA1/2. For some, however, a body of clinical evidence is beginning to emerge in several cancer histologies.
PALB2 appears to be a core HR gene, with clinical responses to PARPi in patients with PALB2 mutations approaching what are typically seen for BRCA alterations in pancreatic, ovarian, prostate, and breast cancer.Citation39–Citation41 Mutations in RAD51C and RAD51D are very rare but do occasionally occur in ovarian cancer. These were also shown to be similarly sensitive to rucaparib as BRCA alterations.Citation37 The sensitivity of other, more frequently occurring HR mutations to PARPi remains less clear. Alterations in ATM and CDK12 occur at relatively higher frequencies in metastatic castration-resistant prostate cancer (mCRPC). However in TOPARB, a phase 2 study of men with mCRPC and aberrations in DNA damage repair genes, only 1 in 12 evaluable patients with an ATM pathogenic/likely pathogenic alteration and 0 among 18 with CDK12 pathogenic/likely pathogenic alterations had an objective response by RECIST1.1 criteria.Citation41 Similarly, in a recent study of olaparib in metastatic breast cancer, responses were only seen in patients with PALB2 and somatic BRCA1/2 alterations,Citation42 but not in patients with CHEK2 and ATM mutations. Germline alterations in BRIP1 carry an increased risk of hereditary breast or ovarian cancer. Of the five HGOC patients with biallelic loss of BRIP1 enrolled in the ARIEL2 study, none had an objective response, although two did have platinum resistant/refractory disease on enrollment.Citation37
Two additional genes not normally associated with HR repair have garnered much recent attention. IDH1 and IDH2 normally function in the citric acid cycle. However, certain alterations in IDH1 and IDH2 are neomorphic and have been found to occur in several cancers, including in up to 70% of low-grade gliomas.Citation43 Recently, IDH1/2 mutations were shown to induce HR deficiency in tumor cells, rendering them sensitive to PARPi.Citation44 These findings translated to the clinic, where in a recent Phase 1 trial of olaparib in IDH1/2-mutant mesenchymal sarcomas, Eder et al found three of five patients with chondrosarcomas had clinical benefit.Citation45
Genomic Scar Assays
The challenges of establishing the clinical utility of PARPi beyond BRCA1/2 using a gene-based approach have led to the development of functional and phenotypic genomic assays for assessing HRD (). Among these, assays that evaluate the level of genomic scarring in the tumor genome are the most clinically advanced. Large stretches of loss of heterozygosity (LOH) constitute one such scar. Foundation Medicine has developed a commercial next-generation sequencing (NGS) test for LOH that has been approved by the FDA as a complementary diagnostic for rucaparib (the FoundationFocus CDxBRCA Assay).Citation36 The test calculates the percentage of LOH across the 22 autosomal chromosomes, excluding regions spanning >90% of a whole chromosome or chromosomal arm as these usually arise through mechanisms such as non-disjunction that do not involve HRD.Citation37 ARIEL2 Part 1 used the Foundation Medicine test to segregate patients with BRCA-wt HGOC into LOH-high and LOH-low bins based on a cut-off of 14% LOH;Citation37 a higher threshold of ≥16% LOH was later defined due to testing in the ARIEL3 study.Citation46 Patients who were LOH-high achieved a 12-month progression-free survival (PFS) of 28% versus 10% for the LOH-low group. Of the 152 evaluable patients who were BRCA-wild-type, 82 (54%) were found to be LOH-high, reflecting the high rate of genomic scarring observed in HGOC.
A second commercial NGS assay that evaluates genomic scarring, myChoice®, has been developed by Myriad Genetics. myChoice® uses a combined score based on LOH, telomere allelic imbalance (TAI), and large-scale state transitions (LST). A combined score of 42 was established as a threshold between HR-deficient and HR-proficient tumors. This threshold was based on a large cohort of BRCA1/2-mutant breast and ovarian cancers where 95% of cases were found to have a myChoice® score at or above 42.Citation47 In a Phase 3 trial evaluating the PARPi niraparib as maintenance therapy in platinum-sensitive recurrent ovarian cancer, patients who were BRCA1/2 wild-type but HR deficiency-positive by myChoice® (ie, HR-deficient) had a better clinical outcome with niraparib versus placebo.Citation48
In addition to ovarian cancer, HR deficiency as assessed by genomic scarring or other functional assay may occur in 24% of primary breast cancers, 15% of metastatic pancreatic, and 13% of metastatic prostate cancers;Citation49 however, the clinical utility of genomic scarring assays in histologies beyond ovarian cancer is undetermined. Of note for mCRPC, BRCA2-altered tumors were found to have significantly higher myChoice® HR deficiency scores than germline ATM-altered or CHEK2-altered cases,Citation50 consistent with the lower efficacy of PARPi with these alterations as described above.
It should be noted that a limitation of genomic scarring as a predictive biomarker of PARPi response is that is irreversible. Consistent with this notion, there was high concordance observed when %LOH was compared between archival and matched freshly collected specimens, with some LOH scores even increasing over time in the freshly collected specimen but none decreasing.Citation35 Genomic scarring assays therefore cannot identify patients who may have developed resistance to PARPi through BRCA1/2 reversion mutations or other mechanisms, suggesting they have highest clinical utility in the treatment-naïve or platinum-sensitive setting.
Assays That Use Mutational Signatures
The cancer genome harbors distinct mutational signatures, each of which reflects a mutational process that may underlie the development of cancer ().Citation51 One of the base substitution signatures identified in breast, ovarian and pancreatic cancer, signature 3, was strongly associated with the presence of BRCA1 and BRCA2 inactivating mutations.Citation51 Davies et al extended this finding using whole-genome sequencing to identify a weighted index of 6 mutational signatures, termed HRDetect, that quantitatively defined BRCA deficiency with high sensitivity.Citation52 Besides signature 3, HRDetect incorporates signatures representing microhomology-mediated indels, the HR deficiency index, rearrangement signature 3, rearrangement signature 5, and base substitution signature 8. Interestingly, nearly 1/3 of the 124 breast cancer cases identified with high HRDetect scores were not BRCA1/2 null or known to be BRCA1 PM, suggesting the potential of HRDetect to identify cases beyond BRCA1/2 that may benefit from PARPi.
Similar to HRDetect, Nguyen et al developed CHORD, a mutational classifier for HR deficiency.Citation49 A drawback of both HRDetect and CHORD is the requirement for WGS, which is not typically performed in clinical sequencing laboratories. To address this, Gulhan et al developed a computational tool, SigMA, that can faithfully identify HR deficiency signature 3 in tumors using only a targeted gene panel.Citation53
RNA Expression Assays
In contrast to DNA-based signatures, which continue to reflect an HR-deficient state even when a reversion event in an HR gene allows a tumor to become HR-proficient again, an RNA-based gene expression signature should in theory be able to accurately assess HR proficiency at any time due to the dynamic nature of RNA expression (). Several gene expression signatures were evaluated in I-SPY 2, a neoadjuvant breast cancer study that included a combination of the PARPi veliparib plus carboplatin.Citation54 A 7-gene signature, PARPi-7 (BRCA1, CHEK2, MAPKAPK2, MRE11A, NBN, TDG, and XPA), and a 77-gene BRCA1ness signature both showed associations with response in the veliparib/carboplatin arm but not the control arm. Interestingly, the concordance between the two signatures was only moderate (64%; k = 0.29). A limitation of the study was the relatively small size of the cohort (n = 115) and the retrospective nature of the analysis. These signatures, while promising, will require further exploration.
Resistance to PARPi Therapy Through HR Gene Reversion Mutations
The best-characterized mechanisms of resistance to PARPi are molecular events that result in the restoration of the open reading frame in BRCA1, BRCA2 and other mutant HR genes. Reversion mutations were first identified in studies of BRCA2-mutant breast or pancreatic cell lines selected to be resistant to PARPi or cisplatin.Citation55,Citation56 In a clinical setting, reversion mutation events have now been observed in breast, pancreatic, ovarian, and prostate cancers.Citation57
Pettitt et al have recently established a public database where researchers who identify reversion events in HR genes may deposit their findings.Citation58 The corresponding publication described the analysis of over 300 reversions, most of which were in BRCA1 and BRCA2. Deletions, as opposed to base substitutions or other genomic events, were the most frequent type of reversion alteration seen. Over half of the deletions in BRCA2 and 47% of deletions in BRCA1 showed evidence of microhomology-mediated end joining (MMEJ), demonstrating that this mechanism of DNA repair plays a prominent role in mediating reversion events. Reversion mutations are not unique to BRCA1/2, having also been identified in PALB2, RAD51C, and RAD51D.Citation59,Citation60
Liquid biopsies continue to be increasingly embraced by both the clinical and research communities, with one more recent example being the identification of reversion events in HR genes in several histologies.Citation31,Citation60–Citation62 In one of the largest such clinical studies to date, Lin et al described reversions identified in circulating free DNA (cfDNA) from a cohort of 97 HGOC patients treated with rucaparib.Citation31 In pretreatment cfDNA, 18% (2/11) of platinum-resistant and 13% (5/38) of platinum-refractory patients had BRCA1/2 reversions compared with 2% (1/48) of platinum-sensitive patients. Patients without pretreatment cfDNA BRCA1/2 reversions had a significantly longer PFS on rucaparib (9.0 versus 1.8 months; P < 0.0001), supporting the potential clinical utility of screening pre-treated BRCA1/2-mutant ovarian cancer patients for reversion mutations using liquid biopsies prior to initiating PARPi therapy. Strikingly, cfDNA captured a multiclonal heterogeneity of reversion events that was not observed in the matched, contemporaneously collected tumor specimens. In one instance, 8 unique BRCA1 reversion events were identified in cfDNA with only 1 of 8 observed in the matched biopsy. A well-recognized limitation for the use of cfDNA is the potential for a false-negative result in patients whose tumors shed very little circulating tumor DNA (ctDNA). Of note, an additional challenge is the detection of copy number alterations such as homozygous deletions in BRCA2, which constitute ~20% of all BRCA2 deleterious mutations in the tumors of men with mCRPC.Citation63
Resistance Beyond HR Gene Reversion Mutations
While genomic reversion mutations constitute the majority of documented resistance to PARPi in patients, non-revertant mechanisms of resistance have also been identified. Among these is the upregulation of ABCB1, which encodes the MDR1 drug efflux pump.Citation57 Upregulation of ABCB1 was first identified as a mechanism of resistance to olaparib and other DNA-damaging agents in BRCA1/2-deficient mouse models.Citation64,Citation65 Evidence supporting the clinical relevance of this mechanism comes from a study of 92 patients with high-grade serous ovarian cancers (HGSOC), where ABCB1 promoter fusion events were associated with ABCB1 upregulation in 8% of evaluable cases.Citation66 PARPi that are poor substrates of MDR1, such as veliparib and niraparib, may be considered for these patients.
A second non-revertant mechanism of PARPi resistance that may translate to the clinic is loss of TP53BP1 function. Loss of TP53BP1 has been shown in vitro to promote the restoration of BRCA-independent HR through loss of DNA end protection.Citation67,Citation68 Waks et al observed biallelic TP53BP1 loss in one BRCA1-mutant metastatic breast cancer patient progressing on platinum, and Johnson et al observed decreased TP53BP1 expression by IHC in the biopsies of four HGSOC platinum progressors.Citation69,Citation70
Protein Biomarkers for PARPi Sensitivity
Homologous Recombination
Development of a phenotypic, or functional, assay for HR deficiency is very attractive due to the challenges of measuring all proteins of interest within the pathway. A functional assay should ideally measure a single event downstream that indicates pathway proficiency of upstream factors. Much interest has been generated in a Rad51 foci assay as a measure of HR proficiency. Rad51 is a recombinase that forms discrete nuclear foci after induction of DNA damage in S/G2 when a sister chromatid is present for recombination (). Rad51 foci formation indicates the ability of a cell to induce HR, up to the stage of Rad51 loading onto the ends of a DSB but does not measure steps downstream of HR loading. Several groups have examined whether impaired Rad51 foci formation after DNA damage may identify cancers with defects in HR; however, more work needs to be done to evaluate if Rad51 foci formation is an effective predictor of PARPi sensitivity. Here, we will review the evidence supporting the correlation between Rad51 foci formation after drug treatment and 1) HR deficiency and/or 2) PARPi sensitivity. Some of the studies cited include a second correlate to define HR deficiency, while earlier studies correlate Rad51 scores with PARPi sensitivity directly.
Rad51 foci formation was examined in patients with sporadic breast cancer in biopsies taken 24 hours post anthracycline-based chemotherapy.Citation71 The percentage of geminin-positive cells also positive for Rad51 foci was used to assign a Rad51 score indicative of HR deficiency. A quarter of cases (15/57; 26%) were defined as Rad51-low (<10% of geminin-positive cells displaying Rad51 foci) and presumed to be HR-deficient. Out of these 15 Rad51-low cases, 33% achieved a pathological complete response compared to 3% of tumors without a Rad51-low score, although there were no differences in overall clinical responses to chemotherapy. Importantly, the baseline biopsies of the same 35 patients were also evaluated and Rad51 was observed in only 2 biopsies, each of which only had 2% of cells with Rad51 foci. This study underscores that the predictive value of this marker may not be found in baseline measurements, as are usually used for diagnostic assays, but in the response of the tumor to treatment, requiring a post-treatment biopsy to assess.
Attempting to correlate phenotypic and genomic markers of HR deficiency, a retrospective study of patients with TNBC treated with rucaparib for two weeks evaluated HR deficiency using two assays: 1) the HRDetect assay on baseline biopsies and 2) impaired Rad51 foci formation on end-of-treatment biopsies.Citation72 HRDetect assay indicated HR-pathway defects in 69% of patient baseline biopsies. End-of-treatment biopsies from these patients were then evaluated by a Rad51 and geminin immunohistochemistry assay and defined as HR-deficient if Rad51 foci were observed in less than 20% of geminin-positive cells. The authors reported 77% of the end-of-treatment biopsies were characterized as Rad51-deficient, although it is not clear whether the biopsy was taken within a window when Rad51 can be reliably detected after rucaparib treatment. The authors reported that 61% of those Rad51-deficient cancers had a detectable HR defect by HRDetect mutational signatures. Whether the Rad51 score correlates with the clinical response to rucaparib was not the focus of the study. Studies using PDX models have investigated the correlation between PARPi sensitivity and the ability to form Rad51 foci ex vivo. The absence of Rad51 foci formation post IR-exposure in cells from PDX models derived from HGSOC tumors correlated with olaparib or veliparib sensitivity.Citation73,Citation74 However, AlHilli et al reported that the Rad51-low score ex vivo post IR-exposure failed to predict the response to niraparib in HGSOC PDX models.Citation75 This study incorporated five HGSOC models and assessed Rad51 in G2/S cells by geminin staining, but even though few if any Rad51 foci were formed after ionizing radiation in PDX models with BRCA2 mutations, the ability to form Rad51 foci was not a good predictor of niraparib response in vivo.
Evaluation of Rad51 at the appropriate time point post-treatment is critical to have some degree of confidence in the correlation with HR proficiency; however, there is great interest in the next steps of establishing a clinically feasible Rad51 foci assay that could be performed before treatment (rather than on a post-treatment biopsy) for potential use as an assay to predict PARPi sensitivity prior to treatment. Castroviejo-Bermejo et al established Rad51 scores in vehicle-treated breast cancer PDXs, and, using a cutoff of Rad51 foci in <10% of geminin-positive cells, the authors were able to correlate Rad51-low scores from vehicle-treated PDXs with olaparib sensitivity, suggesting the enticing possibility of a baseline olaparib sensitivity assay.Citation76 In other studies, including those cited above, the ability to detect significant differences in Rad51 foci in baseline tumors was not possible due to the low levels of endogenous DNA damage in baseline tumor specimens.Citation71,Citation72 In addition, some tumors have low levels of geminin-positive cells due to slow proliferation rate. The PDX models used in Castroviejo-Bermejo et al were reported by the authors to have high levels of endogenous DNA damage, which was correlated to γH2AX signal.Citation76 The presence of γH2AX as an independent marker of DSBs in actively proliferating (ie, geminin-positive) cells indicated that the lack of Rad51 foci formation in olaparib-sensitive PDXs was not due to a general lack of DNA damage induction, but instead an indication of HR deficiency. The low levels of endogenous DNA damage usually observed in untreated PDX tissue (and in human tumor biopsies) is a critical limitation in the ability to evaluate significant differences between Rad51 scores in untreated PARPi-sensitive and -resistant models. This approach is not suitable for tumors with low proliferation rates or low endogenous DNA damage.
Major differences across the studies to date of Rad51 as a marker of HR deficiency and PARPi sensitivity include the method of detection used (IHC and immunofluorescence assays), the resolution of the microscopy, and the reliability of detection of Rad51 foci in FFPE samples. Validation of the Rad51 antibodies used across the studies is needed to specify the readout of Rad51 versus paralogs such as Rad51C and Rad51D. For clinical utility of the assay, it is important to have a thorough understanding of the baseline variability of Rad51 expression within and across histologies and to understand the minimum number of cells required to make a meaningful measurement. The quantitative biological cut-off of a true Rad51-Low score may be dependent on tissue type, proliferation rate, and whether the entire population or only the S/G2 population of cells are evaluated using geminin co-expression.Citation77 Despite these considerations, the most relevant measurement expected to reflect HR proficiency is not the baseline Rad51 but rather the induction of Rad51 foci after DSB-inducing genotoxic insults. This post-treatment measurement requires biopsy tissue to be taken at the appropriate time point expected to produce an HR response, or may lead to reporting of false negatives. The need for post-treatment biopsies at appropriate time points presents a significant challenge for the use of this assay as a selection criterion.
Currently, further clinical validation and retrospective studies are needed to determine whether the Rad51 foci assay can predict PARPi sensitivity in clinical practice. Towards these efforts, Meijer et al have developed the Repair Capacity (RECAP) test in which fresh tissue biopsies from patients with metastatic breast cancer lesions were irradiated with 5Gy ex vivo and cultured for 2 hours.Citation78 The core needle or punch biopsies were then formalin-fixed, paraffin-embedded and evaluated for the presence of Rad51 (HR-proficient) or absence (HR-deficient) in at least 30 geminin-positive cells. The authors demonstrated that, while technically challenging, this approach may be feasible in some specialized laboratories. Adaptation of this approach has been applied to surgically resected ovarian carcinoma specimens treated with ex vivo irradiation.Citation79 Other ex vivo studies are underway to address the feasibility of assessing patient-derived organoids as a platform to profile DNA repair defects using the approach of combining a panel of multiple genomic and functional assays. Hill et al reported that while genomics assays alone could not predict therapeutic response, a combination of genomic and functional HR deficiency assays correlated with sensitivity to PARPi in organoids derived from high-grade serous ovarian cancers.Citation80 This platform may provide an alternative method of assessing HR deficiency using multiple markers in addition to Rad51 foci induction after ionizing radiation.
Replication Stress
In addition to its role in HR, Rad51 also plays a known role in replication fork protection. Fork protection allows for the recovery and restart of replication in response to genomic insults and chemotherapy. Rad51 localizes at stalled replication forks to prevent fork collapse or fork degradation via processing by MRE11 exonuclease.Citation81,Citation82 While PARPi have remarkable promise in cancers with HRD, the role of other proteins involved in replication stress may be an additional, critical component of cellular response to PARP inhibition.Citation83 One notable marker that may have strong predictive value and can be robustly detected by immunohistochemistry is SLFN11, a marker of replication stress that has emerged as a potential predictive marker of sensitivity to PARPi. SLFN11 is a nuclear helicase that is recruited to ssDNA regions of stalled replication forks by RPA1 and is thought to block replication by loosening and opening chromatin in an ATPase-dependent mechanism.Citation84 Genetic loss of SLFN11 at the transcript level among cell lines in the NCI-60 cell-line panel was reported as being highly resistant to talazoparib.Citation85,Citation86 SLFN11 has been identified in independent studies as a determinant of response to PARPi in small cell lung cancer (SCLC), in the absence of HR deficiency using both cell lineCitation87 and PDXCitation88 experiments. We and others have developed an assay to evaluate SLFN11 in FFPE tissue from baseline biopsies in support of further studies to retrospectively evaluate its expression related to clinical response to PARPi.Citation89 There are currently several advantages to the SLFN11 assay including the ability to measure using a semi-quantitative IHC assay in a baseline (pre-treatment) biopsy using routine clinical practices. Further investigation is required to fully understand the tissue-specific expression pattern of SLFN11, which is not expressed in normal colon, breast, bladder and prostate tissue.Citation89 Retrospective studies of clinical specimens will be needed to establish the correlation of SLFN11 expression (either loss or re-expression) and PARPi sensitivity and resistance across tumor types.
HR Deficiency Testing in the Clinic
In the clinical setting, response to first-line platinum chemotherapy for patients with advanced HGSC has traditionally been utilized as a surrogate predictive biomarker of benefit from PARPi therapy, supported by trial-level data that reported platinum-sensitivity better predicted PARPi benefit than tissue or blood HR deficiency testing.Citation90 There is not currently a single standardized test to detect HR deficiency—to date, only two testing strategies have been recognized and approved by the FDA as companion diagnostic tests (CDx) specifically for patient treatment selection: myChoice® CDx for the use of olaparib and niraparib in HGSC, and FoundationOne CDx for the use of olaparib in patients with HR repair gene-mutated (both germline or somatic) metastatic castration-resistant prostate cancer (mCRPC). Both of these tests were prospectively validated but have not been compared head-to-head in clinical trials. Both putatively determine HR deficiency using some combination of gene-based testing for BRCA1/2 mutations and LOH assessment, although myChoice® adds telomere allelic imbalance and large-scale state transitions to its composite score. The challenges with both tests are also similar: examining blood germline defects does not capture somatic alterations in a tumor tissue specimen that include genomic information besides HR deficiency, initial biopsy or specimens from debulking surgery are a single snapshot in time (and usually archival) and re-biopsy is needed to evaluate HR changes over time, and also adequate tissue and correct specimen handling are needed. Additionally, these concerns are all irrespective of financial challenge associated with equitable access to these tests globally for patients with reproductive malignancies. We agree with Ngoi et al that the key element is being able to identify patients with HGSC that are HR deficient given the promise of benefit with PARPi therapy in front-line maintenance therapy and their recommendation of selecting targeted germline testing and/or tumor BRCA1/2 mutation testing first line, and subsequently the consideration of HR deficiency assays in patients who are wild-type for BRCA.Citation90
Separately, both preclinical and preliminary clinical data have shown that patients who have mutations in HR repair genes other than BRCA1/2 can benefit from PARPi therapy.Citation42,Citation91,Citation92 The prevalence of these mutations across non-reproductive solid tumors has been reported to be less than 20%,Citation93 and the rarity of these alterations makes developing clinical trials with sufficient statistical power to demonstrate the benefit of PARPi in these populations a challenge. Multiple companies have off-the-shelf targeted sequencing panels that include HR repair genes “beyond” BRCA1/2, and summarizes the use of these non-BRCA1/2 HR repair genes as eligibility criteria in currently recruiting PARPi clinical trials listed on clinicaltrials.gov.
Table 1 Currently Accruing Clinical Trials of PARPi Using Non-BRCA1/2 Biomarkers of HR Deficiency to Select Patients
Selection of patients using cfDNA is less common than tissue-based approaches to date, despite offerings from FoundationOne (FoundationOne Liquid CDx) and newer entrants to the field such as the Resolution HR deficiency (Resolution Bioscience) liquid biopsy assay for detecting sequence variation in genes related to HR deficiency, which received FDA breakthrough device designation in May of 2019.Citation94 However, the field of liquid biopsy continues to evolve: although these approaches remain experimental at this time, detection of cfDNA and reliable identification of BRCA mutations appear technically possible.Citation60 HR deficiency is dynamic, so the possibility of sampling a tumor serially could have clinical implications beyond diagnosis such as detecting treatment resistance. Additionally, issues such as residual disease, presence of subclonal populations, and clonal hematopoiesis are considerations that have been identified as contributing to technical issues relating to discordance between tumor and tissue sampling.Citation95
lists some of the currently ongoing clinical trials of PARPi, either as monotherapy or in combination, whose eligibility requirements specifically target HR repair gene alterations other than BRCA1/2. While the genes chosen to select patients in these trials vary, few utilize genomic scar measures. None of the trials identified by our search as currently recruiting (clinicaltrials.gov, accessed September 2021) utilize non-genomic platforms to determine eligibility, and we were only able to identify two reported studies that have trialed protein or RNA methodology. A first-in-human trial of PARP/tankyrase inhibitor E7449 utilized a 414 gene expression biomarker panel analyzed via a customized RNA microarray analysis in archival tissue.Citation96 Positively correlated and negatively correlated genes for drug sensitivity generated a single prediction score that identified patients achieving both tumor response prolonged stabilization of disease, irrespective of their BRCA mutation status. A further randomized, double-blind Phase II study of paclitaxel versus olaparib plus paclitaxel in advanced gastric cancer enrolled patients with low or undetectable ATM protein levels, based on the premise of improved clinical outcome in low expressors.Citation97 The study population was enriched to 50% (low/undetectable ATM by IHC on archival biopsy from either primary or metastatic lesions), and randomized 1:1; however, the study did not achieve its primary endpoint of improved PFS in ATM-low patients and the combination improved overall survival for the overall population as well as the ATM low expressing patients. Ultimately, interpreting the relevance of HR-related mutations is likely still to be dependent on the clinical and histological context of a patient, as few of these mutations predict for PARPi benefit outside of reproductive cancers.Citation34,Citation92
Conclusions
The current “revolutions” in genomics and big data will further evolve as the cost of sequencing continues to fall and its output continues to change patient care. More patients will undergo panel testing for pathogenic mutations and whole-exome sequencing, eventually enabling identification of small subpopulations who will benefit from highly targeted therapies. cfDNA sequencing may provide more insight into the full spectrum of metastatic disease than is available from a single-site biopsy. The relevance of HR deficiency testing, however, remains unclear: HR-proficient patients with HGSC have derived benefit from PARPi therapy if not to the same extent as HR-deficient,Citation46,Citation90,Citation98 suggesting that the quest for a gold standard HR deficiency measure is ongoing.
Disclosure
Dr Chris Karlovich is an ex-employee of Clovis Oncology, which has developed the PARP inhibitor rucaparib, during the conduct of the study. Andrea Regier Voth, Ralph E Parchment, Deborah Wilsker and Chris Karlovich received grants from the National Institutes of Health/National Cancer Institute Contract Number HHSN261201500003I, during the conduct of the study. The authors declare no other potential conflicts of interest.
Additional information
Funding
References
- Chabner BA, Roberts TG Jr. Timeline: chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5(1):65–72. doi:10.1038/nrc1529
- Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353(2):172–187. doi:10.1056/NEJMra044389
- Falzone L, Salomone S, Libra M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front Pharmacol. 2018;9:1300. doi:10.3389/fphar.2018.01300
- Tsimberidou AM. Targeted therapy in cancer. Cancer Chemother Pharmacol. 2015;76(6):1113–1132. doi:10.1007/s00280-015-2861-1
- Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12:14.
- Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11(1):39–43. doi:10.1016/0006-291x(63)90024-x
- Curtin N. PARP inhibitors for anticancer therapy. Biochem Soc Trans. 2014;42(1):82–88. doi:10.1042/BST20130187
- Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283(5747):593–596. doi:10.1038/283593a0
- O’Sullivan Coyne G, Chen AP, Meehan R, Doroshow JH. PARP inhibitors in reproductive system cancers: current use and developments. Drugs. 2017;77(2):113–130. doi:10.1007/s40265-016-0688-7
- O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547–560. doi:10.1016/j.molcel.2015.10.040
- Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer. 2015;15(3):166–180. doi:10.1038/nrc3891
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi:10.1038/nature08467
- Friedberg EC. A brief history of the DNA repair field. Cell Res. 2008;18(1):3–7. doi:10.1038/cr.2007.113
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73(1):39–85. doi:10.1146/annurev.biochem.73.011303.073723
- Hakem R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008;27(4):589–605. doi:10.1038/emboj.2008.15
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi:10.1016/j.molcel.2010.09.019
- Bader AS, Hawley BR, Wilczynska A, Bushell M. The roles of RNA in DNA double-strand break repair. Br J Cancer. 2020;122(5):613–623. doi:10.1038/s41416-019-0624-1
- McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. doi:10.1158/0008-5472.CAN-06-0140
- Caron MC, Sharma AK, O’Sullivan J, et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat Commun. 2019;10(1):2954. doi:10.1038/s41467-019-10741-9
- Chan CY, Tan KV, Cornelissen B. PARP inhibitors in cancer diagnosis and therapy. Clin Cancer Res. 2021;27(6):1585–1594. doi:10.1158/1078-0432.CCR-20-2766
- Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336(6082):728–732. doi:10.1126/science.1216338
- Langelier MF, Zandarashvili L, Aguiar PM, Black BE, Pascal JM. NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat Commun. 2018;9(1):844. doi:10.1038/s41467-018-03234-8
- Liu X, Shi Y, Maag DX, et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a bona fide PARP inhibitor. Clin Cancer Res. 2012;18(2):510–523. doi:10.1158/1078-0432.CCR-11-1973
- Murai J, Pommier Y. PARP trapping beyond homologous recombination and platinum sensitivity in cancers. Annu Rev Cancer Biol. 2019;3(1):131–150. doi:10.1146/annurev-cancerbio-030518-055914
- Gourley C, Balmana J, Ledermann JA, et al. Moving from Poly (ADP-Ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J Clin Oncol. 2019;37(25):2257–2269. doi:10.1200/JCO.18.02050
- Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. doi:10.1158/0008-5472.CAN-12-2753
- Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585(1):1–6. doi:10.1016/j.febslet.2010.11.024
- Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152–1158. doi:10.1126/science.aam7344
- Miller RE, Leary A, Scott CL, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020;31(12):1606–1622. doi:10.1016/j.annonc.2020.08.2102
- Radhakrishnan SK, Jette N, Lees-Miller SP. Non-homologous end joining: emerging themes and unanswered questions. DNA Repair (Amst). 2014;17:2–8. doi:10.1016/j.dnarep.2014.01.009
- Lin KK, Harrell MI, Oza AM, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9(2):210–219. doi:10.1158/2159-8290.CD-18-0715
- Venkitaraman AR. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev Pathol. 2009;4:461–487. doi:10.1146/annurev.pathol.3.121806.151422
- den Brok WD, Schrader KA, Sun S, et al. Homologous recombination deficiency in breast cancer: a clinical review. Jco Precis Oncol. 2017;1. doi:10.1200/Po.16.00031
- Hoppe MM, Sundar R, Tan DSP, Jeyasekharan AD. Biomarkers for homologous recombination deficiency in cancer. J Natl Cancer Inst. 2018;110(7):704–713. doi:10.1093/jnci/djy085
- Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87. doi:10.1016/S1470-2045(16)30559-9
- U.S. Food & Drug Administration. List of cleared or approved companion diagnostic devices (in vitro and imaging tools). Available from: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools. Accessed July 2021.
- Swisher EM, Kwan TT, Oza AM, et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat Commun. 2021;12(1):2487. doi:10.1038/s41467-021-22582-6
- Sharma P, Barlow WE, Godwin AK, et al. Impact of homologous recombination deficiency biomarkers on outcomes in patients with triple-negative breast cancer treated with adjuvant doxorubicin and cyclophosphamide (SWOG S9313). Ann Oncol. 2018;29(3):654–660. doi:10.1093/annonc/mdx821
- Bhangoo MS, Costantini C, Clifford BT, et al. Biallelic deletion of PALB2 occurs across multiple tumor types and suggests responsiveness to Poly (ADP-ribose) polymerase inhibition. Jco Precis Oncol. 2017;1. doi:10.1200/Po.17.00043
- Reiss KA, Mick R, O’Hara MH, et al. Phase II study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic variant in BRCA1, BRCA2, or PALB2. J Clin Oncol. 2021;39(22):2497–2505. doi:10.1200/JCO.21.00003
- Mateo J, Porta N, Bianchini D, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21(1):162–174. doi:10.1016/S1470-2045(19)30684-9
- Tung NM, Robson ME, Ventz S, et al. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol. 2020;38(36):4274–4282. doi:10.1200/JCO.20.02151
- Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi:10.1056/NEJMoa0808710
- Sulkowski PL, Corso CD, Robinson ND, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9(375). doi:10.1126/scitranslmed.aal2463
- Eder JP, Doroshow DB, Do KT, et al. Clinical efficacy of olaparib in IDH1/IDH2-mutant mesenchymal sarcomas. Jco Precis Oncol. 2021;5:466–472. doi:10.1200/Po.20.00247
- Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. doi:10.1016/S0140-6736(17)32440-6
- Telli ML, Timms KM, Reid J, et al. Homologous Recombination Deficiency (HRD) Score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22(15):3764–3773. doi:10.1158/1078-0432.CCR-15-2477
- Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. doi:10.1056/NEJMoa1611310
- Nguyen L, Martens JWM, Van Hoeck A, Cuppen E. Pan-cancer landscape of homologous recombination deficiency. Nat Commun. 2020;11(1):5584. doi:10.1038/s41467-020-19406-4
- Lotan TL, Kaur HB, Salles DC, et al. Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod Pathol. 2021;34(6):1185–1193. doi:10.1038/s41379-020-00731-4
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
- Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23(4):517–525. doi:10.1038/nm.4292
- Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019;51(5):912–919. doi:10.1038/s41588-019-0390-2
- Wolf DM, Yau C, Sanil A, et al. DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. NPJ Breast Cancer. 2017;3(1):31. doi:10.1038/s41523-017-0025-7
- Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi:10.1038/nature06633
- Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. doi:10.1038/nature06548
- Dias MP, Moser SC, Ganesan S, Jonkers J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol. 2021. doi:10.1038/s41571-021-00532-x
- Pettitt SJ, Frankum JR, Punta M, et al. Clinical BRCA1/2 reversion analysis identifies hotspot mutations and predicted neoantigens associated with therapy resistance. Cancer Discov. 2020;10(10):1475–1488. doi:10.1158/2159-8290.CD-19-1485
- Kondrashova O, Nguyen M, Shield-Artin K, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7(9):984–998. doi:10.1158/2159-8290.CD-17-0419
- Goodall J, Mateo J, Yuan W, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017;7(9):1006–1017. doi:10.1158/2159-8290.CD-17-0261
- Christie EL, Fereday S, Doig K, Pattnaik S, Dawson SJ, Bowtell DDL. Reversion of BRCA1/2 germline mutations detected in circulating tumor DNA from patients with high-grade serous ovarian cancer. J Clin Oncol. 2017;35(12):1274–1280. doi:10.1200/JCO.2016.70.4627
- Weigelt B, Comino-Mendez I, de Bruijn I, et al. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin Cancer Res. 2017;23(21):6708–6720. doi:10.1158/1078-0432.CCR-17-0544
- Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. doi:10.1056/NEJMoa1506859
- Jaspers JE, Sol W, Kersbergen A, et al. BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res. 2015;75(4):732–741. doi:10.1158/0008-5472.CAN-14-0839
- Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. doi:10.1073/pnas.0806092105
- Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–494. doi:10.1038/nature14410
- Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–695. doi:10.1038/nsmb.1831
- Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–254. doi:10.1016/j.cell.2010.03.012
- Waks AG, Cohen O, Kochupurakkal B, et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann Oncol. 2020;31(5):590–598. doi:10.1016/j.annonc.2020.02.008
- Johnson N, Johnson SF, Yao W, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110(42):17041–17046. doi:10.1073/pnas.1305170110
- Graeser M, McCarthy A, Lord CJ, et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010;16(24):6159–6168. doi:10.1158/1078-0432.CCR-10-1027
- Chopra N, Tovey H, Pearson A, et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat Commun. 2020;11(1):2662. doi:10.1038/s41467-020-16142-7
- Parmar K, Kochupurakkal BS, Lazaro JB, et al. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin Cancer Res. 2019;25(20):6127–6140. doi:10.1158/1078-0432.CCR-19-0448
- Shah MM, Dobbin ZC, Nowsheen S, et al. An ex vivo assay of XRT-induced Rad51 foci formation predicts response to PARP-inhibition in ovarian cancer. Gynecol Oncol. 2014;134(2):331–337. doi:10.1016/j.ygyno.2014.05.009
- AlHilli MM, Becker MA, Weroha SJ, et al. In vivo anti-tumor activity of the PARP inhibitor niraparib in homologous recombination deficient and proficient ovarian carcinoma. Gynecol Oncol. 2016;143(2):379–388. doi:10.1016/j.ygyno.2016.08.328
- Castroviejo-Bermejo M, Cruz C, Llop-Guevara A, et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol Med. 2018;10(12):Dec. doi:10.15252/emmm.201809172
- Wilsker DF, Barrett AM, Dull AB, et al. Evaluation of pharmacodynamic responses to cancer therapeutic agents using DNA damage markers. Clin Cancer Res. 2019;25(10):3084–3095. doi:10.1158/1078-0432.CCR-18-2523
- Meijer TG, Verkaik NS, van Deurzen CHM, et al. Direct ex vivo observation of homologous recombination defect reversal after DNA-damaging chemotherapy in patients with metastatic breast cancer. Jco Precis Oncol. 2019;3. doi:10.1200/Po.18.00268
- van Wijk LM, Vermeulen S, Meijers M, et al. The RECAP test rapidly and reliably identifies homologous recombination-deficient ovarian carcinomas. Cancers. 2020;12(10):2805. doi:10.3390/cancers12102805
- Hill SJ, Decker B, Roberts EA, et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 2018;8(11):1404–1421. doi:10.1158/2159-8290.CD-18-0474
- Zellweger R, Dalcher D, Mutreja K, et al. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. 2015;208(5):563–579. doi:10.1083/jcb.201406099
- Berti M, Teloni F, Mijic S, et al. Sequential role of RAD51 paralog complexes in replication fork remodeling and restart. Nat Commun. 2020;11(1):3531. doi:10.1038/s41467-020-17324-z
- Le Page C, Amuzu S, Rahimi K, Gotlieb W, Ragoussis J, Tonin PN. Lessons learned from understanding chemotherapy resistance in epithelial tubo-ovarian carcinoma from BRCA1and BRCA2mutation carriers. Semin Cancer Biol. 2021;77:110–126. doi:10.1016/j.semcancer.2020.08.005
- Murai J, Tang SW, Leo E, et al. SLFN11 blocks stressed replication forks independently of ATR. Mol Cell. 2018;69(3):371–384e6. doi:10.1016/j.molcel.2018.01.012
- Murai J, Thomas A, Miettinen M, Pommier Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol Ther. 2019;201:94–102. doi:10.1016/j.pharmthera.2019.05.009
- Murai J, Feng Y, Yu GK, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534–76550. doi:10.18632/oncotarget.12266
- Lok BH, Gardner EE, Schneeberger VE, et al. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res. 2017;23(2):523–535. doi:10.1158/1078-0432.CCR-16-1040
- Stewart CA, Tong P, Cardnell RJ, et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget. 2017;8(17):28575–28587. doi:10.18632/oncotarget.15338
- Takashima T, Sakamoto N, Murai J, et al. Immunohistochemical analysis of SLFN11 expression uncovers potential non-responders to DNA-damaging agents overlooked by tissue RNA-seq. Virchows Arch. 2021;478(3):569–579. doi:10.1007/s00428-020-02840-6
- Ngoi NYL, Tan DSP. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: do we need it? ESMO Open. 2021;6(3):100144. doi:10.1016/j.esmoop.2021.100144
- Hodgson DR, Dougherty BA, Lai Z, et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br J Cancer. 2018;119(11):1401–1409. doi:10.1038/s41416-018-0274-8
- Wagener-Ryczek S, Merkelbach-Bruse S, Siemanowski J. Biomarkers for homologous recombination deficiency in cancer. J Pers Med. 2021;11(7):612. doi:10.3390/jpm11070612
- Heeke AL, Pishvaian MJ, Lynce F, et al. Prevalence of homologous recombination-related gene mutations across multiple cancer types. Jco Precis Oncol. 2018;2018. doi:10.1200/PO.17.00286
- Resolution liquid biopsy assay receives breakthrough device designation from FDA. Available from: https://www.resolutionbio.com/company/press/2019.05.30_resolution_fda_btd.html. Accessed January 27, 2022.
- Williams PM, Forbes T, Lund SP, et al. Validation of ctDNA quality control materials through a precompetitive collaboration of the foundation for the National Institutes of Health. Jco Precis Oncol. 2021;5. doi:10.1200/PO.20.00528
- Plummer R, Dua D, Cresti N, et al. First-in-human study of the PARP/tankyrase inhibitor E7449 in patients with advanced solid tumours and evaluation of a novel drug-response predictor. Br J Cancer. 2020;123(4):525–533. doi:10.1038/s41416-020-0916-5
- Bang YJ, Im SA, Lee KW, et al. Randomized, double-blind phase ii trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol. 2015;33(33):3858–3865. doi:10.1200/JCO.2014.60.0320
- Del Campo JM, Matulonis UA, Malander S, et al. Niraparib maintenance therapy in patients with recurrent ovarian cancer after a partial response to the last platinum-based chemotherapy in the ENGOT-OV16/NOVA trial. J Clin Oncol. 2019;37(32):2968–2973. doi:10.1200/JCO.18.02238