Abstract
Background:
Malignant peripheral nerve sheath tumors (MPNSTs) are rare entities and MPNSTs of intracranial nerves are even more sporadic. MPNSTs present diagnosis and treatment challenges since there are no defined diagnosis criteria and no established therapeutic strategies.
Methods:
We reviewed literature for MPNST-related articles. We found 45 relevant studies in which 60 cases were described.
Results:
We identified 60 cases of intracranial nerve MPNSTs. The age ranged from 3 to 75 years old. Male to female ratio was 1.5:1. The most involved cranial nerves (CNs) were CN VIII (60%), CN V (27%), and CN VII (10%). Most of the MPNSTs reported (47%) arose sporadically, 40% arose from a schwannoma, 8% arose from a neurofibroma, and 6% arose from an unspecified nerve tumor. Twenty patients had a history of radiation exposure, four patients had neurofibromatosis type 1 (NF1), four patients had neurofibromatosis type 2 (NF2), and NF2 was suspected in two other patients. Twenty-two patients were treated with radiotherapy and presented a higher survival rate. Seventy-two percent of patients died of their disease while 28% of patients survived. One-year survival rate was 33%. Forty-five percent of tumors recurred and 19% of patients had metastases.
Conclusion:
MPNSTs involving CNs are very rare. Diagnosis is made in regards to the histological and pathological findings. Imaging may help orient the diagnosis. A preexisting knowledge of the clinical situation is more likely to lead to a correct diagnosis. The mainstay of treatment is radical surgical resection with adjuvant radiotherapy. Since these tumors are associated with a poor prognosis, a close follow-up is mandatory.
Introduction
Malignant peripheral nerve sheath tumor (MPNST) is a malignant tumor arising from or differentiating toward cells of the nerve sheath. Most of these tumors arise within a normal peripheral nerve or from preexisting nerve sheath tumors, usually intraneural or plexiform neurofibromas. Transition from a schwannoma is a much rarer phenomenon.Citation1 An association between therapeutic irradiation and the development of MPNSTs is well established.Citation2 Indeed, cases have been published describing malignant transformation of benign tumors, mainly schwannomas, after a history of radiation exposure.
While accounting for 5% to 10% of all soft tissue sarcomas,Citation3 MPNSTs are rare tumors with an incidence of 0.001% in the general population.Citation4 Intracranial MPNSTs are aggressive tumors that are even more sporadic; literature is therefore limited to isolated case reports and small series. MPNSTs remain a diagnosis challenge since they lack specific criteria radiologicallyCitation5 and pathologically.Citation6 Clinical evolution is central to making a proper diagnosis.
Total surgical resection is the standard treatment for MPNSTs of all body sites.Citation7,Citation8 While the foremost treatment is surgical, difficult access to cranial nerves (CNs) does not always allow for complete tumor resection.Citation9 Adjuvant therapies become even more important in this context. Radiotherapy is the most widely used adjuvant treatment. While development of MPNST may be associated with a history of radiation therapy in some cases, a longer survival rate has been reported with adjuvant radiotherapy in the treatment of MPNST.Citation7 Even with aggressive treatment regimens, the prognosis for intracranial nerve MPNSTs remains poor, being poorer than for MPNSTs in all other body sites.Citation7,Citation8
The scope of this paper is to review all cases of MPNSTs of intracranial nerves described in the literature in order to identify the etiology, the histopathology, the immunohistochemistry, and to discuss the differential diagnosis, treatments, metastasis possibilities, the prognosis, and overall survival.
Materials and methods
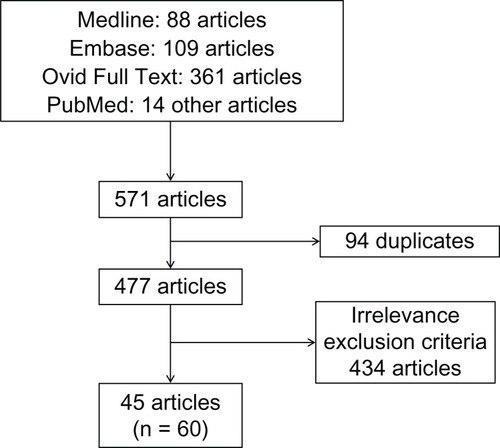
We performed a Medline, an Embase, an Evidence-Based Medicine Reviews/Cochrane, and an Ovid Full Text databases and PubMed search for MPNST-related articles. No limitations regarding language or time of publication were imposed. We used the keywords: “malignant peripheral nerve sheath tumor,” “cranial nerve,” “intracranial,” “malignant schwannoma,” “malignant neurofibroma,” “vestibular schwannoma,” “trigeminal schwannoma,” “malignancy,” and “malignant transformation.” We considered cases where MPNSTs involved intracranial CNs. Studies concerning head and neck MPNSTs without specifying the exact location of the tumor were excluded. We also excluded cases of MPNSTs arising from extracranial CNs and brain parenchyma. We found 31 relevant studies through Medline, Embase, and Ovid Full Text (27 articles, two reviews, and two congress abstracts) and 14 other studies through PubMed () in which 60 cases of intracranial MPNSTs were reported. Two of these articles were reviews (Chen et al and Scheithauer et al).Citation9,Citation10 In one article (Bari et al),Citation11 a congress abstract (Kurita et al)Citation12 not found elsewhere was mentioned. We analyzed information concerning the clinical aspects, the histopathology, the treatment, the rate of recurrence, and the follow-up.
Figure 1 Flow chart representing the review methodology.
Results
We found 60 cases of MPNSTs of intracranial CNs (). The mean age was 39.3 ± 4.95 (3–75) years. The male to female ratio was 1.5:1. The most involved CN was CN VIII (60%), followed by CN V (27%) and CN VII (10%). Among the 53 cases where precursors of lesions were mentioned, we found that 47% of MPNSTs reported arose sporadically, 40% arose from a malignant transformation of schwannoma, 8% arose from a neurofibroma, and 6% arose from an unspecified nerve tumor (). Four patients had neurofibromatosis type 1 (NF1), four patients had neurofibromatosis type 2 (NF2), and NF2 was suspected in two other patients. Otherwise, 20 patients had a history of radiation exposure; 17 of which presented a malignant transformation of a previously benign tumor.
Table 1 Review of published cases of intracranial cranial nerve MPNSTs
Table 2 Results of the 60 reported cases of intracranial MPNSTs associated with cranial nerves
Because of the different CNs involved, clinical presentation is variable in these patients. Clinical symptoms are related to the location and the extension of the tumor.
Of the 28 patients who presented a malignant transformation of a former benign entity (), 17 patients (61%) had previous radiation exposure: eleven had stereotactic radiosurgery (SRS) and six had conventional radiotherapy. Among those 17 patients, seven (41%) were retreated with radiotherapy. The mean time to malignancy was 6.1 years (1 month to 19 years). Three patients were alive at the one-year follow-up. Nineteen of these patients died with a mean survival rate of 6.3 months (1–20 months).
Table 3 Malignant transformation of benign tumors
In regards to the pathological findings, the predominant histological type was spindle cells followed by rarer epithelioid cells and rhabdomyoblastic cells. All of the histological types found are presented in . One tumor presented four different histological types, two tumors presented three histological types, and seven tumors presented two histological types.
Immunohistochemistry findings are summarized in . S100 was the most used marker; p53, Ki67, and MIB-1 appear to have been used more frequently in the last few years. A lot of other markers have been tested heterogeneously, among them: vimentin, desmin, myoglobin, protein gene product 9.5 (PGP 9.5), glial fibrillary acidic protein, CD99, p75, bcl-2, epithelial membrane antigen (EMA), collagen-IV, and synaptophysin.
Table 4 Immunohistochemistry results reported in MPNST cases
Concerning treatment, surgery is the primary treatment; only one patient was not treated with surgery because of his advanced stage disease at presentation. Gross total surgery was preferred but could not be achieved in all patients according to the localization and the extension of the tumor. Radiotherapy was used in 22 patients, doses ranging from 45–70 Gy, and SRS was used in six of them. Five patients received chemotherapy regimens after surgical resection. Only one was treated with combined radiotherapy and chemotherapy.
Recurrence occurred in 45% of cases and metastases were found in 19% of cases. All metastases except one involved the cervical, thoracic, and lumbar spine and were pial-based or dural-based. The metastasis that did not involve the spine was found in the pons. It was specified in three cases that metastasis happens by cerebrospinal fluid (CSF) dissemination. At follow-up, 36 patients had died and 14 patients were alive with or without complications (see ). Interestingly, patients who were treated with adjuvant radiotherapy had a one-year survival rate of 65% compared to 12% for patients who did not receive radiotherapy. Among the 42 cases in which length of follow-up was mentioned, the one-year survival rate was 33%. Complications were not clearly enunciated in most of the cases reported, but they consisted mainly of CNs dysfunction and residual pain. Two patients that were alive at follow-up were receiving supportive care and three patients showed no complications. Among the patients who were alive, follow-up ranged from 7 to 60 months. Among the 36 patients who died, the mean survival rate was 9.2 months (1–84 months).
Discussion
MPNST defines a spindle cell sarcoma arising from a nerve or a nerve sheath tumor that demonstrates neural differentiation. The World Health Organization (WHO) created this terminology to replace these previous heterogeneous terminologies: neurofibrosarcoma, neurogenic sarcoma, malignant neurilemoma, malignant schwannoma, and malignant neurofibroma.
Etiology
MPNSTs arise within a normal peripheral nerve or from pre-existing nerve sheath tumors, usually intraneural or plexiform neurofibromas, and more rarely from a schwannoma.
MPNSTs have three recognized etiologies: tumors may occur in association with neurofibromatosis, may be the consequence of previous radiation therapy, or may occur sporadically.Citation13 However, intracranial MPNSTs more frequently arise from schwannomas than neurofibromas. Indeed, intracranial neurofibromas are exceptionally rare while most intracranial nerve sheath tumors are schwannomas.
Radiation-induced
An association between therapeutic irradiation and the development of MPNSTs is well-established. Foley et al suggested that radiation-induced MPNSTs result in proliferation and cytologic atypia in Schwann cells and chromosomic injury caused by ionizing radiation.Citation2 As reported in the results, eleven cases of malignant brain tumor occurred after stereotactic radiation, for vestibular schwannoma mainly (nine cases). SRS or fractioned stereotactic radiotherapy allows for the administration of precisely directed, high-dose irradiation. In recent years, it has become a commonly used modality in the treatment of benign nerve sheath tumors. Since malignant transformation of such tumors remains a rare phenomenon, SRS continues to be used as it is an effective treatment for brain tumors. Indeed, the rate of local control constitutes 92% to 98.5%, whereas the associated risk of acute adverse effects is extremely low.Citation12 The opponents of radiosurgery, however, discuss the possibility of the formation of new tumors or the malignant transformation of benign tumors. Some authors advocate careful weighing of the risks and benefits, especially in patients affected by NF1.Citation14
The relation between SRS and malignant transformation of benign tumors remains unclear since radiosurgery is frequently used after incomplete surgical resection if residual neoplasm shows regrowth and there are no specific histological criteria for the identification of the irradiation-induced malignant transformation of benign intracranial tumors.Citation15 However, in 1948, Cahan et alCitation16 first outlined the criteria of a radiation-induced tumor: (1) a second tumor occurs within the radiation field and it was not present at the time of irradiation; (2) a latency period is required between radiotherapy and tumor development (several years); (3) a histological difference must exist between the primary and the new tumor; and (4) the patient should not have any genetic predisposition for cancer development. Indeed, a latency period is suggestive of a malignant transformation since malignant tumors progress rapidly.Citation16 Kubo et al mentioned a usual time elapsed from treatment of 24 months or more when considering malignant progression.Citation15 Besides, the coexistence of benign schwannomatous cells and the malignant component might confirm the hypothesis of malignant transformation. We reported 28 cases of malignant transformation of benign tumors, 17 (61%) of which had a history of radiation exposure, and eleven had received SRS. Primary diagnosis was mentioned in 15 patients; ten of them had primary surgery with a pathological diagnosis of a benign tumor before receiving radiation therapy, and five patients received empiric radiotherapy after a benign tumor was diagnosed based on imaging.
Diagnosis
Clinical signs
Clinical features depend on the location and the nerve involved. We found that the intracranial CNs affected most often by MPNSTs were CN VIII, followed by CN V and CN VII (see ). This is possibly related to the thickness of the nerve root and the prevalence of benign intracranial schwannomas.
Involvement of the vestibulocochlear nerve is associated with hearing loss, deafness, tinnitus, unsteadiness, imbalance, vertigo, and facial numbness or weakness secondary to compression or invasion of the trigeminal and facial nerves, respectfully. When the trigeminal nerve was involved, patients usually complained of facial pain, sensory paresthesias, diminished corneal reflex, and dysfunction of the muscles of mastication. Further growth of trigeminal nerve MPNST causes ophthalmoplegia and lower CNs involvement.Citation17 Otherwise, patients can present with CN deficits along with intracranial hypertension and cerebellar ataxia.Citation9,Citation10
MPNSTs are highly malignant tumors that grow rapidly along nerves, infiltrate surrounding tissues, and develop hematogenous metastasis. Alone, this rapid evolution may differentiate MPNST from its benign counterpart. To illustrate this statement, Bowers et al found that symptom onset to diagnosis is usually longer than 2 to 3 years in benign CN V schwannoma compared to 2.8 months for MPNST of CN V.Citation18 In regards to these metastases, although MPNSTs have been known to develop hematogenous distant metastases, among the eleven reported cases of CN MPNSTs that developed metastases, it was specified in three of these cases that metastases were a consequence of CSF dissemination. Additionally, as metastases were either pial-based or dural-based and localized in the spinal cord in ten cases, it is likely that metastases from primary CN MPNSTs mostly arose from CSF dissemination.
Pathology
The diagnosis of MPNST is controversial due to its lack of specific morphological criteria and immunohistochemical or molecular tests. Among sarcomas, a diagnosis of MPNST is often one of exclusion. Because no cytologic feature or set of features appears to be specific, a pathological diagnosis of MPNST remains challenging. However, a preexisting knowledge of the clinical situations in which MPNST typically occurs is more likely to lead to a correct diagnosis. Tumor characteristics suggestive of MPNST are presented in .
Table 5 Tumor characteristics suggestive of an MPNST
Immunohistochemical features
Neural markers such as S100, CD56, and PGP 9.5 have been considered sensitive markers for peripheral nerve sheath tumors.Citation19 Although S100 was regarded as the best marker, it is positive in only about 50% to 90% of the MPNSTs;Citation20 only scattered, if any, tumor cells are positive in high grade tumors. In fact, the lack of S100 protein immunoreactivity in the malignant areas indicates the possible loss of S100 protein synthesis potentiality through dedifferentiation. Additionally, the association of S100 α expression with malignancy, instead of S100 β has also been suggested.Citation21 In our review, 82% of tumors tested with S100 expressed some level of positivity. The use of EMA, vimentin, neuron-specific enolase, synaptophysin, muscle-specific actin, desmin, S100 protein, myogenin, CD34, CD45, CD117, and cytokeratins may help to establish the proper diagnosis. According to the six cases presenting tumors with rhabdomyoblastic cells, also called malignant triton tumors, these tumors are negative for S100 but positive for desmin and/or myoglobin.
Otherwise, CD56 and PGP 9.5 are sensitive but are not specific to MPNSTs. Studies have recently suggested that nestin, a protein marker for neural stem cells, is more sensitive to MPNSTs than other neural markers such as S100, CD56, and PGP 9.5.Citation22 None of the cases we reported were tested with nestin, but it could be an interesting avenue to explore in the future.
Increased proliferation marker MIB1, an antibody against Ki-67 antigen, is a significant adverse prognosis factor.Citation23 In the cases we reviewed, patients with tumors associated to increased MIB1 had a shorter survival rate compared to all other cases. Expression of p53 does not seem to be a prognosis factor, but it may help differentiate MPNSTs from neurofibromas since it is frequently expressed in MPNST but not in neurofibromas.Citation24
In regards to the markers mentioned, a systematic battery of stains cannot be established, since no stain is sensitive and specific enough to confirm a diagnosis of MPNST alone. Immunohistochemistry analysis can help to determine the diagnosis in combination with clinical, pathological and radiological features. Although S100 has been widely used, vimentin has also been used for testing relatively often. Desmin and myoglobin should be used if rhabdomyoblastic cells are visualized. MIB1 can be of interest as a prognosis factor. In cases in which the malignant transformation of a previously benign tumor is suspected, p53 should be used. Otherwise, we should also start to use nestin for testing.
Differentiation patterns
Frequent histologic findings include (1) fascicles of alternating hypo- and hyper-cell areas, (2) a diffuse growth pattern of asymmetrical and fusiform spindle cells with palisades or rosette-like arrangements of the wavy or comma-shaped hyperchromatic nuclei, (3) perineural/intraneural spread when nerves are involved, (4) subendothelial infiltration into vessel walls, and (5) large areas of geographic-like necrosis.Citation6,Citation19,Citation25 In our review, spindle cells were identified in 83% of MPNSTs, epithelioid cells were found in 13% of cases, and heterologous differentiation was found in 34% of cases. Almost half of the cases (22 of the 46 cases) where pathological evaluation was made presented epithelioid or heterologous differentiation. However, we have to be careful with this result since more cases presenting those less frequent differentiation types may have been published compared to the classical presentation of cases presenting spindle cells.Citation26
According to the WHO, there are four histological types of MPNST: epithelioid MPNST, MPNST with mesenchymal differentiation, melanotic MPNST, and MPNST with glandular differentiation. The first has predominantly large epithelioid cells and the others contain gland-forming epithelium.Citation27 The epithelioid subtype of MPNST has a distinctive differential diagnosis. These tumors more frequently affect superficial sites and express S100 protein strongly and diffusely.Citation25
Rodriguez et al suggest updates to the WHO classification of tumors of cranial and paraspinal nerves, in particular MPNST, because of differences in their evolution and differential diagnosis.Citation25 They proposed seven categories: low-grade MPNST, high-grade MPNST, epithelioid MPNST, MPNST with divergent differentiation, MPNST ex-schwannoma, malignant melanotic schwannoma, and perineural MPNST.Citation25 Considering the cases we reported, MPNSTs with divergent differentiation include tumors in which were found rhabdomyoblastic cells (malignant triton tumors), round cells, lymphoblastic cells, osteoclastic giant cells, and with bone formation. Additionally, we found that a specific category for ex-schwannoma is pertinent since these tumors are quite prevalent among intracranial MPNSTs.
Grading of MPNST
Standardized grading systems for MPNSTs are presently lacking. The French system (Fédération Nationale des Centres de Lutte Contre le Cancer [FNCLCC]) and the WHO grading system are the most commonly used systems for grading soft tissue sarcomas. However, these systems have no prognostic value for some histological subtypes, including MPNSTs.Citation24,Citation28 The WHO grading system may need refinement for MPNST grading, particularly in high-grade categories. Some authors have advocated the use of a practical approach to MPNSTs by dividing tumors into low grade (15%) and high grade (85%).Citation24,Citation25
The WHO grading scheme published in 2007 ranks central nervous system tumors from grade I to grade IV in order to predict a response to therapy and outcome. Grade I applies to lesions with low proliferative potential. Grade II tumors are generally infiltrative and often progress to higher grades of malignancy. Grade III tumors show histological evidence of malignancy, including nuclear atypia and brisk mitotic activity. Grade IV is assigned to cytologically malignant, mitotically active, necrosis-prone tumors typically. Grade II to IV characterize malignant tumors such as MPNSTs.Citation26
The FNCLCC system has three grades (1, 2, and 3) and incorporates three histologic parameters: tissue differentiation, mitotic activity and necrosis.Citation28
These two grading systems allow separation of high-grade categories. It would take more powerful studies to evaluate their prognostic value in MPNSTs and specifically in intracranial MPNSTs considering the small number of cases reported.
Differential diagnosis
The spectrum of differential diagnosis is wide and includes neurofibroma, schwannoma, perineuroma, fibrosarcoma, synovial sarcoma, spindle cell melanoma, spindle cell rhabdomyosarcoma, follicular dendritic cell sarcoma, and ectopic thymoma.Citation29
The distinction between neurofibroma and low-grade MPNST can be challenging particularly in the setting of an NF1 patient. Although hypercellularity, nuclear enlargement, and hyperchromasia have been associated with low-grade MPNST arising in neurofibroma,Citation9 the lack of objective criteria makes malignant diagnosis difficult to establish. The development of a fascicular pattern of growth can be a feature of malignant transformation. MPNSTs tend to show increased MIB1 and p53, while p16 and p27 expression is typically present in neurofibromas.Citation25
Schwannoma can display focal nuclear atypia, but it is encapsulated and diffusely positive; the S100 protein appears as diffusely positive. One of the most useful distinctions from benign schwannoma is the partial or complete loss of S100 expression in MPNST.Citation25
Synovial sarcoma can also be focally positive for S100, but it can be distinguished from MPNST by its more rounded and uniform nuclei, and by the expression of transducin-like enhancer protein (TLE1) (diffuse and strong), bcl-2, CK7, and CK19. Additionally, CD34 expression is not seen in synovial sarcoma. Morphologically, pleomorphic cells are essentially never present in synovial sarcoma, contrary to MPNST.Citation25
Otherwise, epithelioid MPNST differential diagnosis includes melanoma, clear cell sarcoma, epithelioid sarcoma, and carcinoma. Melanoma is diffusely positive for S100, but it is infiltrative and more diffusely pleomorphic with occasional positivity to melanocytic markers: HMB45 and Melan-A. The lack of these markers in epithelioid MPNST helps in its distinction from melanoma and clear cell sarcoma and the absence of cytokeratin expression distinguishes MPNST from carcinoma and epithelioid sarcoma.Citation25
Imaging
Magnetic resonance imaging (MRI) does not allow for the confident distinction of MPNSTs from benign peripheral nerve sheath tumors (BPNSTs). At a size greater than 5 cm, infiltrative margins and marked signal heterogeneity may characterize an MPNST rather than a BPNST.Citation5 Adjacent structures compression and local invasion are more likely to be seen in MPNSTs. MRI is useful to follow progression of MPNSTs and to assess post-surgical status. Otherwise, since the metastases reported are relatively frequent and almost all involve the spine, spine MRI should be performed when the diagnosis is suspected even without symptoms.
Computed tomography scan sets in a bone window can show bone invasion.Citation14
Treatment
Surgery
The mainstay of therapy for MPNSTs is surgical resection with the goal of achieving complete removal with negative margins. Indeed, Anghileri et al and Zou et al have found that complete resection of the tumor correlates significantly with longer overall survival and inversely with local recurrence.Citation7,Citation8
Since cases of intracranial MPNSTs are limited, their management is based on the experience gained from the management of peripheral MPNSTs. The management of intracranial MPNSTs is therefore firstly surgical. Gross total resection of intracranial MPNST has been shown to significantly influence overall survival.Citation30 The treatment goal should be complete surgical tumor resection with preservation of neurological function. However, as MPNSTs can extend considerably along nerves and given the location of CN MPNSTs, complete surgical resection can be challenging and is not always feasible.
A staged surgical approach has been described by Karami et al in a case of MPNST of the vestibulocochlear nerve and brainstem, where gross total resection would likely have not been technically possible.Citation14 It was used to facilitate tumor debulking with the preservation of CN function. The goal of these interventions was to slow the disease’s progression and associated neurological sequelae. This surgical approach clashes with the aggressive complete resection recommended for peripheral MPNSTs.Citation14 No conclusion can be drawn from this single case, but regarding the often challenging surgical total resection of intracranial MPNST, this approach combined with adjuvant therapies as described can be an interesting alternative.
Adjuvant therapies
The role of adjuvant radiotherapy remains controversial since radiation may be implicated in the pathogenesis of MPNSTs, as previously discussed. Radiation therapy appears to be an important adjunct to surgery in improving local control of extracranial MPNSTs.Citation31 In a review of intracranial MPNSTs performed by Gousias et al, the beneficial prognostic significance of adjuvant radiotherapy on overall survival for intracranial MPNST was found in the univariate analysis but not in the multivariate analysis.Citation32 The majority of patients received a dose of 50–60 Gy. In our review, patients that were treated with radiotherapy had a longer survival rate.
Six cases had adjuvant radiotherapy with stereotactic guidance.Citation14,Citation30,Citation32–Citation35 This strategy provides the possibility to apply higher radiation doses even with nearby sensitive structures such as the brainstem. The case presented by Karami et al showed stabilization of this aggressive tumor with the combined use of conventional fractioned radiotherapy and stereotactic gamma knife radiosurgery.Citation14
Treatment using chemotherapy is controversial.Citation36 Numerous chemotherapy regimens have been used against MPNST but the clinical benefits have been variable and inconclusive. When combined with surgery and radiotherapy, slight or absent improvement of survival has been observed.Citation37
Prognosis and overall survival
For MPNST of the head and neck, a 5-year cause-specific mortality rate of 56% to 67% has been reported.Citation7,Citation8 Gousias et al recently reported a median overall survival of 9 months among 43 patients with intracranial MPNSTs. They found that gross total resection was the only independent predictor of prolonged overall survival.Citation30
In our review, among the 50 cases where outcome was mentioned, 72% patients had died and 28% were alive at follow-up. The survival rate was low; the one-year survival rate being 33%. Among the patients that were alive at follow-up (see ), only three showed no complications. For the other patients, complications were mainly CN dysfunction and residual pain. It was not always clear, but these complications may be consequences of the surgical intervention or radiotherapy treatments.
Firstly, we observed that patients that had a history of radiation exposure had a lower survival rate compared to patients without a history of radiation exposure. We also found that patients treated with complete surgical resection of their tumors developed less recurrences compared to patients who underwent subtotal surgical resection. Additionally, patients who received adjuvant radiotherapy had a higher survival rate. Indeed, among the 28 patients who did not receive radiotherapy for which survival was mentioned, 22 (79%) had died and six (21%) were alive at follow-up ranging from 1 to 39 months. The one-year survival rate was 12% for these 28 patients. Comparatively, among the 20 patients treated with radiotherapy for which survival was mentioned, 13 (65%) had died and seven (35%) were alive at follow-up ranging from 1 to 84 months. The one-year survival rate was 65% for these 20 patients. According to these survival data, we can conclude that radiotherapy prolongs survival in patients presenting with intracranial MPNSTs.
Follow-up
Immediate post-operative follow-up usually includes primarily a clinical exam where CN function is assessed to rule out any deficits. An MRI that allows for the detection of residual tumor should also be part of the follow-up.
Since CN MPNSTs are associated with a low survival rate and a high rate of recurrence and metastasis, we advocate a close clinical follow-up to highlight any new symptoms that could be signs of recurrence or consequences of radiation therapy. In regards to imaging, we believe a close follow-up is mandatory. During the first year, we recommend a 3-month clinical and radiological follow-up. Our recommendations upon treatment and follow-up are presented in .
Table 6 Recommendations upon treatment and follow-up of patients presenting an intracranial MPNST
Conclusion
CN MPNSTs are a rare entity. Compared to MPNSTs from all body sites, it appears that intracranial MPNSTs arise more frequently from schwannomas and are more frequently associated with radiation exposure. The association between intracranial MPNSTs and NF2 is as strong as its association with NF1, if not even stronger when we consider cases where NF2 was suspected. CN VIII is the most involved CN.
Histopathological diagnosis remains challenging due to its lack of specific morphological criteria. Knowledge of the clinical presentation and evolution is more likely to lead to a correct diagnosis. Investigations should also include brain and spine MRIs to visualize the tumor and rule out the presence of metastasis. Radical surgical resection and adjuvant radiotherapy are the cornerstones of the management of these tumors. The location of intracranial tumors may limit surgical options, which may lead to subtotal resection. In these cases, a staged surgical approach may present an interesting alternative. Although an association between therapeutic irradiation and the development of MPNSTs has been established, adjuvant radiotherapy (standard radiation therapy and stereotactic radiotherapy) plays an important role in the therapeutic strategy as it prolongs the survival rate. MPNSTs involving CNs are aggressive tumors. A close clinical and radiological follow-up is crucial, (see ).
Disclosure
The authors report no conflicts of interest in this work.
References
- WoodruffJMSeligAMCrowleyKAllenPWSchwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumorAm J Surg Pathol19941898828958067509
- FoleyKMWoodruffJMEllisFTPosnerJBRadiation-induced malignant and atypical peripheral nerve sheath tumorsAnn Neurol1980743113187377756
- KransdorfMJMalignant soft-tissue tumors in a large referral population: distribution of diagnoses by age, sex, and locationAJR Am J Roentgenol199516411291347998525
- WaneboJEMalikJMVandenBergSRWaneboHJDriesenNPersingJAMalignant peripheral nerve sheath tumors. A clinicopathologic study of 28 casesCancer1993714124712538435801
- WoertlerKTumors and tumor-like lesions of peripheral nervesSemin Musculoskelet Radiol201014554755821072731
- WakelyPEJrAliSZBishopJAThe cytopathology of malignant peripheral nerve sheath tumor: a report of 55 fine-needle aspiration casesCancer Cytopathol2012120633434122434579
- AnghileriMMiceliRFioreMMalignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institutionCancer200610751065107416881077
- ZouCSmithKDLiuJClinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcomeAnn Surg200924961014102219474676
- ChenLMaoYChenHZhouLFDiagnosis and management of intracranial malignant peripheral nerve sheath tumorsNeurosurgery2008624825832 discussion 83218496188
- ScheithauerBWErdoganSRodriguezFJMalignant peripheral nerve sheath tumors of cranial nerves and intracranial contents: a clinicopathologic study of 17 casesAm J Surg Pathol200933332533819065105
- BariMEForsterDMKemenyAAMalignancy in a vestibular schwannoma. Report of a case with central neurofibromatosis, treated by both stereotactic radiosurgery and surgical excision, with a review of the literatureBr J Neurosurg200216328428912201399
- KuritaPoster presentation at the Third Congress of International Stereotactic Radiosurgery MeetingMadrid1997
- LafeminaJQinLXMoracoNHOncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumorsAnn Surg Oncol2013201667222878618
- KaramiKJKelkarPSVerdonMPGrillsISBojrabDIPieperDRMalignant peripheral nerve sheath tumor of the vestibulocochlear nerve and brainstem: multimodality treatment with survival of 27 months. A case report and review of the literatureNeurosurgery2011695E1152E1165 discussion E116521577168
- KuboOChernovMIzawaMMalignant progression of benign brain tumors after gamma knife radiosurgery: is it really caused by irradiation?Minim Invasive Neurosurg200548633433916432782
- CahanWGWoodardHQHiginbothamNLStewartFWColeyBLSarcoma arising in irradiated bone: report of eleven cases. 1948Cancer19988218349428476
- RodríguezJAHedgesTR3rdHeilmanCBStromingerMBLaverNMPainful sixth cranial nerve palsy caused by a malignant trigeminal nerve sheath tumorJ Neuroophthalmol2007271293117414869
- BowersCATausskyPDuhonBSChinSSCouldwellWTMalignant peripheral nerve sheath tumour of the trigeminal nerve: case report and literature reviewBr J Neurosurg201125675075321513444
- GuoALiuAWeiLSongXMalignant peripheral nerve sheath tumors: differentiation patterns and immunohistochemical features – a mini-review and our new findingsJ Cancer2012330330922773934
- StasikCJTawfikOMalignant peripheral nerve sheath tumor with rhabdomyosarcomatous differentiation (malignant triton tumor)Arch Pathol Lab Med2006130121878188117149968
- AkimotoJItoHKudoMPrimary intracranial malignant schwannoma of trigeminal nerve. A case report with review of the literatureActa Neurochir (Wien)2000142559159510898369
- ShimadaSTsuzukiTKurodaMNestin expression as a new marker in malignant peripheral nerve sheath tumorsPathol Int2007572606717300669
- KarMDeoSVShuklaNKMalignant peripheral nerve sheath tumors (MPNST) – clinicopathological study and treatment outcome of twenty-four casesWorld J Surg Oncol200645516923196
- CunhaKSCarusoACFariaPAMalignant peripheral nerve sheath tumors: clinicopathological aspects, expression of p53 and survivalClinics (Sao Paulo)201267896396822948467
- RodriguezFJFolpeALGianniniCPerryAPathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problemsActa Neuropathol2012123329531922327363
- LouisDNOhgakiHWiestlerODThe 2007 WHO classification of tumours of the central nervous systemActa Neuropathol200711429710917618441
- ZiadiASalibaIMalignant peripheral nerve sheath tumor of intracranial nerve: a case series reviewAuris Nasus Larynx201037553954520399579
- CoindreJMGrading of soft tissue sarcomas: review and updateArch Pathol Lab Med2006130101448145317090186
- FisherCSpindle cell sarcomasSurg Pathol Clin201143721744
- GousiasKBoströmJKovacsANiehusmannPWagnerIKristofRFactors of influence upon overall survival in the treatment of intracranial MPNSTs. Review of the literature and report of a caseRadiat Oncol2010511421106096
- WongWWHiroseTScheithauerBWSchildSEGundersonLLMalignant peripheral nerve sheath tumor: analysis of treatment outcomeInt J Radiat Oncol Biol Phys19984223513609788415
- MrakREFlaniganSCollinsCLMalignant acoustic schwannomaArch Pathol Lab Med199411855575618192564
- HanabusaKMorikawaAMurataTTakiWAcoustic neuroma with malignant transformation. Case reportJ Neurosurg200195351852111565878
- GonzalezLFLekovicGPEschbacherJCoonsSSpetzlerRFA true malignant schwannoma of the eighth cranial nerve: case reportNeurosurgery2007612E421E422 discussion E42217762727
- Tanbouzi HusseiniSPiccirilloETaibahAPatiesCTRizzoliRSannaMMalignancy in vestibular schwannoma after stereotactic radiotherapy: a case report and review of the literatureLaryngoscope2011121592392821520103
- GrobmyerSRReithJDShahlaeeABushCHHochwaldSNMalignant Peripheral Nerve Sheath Tumor: molecular pathogenesis and current management considerationsJ Surg Oncol200897434034918286466
- MorettiVMCrawfordEAStaddonAPLackmanRDOgilvieCMEarly outcomes for malignant peripheral nerve sheath tumor treated with chemotherapyAm J Clin Oncol201134441742120838322
- KudoMMatsumotoMTeraoHMalignant nerve sheath tumor of acoustic nerveArch Pathol Lab Med19831072931976687792
- DucatmanBSScheithauerBWPiepgrasDGReimanHMMalignant peripheral nerve sheath tumors in childhoodJ Neurooncol19842324186438279
- MatsumotoMSakataYSanpeiKOnagiATeraoHKudoMMalignant schwannoma of acoustic nerve: a case reportNo Shinkei Geka199018159622406637
- HorieYAkagiSTaguchiKMalignant schwannoma arising in the intracranial trigeminal nerve. A report of an autopsy case and a review of the literatureActa Pathol Jpn1990403219252193485
- McLeanCALaidlawJDBrownbillDSGonzalesMFRecurrence of acoustic neurilemoma as a malignant spindle-cell neoplasm. Case reportJ Neurosurg1990736946502230979
- HanDHKimDGChiJGParkSHJungHWKimYGMalignant triton tumor of the acoustic nerve. Case reportJ Neurosurg1992768748771564550
- MaedaMJosakiTBabaSMuroHShirasawaHIchihashiTMalignant nerve sheath tumor with rhabdomyoblastic differentiation arising from the acoustic nerveActa Pathol Jpn19934341982038493870
- GruberBPetchenikLWilliamsMThomasCLukenMGMalignant vestibular schwannomaSkull Base Surgery19944422723117171176
- NorenGLong-term complications following gamma knife radiosurgery of vestibular schwannomasStereotact Funct Neurosurg199870suppl 165739782237
- ComeyCHMcLaughlinMRJhoHDMartinenzAJLunsfordLDDeath from a malignant cerebellopontine angle triton tumor despite stereotactic radiosurgeryJ Neurosurg1998896536589761063
- HigamiYShimokawaIKishikawaMMalignant peripheral nerve sheath tumors developing multifocally in the central nervous system in a patient with neurofibromatosis type 2Clin Neuropathol1998171151209561334
- SaitoTOkiSMikamiT[Malignant peripheral nerve sheath tumor with divergent cartilage differentiation from the acoustic nerve: case report]No To Shinkei20005273473911002486
- SonEIKimIMKimSPVestibular schwannoma with malignant transformation: a case reportJ Korean Med Sci200116681782111748371
- StarkAMBuhlRHugoHHMehdornHMMalignant peripheral nerve sheath tumours-report of 8 cases and review of the literatureActa Neurochir (Wien)200114335736411437289
- ShinMUekiKKuritaHKirinoTMalignant transformation of a vestibular schwannoma after gamma knife radiosurgeryLancet2002360932930931012147377
- UedaRSaitoRHoriguchiTNakamuraYIchikizakiKMalignant peripheral nerve sheath tumor in the anterior skull base associated with neurofibromatosis type 1—case reportNeurol Med Chir (Tokyo)200444384214959936
- WilkinsonJSReidHArmstrongGRMalignant transformation of a recurrent vestibular schwannomaJ Clin Pathol20045710711214693851
- FisherBJDennisKEMalignant epithelioid cranial nerve sheath tumor: case report of a radiation responseJ Neurooncol200678217317716557352
- MaireJPHuchetAMilbeoYTwenty years’ experience in the treatment of acoustic neuromas with fractionated radiotherapy: a review of 45 casesInt J Radiat Oncol Biol Phys200666117017816904521
- KozicDNagulicMSamardzicMOstojicJRasulicLCvetkovic-DozicDIntrapontine malignant nerve sheath tumor: MRI and MRS featuresActa Neurol Belg2008108677118795600
- ChibarroSHermanPPovlikaMGeorgeBMalignant trigeminal schwannoma extending into the anterior skull baseActa Neurochir (Wien)200815059960418473115
- AkamatsuYMurakamiKWatanabeMJokuraHTominagaTMalignant peripheral nerve sheath tumor arising from benign vestibular schwannoma treated by gamma knife radiosurgery after two previous surgeries:a case report with surgical and pathological observationsWorld Neurosurg201073675175420934169
- DemetriadesAKSaundersNRosePMalignant transformation of acoustic neuroma/vestibular schwannoma 10 years after gamma knife stereotactic radiosurgerySkull Base201020538138721359005
- FardMAMontgomeryEMillerNRComplete, pupil-sparing third nerve palsy in a patient with a malignant peripheral nerve sheath tumorArch Ophthalmol2011129681381421670356
- PuataweepongPJanwityanujitTLarbcharoensubNDhanachaiMRadiation-induced peripheral malignant nerve sheath tumor arising from vestibular schwannoma after linac-basedstereotactic radiation therapy: a case report and review of literaturesCase Rep Med2012201264819122829840
- NakayamaYWatanabeMSuzukiKA case of malignant peripheral nerve sheath tumor originated in the trigeminal nervePoster presentation at the 53rd Annual Meeting of the Japanese Society of Neuropathology
- NewellKPollackABilateral Cranial Nerve VII–VIII Malignant Peripheral Nerve Sheath Tumors Mimicking Neurofibromatosis Type 2J Neuropathol Exp Neurol2012716168
- YanamadalaVWilliamsonRWFuscoDJMalignant Transformation of a Vestibular Schwannoma After Gamma Knife RadiosurgeryWorld Neurosurg2012 pii: S1878-8750(12)00434-2.
- MarkouKEimerSPerretCUnique case of malignant transformation of a vestibular schwannoma after fractionated radiotherapyAm J Otolaryngol201233116817321696856
- WeiCHeman-AckahSENewmanKTemporal bone histopathology case of the month: Malignant peripheral nerve sheath tumor arising within vestibular schwannomaOtol Neurotol2012339e83e8422935807
- VoorhiesJHattabEMCohen-GadolAAMalignant Peripheral Nerve Sheath Tumor of the Abducens Nerve and a Review of the LiteratureWorld Neurosurg2012 pii: S1878-8750(12)00901-1