
Abstract
Drug-induced kidney injury accounts for 20% of community- and hospital-acquired cases of acute kidney injury (AKI). The incidence is higher among older individuals, who often have co-existing morbidities and are exposed to more diagnostic procedures and therapies. While demographic and clinical components have been identified as risk factors, the proposed cellular mechanisms of drug-induced kidney injury are numerous and complicated. There are also limitations recognized in the use of traditional biomarkers, such as serum creatinine and blood urea nitrogen, to provide high sensitivity, specificity, and timeliness to identification of drug-induced kidney injury. Therefore, novel biomarkers are currently being investigated, identified, developed, and validated for their performance over the traditional biomarkers. This review will provide an overview of drug-induced kidney injury and will discuss what is known regarding “omic” (proteomic, genomic, transcriptomic, and metabolomic) biomarker strategies for drugs known to induce nephrotoxicity.
Overview of Drug-Induced Kidney Injury
Acute kidney injury (AKI) presents as an abrupt loss of kidney function leading to accumulation of waste products, electrolyte and metabolic disturbances, and fluid status changes. AKI is classified by changes in serum creatinine (SCr) and/or urine output over defined time periods with the severity of injury based on the degree of biomarker change. Acute kidney injury (AKI) is defined as an increase in SCr by ≥0.3 mg/dL within 48 h, ≥50% increase in SCr within 7 days, or urine output < 0.5 mL/kg/h for 6 h.Citation1 The severity of AKI is staged by the magnitude of change in SCr or urine output. Stage 1 AKI represents a 1.5 to 1.9-fold increase in SCr or reduction in urine output to <0.5 mL/kg/h for 6–12 h.Citation1 Stage 2 AKI is defined by a 2 to 2.9-fold increase in SCr or reduction in urine output to <0.5 mL/kg/h for ≥12 h.Citation1 Stage 3 AKI represents a 3-fold increase in SCr or increase to ≥4 mg/dL or initiation of renal replacement therapy.Citation1 AKI is associated with high mortality, extended length of hospital stay, high rates of renal non-recovery events, and high SCr at hospital discharge.Citation2,Citation3 Additionally, mortality increases with severity of AKI.Citation2 Consequently, strategies to improve early detection of AKI and subsequent intervention are essential to improve patient outcomes.
The epidemiology of AKI has been evaluated in several large observational cohort studies of hospitalized patients. The AKI-EPI study, an international cross-sectional study of 1802 critically ill patients demonstrated that 57.3% of these patients developed AKI, with 18.4% developing stage 1, 8.9% stage 2, and 30% stage 3.Citation2 Drug-induced kidney disease (DIKD) occurred in 14.4% of patients in the AKI-EPI study cohort compared to 26% in the PICARD study cohort.Citation2,Citation3 The most common implicated medications included diuretics (32.4%), non-steroidal anti-inflammatory drugs (11.9%), aminoglycosides (6.8%), glycopeptides (1.4%), and contrast media (2.1%).Citation2
Drug-induced kidney disease can be classified as Type A and B reactions. Type A reactions are usually dose-dependent reactions and predictable based on the known pharmacology of the drug.Citation4 On the other hand, Type B reactions are dose independent and idiosyncratic.Citation4 Historically, the definition of DIKD in published studies has been variable and has included 0.3 to 0.5 mg/dL increases in SCr and/or a 50% rise in SCr. However, as DIKD represents a spectrum of different pathophysiologic mechanisms causing acute, sub-acute, and chronic insult to different locations along the nephron, not all types of kidney injury result in a change in SCr.
In 2015, DIKD was defined using four standardized phenotypes: AKI, glomerular injury, tubular disorders, and nephrolithiasis.Citation5 The biomarkers for the respective phenotypes were SCr or urine output for AKI, proteinuria or hematuria for glomerular injury, hypophosphatemia, glucosuria or hyperchloremic metabolic acidosis in addition to a potassium disorder for tubular disorders, and crystalluria for nephrolithiasis. The AKI phenotype definition was further refined as either a rise or decline in SCr in relation to drug initiation or withdrawal/dose reduction, respectively. A decline in SCr after drug withdrawal or dose reduction was added to address clinical scenarios in which baseline SCr was unknown or the drug was administered as an outpatient, preventing the clinical observation of the peak SCr. The severity of AKI was subsequently defined as stage 2 when there was a doubling or greater of SCr after drug exposure or 50% reduction in SCr after drug withdrawal or dose reduction. A goal of the phenotype standardization process was improving specificity of case diagnoses given the multifactorial nature of AKI due to drugs.
Secondary criteria were also defined to further classify sub-phenotypes. For example, the AKI phenotype includes acute tubular necrosis and acute interstitial nephritis. Secondary criteria such as fractional excretion of sodium or urinary casts help to differentiate injuries.Citation2,Citation3
In the oncology population, cisplatin and ifosfamide are common nephrotoxins causing AKI or tubular disorders. Targeted cancer therapeutics, such as anti-angiogenesis drugs that specifically inhibit vascular endothelial growth factor (VEGF) and tyrosine kinase, have been associated with hypertension, proteinuria, AKI, and thrombotic microangiopathy.Citation6 Anti-cytotoxic T lymphocyte-associated protein 4 (anti-CTLA-4) and anti-programmed death 1 (anti-PD-1) receptor antibodies target immune checkpoints, which enhance tumor-directed immune responses to destroy cancers. Checkpoint inhibitors are known to cause immune-related adverse effects, with recent reports indicating latent onset AKI due to acute tubulointerstitial nephritis.Citation7,Citation8 Several recent reviews have highlighted the kidney complications of targeted cancer chemotherapeutics.Citation9–Citation14
Biomarkers to inform early and more sensitive detection of DIKD are needed. These biomarkers should also be specific enough to identify the sites of injury and related to the phenotype of the injury. It is plausible that the presence of combined DIKD phenotypes, due to single or drug combinations may require a panel of biomarkers for diagnosis. Omic strategies, such as proteomics, genomics, transcriptomics, and metabolomics, will undoubtedly aid our understanding of the pathophysiology of AKI and DIKD and improve screening, detection, and disease prognostication through novel biomarker discovery and a systems-based approach. The following sections will highlight studies that have employed omic techniques using cells, kidney tissue, urine, and blood to identify potential biomarkers for DIKD. For each section, we conducted a literature search of in-vitro, animal and human nephrotoxicity omic studies published within the last 10 years using an array of shot-gun and targeted approaches. Studies selected for inclusion were those that identified novel targets or approaches, evaluated drugs that had less published data, and/or contained significant or new information pertaining to human data.
Proteomics
Protein Biomarkers for Nephrotoxicants
Shot-gun and targeted proteomic studies have been conducted as a tool for understanding mechanisms and to identify biomarkers of nephrotoxicity. The following section () will highlight proteomic studies for several common nephrotoxic drugs. The proteomic methods employed in these studies include both traditional and novel methods, as highlighted in the Supplemental material. Human kidney cells (HEK293, HK-2), animal urine and kidney tissue, and human urine are accessible tools to evaluate proteomics of DIKD. Confirmation and/or diagnoses of kidney injury in animal and human studies use traditional elevations in SCr and blood urea nitrogen (BUN).
Table 1 Proteomic Changes for Specific Nephrotoxicants
Acyclovir
Acyclovir results in nephrotoxicity manifesting as AKI, acute tubulointerstitial nephritis, and nephrolithiasis. Broad proteomics of acyclovir nephrotoxicity was studied in the Institute of Cancer Research (ICR) mouse kidney tissue after administration of 600 mg/kg doses daily for 9 days, and demonstrated reductions in VEGF and its receptor, suggesting mechanisms consistent with apoptosis and tissue repair.Citation15 Furthermore, matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) identified up-regulation of glyoxalase 1, peroxiredoxin 1, and αB-crystallin in the oxidative stress pathway. Major histocompatibility complex (MHC) class II antigen and fibroblast growth factor receptor 1-IIIb were also increased.Citation15 An observed reduction in cytochrome c oxidase subunit Vb suggested mitochondrial damage.
Amphotericin B
Amphotericin B causes nephrotoxicity through vasoconstriction and cell membrane alterations. The performance of neutrophil gelatinase-associated lipocalin (NGAL) in urine as a biomarker of nephrotoxicity was evaluated in the urine of human patients receiving either the deoxycholate or liposomal formulation.Citation16 Overall performance of urinary NGAL by area under the curve (AUC) for detection of AKI (on day 5, based on criteria of SCr ≥1.5 x baseline) was 0.68 (95% CI 0.41 to 0.95), suggesting a poor biomarker performance test result. However, for the deoxycholate formulation, the AUC rose to 0.89 (95% CI 0.67 to 1.00), demonstrating a good biomarker performance test result. AKI was found to be detected 3 days earlier with urinary NGAL versus SCr supporting the use of urinary NGAL as a potential early nephrotoxicity biomarker in patients receiving amphotericin B.Citation16
Platinum Compounds
Cisplatin and carboplatin are chemotherapeutic drugs that cause nephrotoxicity, especially to proximal tubule cells. Proteomic evaluations have been conducted for cisplatin, but are limited for carboplatin. This section will focus on in vitro, animal and human publications with less commonly described biomarkers for platinum nephrotoxicity using targeted and shot-gun proteomic approaches.
Two in vitro studies were identified that assessed proteomics of kidney injury biomarkers for platinum compounds. In a study by Perez et al, exposure of HK-2 cells to carboplatin or cisplatin for 24 h resulted in changes in key proteins involved in ATP synthesis, cellular respiration, oxidative stress, cytoprotection, and apoptosis.Citation17 () Wilmes et al evaluated epithelial barrier function and proteomics in the human renal proximal tubule cell line RPTEC/TERT1.Citation18 Results showed that barrier function in the cell monolayers was intact while cell stress, as assessed by increases in supernatant lactate, was increased. Proteomic analysis through differential peptide labeling (iTRAQ)/HPLC-MS demonstrated changes to proteins that were involved in mitochondrial and DNA damage, and oxidative stress responses.
Montoro-Molina et al used a targeted approach to demonstrate glutamyl aminopeptidase, an enzyme in rodent renal tubules that can be shed in urine during kidney injury, was increased when Wistar rats were exposed to cisplatin 7 mg/kg.Citation19 Zhang et al demonstrated that nestin, hamartin, T-kininogen, and fetuin-A were increased in the urine of Sprague–Dawley rats treated with cisplatin compared to ischemia–reperfusion injury by employing MALDI TOF LC-MS/MS and iTRAQ.Citation20
Organic anion transporter 5 (Oat5) is a transporter found on the apical membrane of proximal tubule cells. Bulacio et al treated male Wistar rats with cisplatin 1 mg/kg to 10 mg/kg and demonstrated Oat5 in the urine was proportional to the cisplatin dose.Citation21 An important finding was the lack of changes to traditional markers of kidney injury (SCr, BUN) at cisplatin doses <10 mg/kg, despite Oat5 changes, suggesting Oat5 may be a more sensitive marker of kidney damage. Transglutaminase 1 (TG1) is a posttranslational modifying enzyme that catalyzes irreversible cross-linking reactions between proteins and thought to aid in recovery of epithelial cell structural proteins after injury. Furukawa et al evaluated the utility of renal TG1 as an early biomarker of kidney injury in ICR mice treated with cisplatin 20 mg/kg/day.Citation22 The authors reported an increase in expression and activity of TG1 in the proximal tubule within 24 h, with no increases in SCr, suggesting the use of this protein as an early indicator of kidney damage.
A recent publication has profiled potential kidney injury proteins in the urine up to 10 days after cancer patients received a single cisplatin dose.Citation23 Several potential biomarkers were significantly increased in the urine. Beta 2-microglobulin was increased 3-fold by day 3, while Kidney Injury Molecule 1 (KIM-1) and trefoil factor 3 (TFF3) were increased 2-fold and calbindin was increased 8-fold by day 10. The authors further defined time-dependent changes in these urinary biomarkers in patients receiving multiple cycles of cisplatin.Citation24 The biomarker changes were distinct. KIM-1 concentrations were elevated after the first cisplatin dose and remained elevated through subsequent cycles. Calbindin was elevated 10 days after the first cisplatin dose but displayed a resistance to change at subsequent doses. TFF3 elevations occurred at 3 days after a cisplatin dose in the first and subsequent doses. These elevations occurred without changes to SCr, suggesting the potential for these urinary biomarkers to detect sub-clinical injury.
Cyclosporine
Chronic cyclosporine nephrotoxicity presents histologically with tubular atrophy and stripped interstitial fibrosis. Lamoureux et al evaluated proteomics in HEK-293 cells exposed to cyclosporine and reported down-regulation of proteins involved in cell adhesion, extracellular matrix, stress response, protein folding, and enzymes using stable isotope labeling in cell culture (SILAC) and LC-MALDI-TOF/TOF ().Citation25 Klawitter et al evaluated proteomic changes in the kidney tissue of rodents (Wistar rats) treated with cyclosporine 10 mg/kg/day for 28 days under either a normal or salt depleted diet.Citation26,Citation27 Cyclosporine treatment in the presence of a low salt diet led to activation of NF-κB pro-inflammatory signaling and decreased glycolysisCitation27 (). Rats treated with cyclosporine under normal diets had proteomic changes in calcium homeostasis, cytoskeleton proteins, response to hypoxia, mitochondrial function, and cell metabolismCitation26 (). Treatment of rats with cyclosporine 45 mg/kg/day for 14 days resulted in a decrease in calbindin and α-2µ-globulin.Citation28 Another study assessed plasma proteomics in Wistar rats after administration of cyclosporine 5 mg/kg/day and 100 mg/kg/day for 7 days.Citation29 Significant kidney dysfunction occurred in the 100 mg/kg/day treatment group, with two proteins (clusterin and α-1-acid glycoprotein) demonstrating up-regulation, while haptoglobin showed down-regulation. The studies demonstrate numerous pathways are implicated in kidney injury secondary to cyclosporine and multiple proteins serve as potential targets for the development of a biomarker panel to detect nephrotoxicity.
Gentamicin
Aminoglycoside antibiotics induce AKI through tubular necrosis and an elevated trough plasma concentration (>2 mg/L) has been identified as a risk factor for nephrotoxicity. Studies have evaluated proteomic signatures in urine samples from rats exposed to gentamicin.Citation30–Citation33 Rouse et al administered gentamicin 150 mg/kg or 300 mg/kg for 3 days to Sprague Dawley rats.Citation31 While many proteins were found to be up-regulated by capillary electrophoresis (CE)-MS, osteopontin, albumin, and clusterin were consistently increased. Ferreira et al administered gentamicin 150 mg/kg to Wistar rats and evaluated urinary proteomics by LC-MS/MS at day 3.Citation30 Novel proteins that were up-regulated included gelsolin, regenerating islet-derived protein IIIβ, and plasminogen activator inhibitor-1 (PAI-1). The study also reported elevations in N-acetyl-β-D-glucosaminidase (NAG), KIM-1, and NGAL. Vincente-Vincente et al performed a kidney injury sensitization study, whereby Sprague-Dawley rats first received uranyl nitrate for up to 21 weeks, with subsequent gentamicin 50 mg/kg/day treatment for 7 days.Citation32 The rats pre-treated with 21 weeks of uranyl nitrate had robust kidney toxicity to gentamicin as assessed by matrix-assisted laser desorption/ionization (MALDI)-TOF proteomics. Albumin, hemopexin, transferrin, and vitamin D binding protein were all elevated in the urine under gentamicin treatment. Zhou et al evaluated KIM-1 in the urine and kidney of Sprague-Dawley rats 72 h after receiving gentamicin (up to 400 mg/kg for 3 days).Citation33 Urinary KIM-1 increased in a time- and dose-dependent manner and the trend was consistent with histopathology evaluations.
The utility of urinary biomarkers for gentamicin nephrotoxicity has recently been evaluated in higher level animals. Cynomolgus monkeys were treated with gentamicin 10 mg/kg to 50 mg/kg for 10 days, with urine assessed for targeted biomarkers (microalbumin, α-1 microglobulin, clusterin, osteopontin, KIM-1, NGAL) over 7 days by LC-MS/MS.Citation34 Urinary KIM-1 and NGAL did not strongly predict gentamicin toxicity. However, microalbumin, α-1 microglobulin, clusterin, and osteopontin were found to be more sensitive than BUN or SCr in detecting injury in the 10 mg/kg gentamicin group at day 11. The urinary biomarkers NGAL, clusterin, NAG, and total protein were evaluated by ELISA assays in beagle dogs receiving gentamicin 80 mg/kg for up to 9 days.Citation35 NGAL, clusterin, and NAG increased in a time-dependent manner after exposure to gentamicin and correlated with severity of histology. While NGAL and NAG showed increases at day 1, clusterin increases were not significant until day 3. These results in higher order animals suggest specific time-dependent increases in the evaluated proteins and demonstrate the utility of applying selective urinary biomarkers in studies assessing nephrotoxins in humans.
Two studies evaluated proteins in kidney tissue from rats treated with gentamicin.Citation36,Citation37 Com et al administered gentamicin 25 mg/kg/day and 75 mg/kg/day to Wistar rats for up to 14 days.Citation36 The most significant changes in the kidney proteome as assessed by LC-MS/MS occurred with the high dose gentamicin (75 mg/kg/day). Down-regulated biomarkers of injury included plasma retinol binding protein 4, glycine amidinotransferase, and peroxiredoxin 1 (). Rat kidney tissue was evaluated by imaging mass spectrometry (IMS) to evaluate the location of proteins that correlated with histopathology of nephrotoxicity after treatment with gentamicin 100 mg/kg/day for 7 days.Citation37 The study reported that transthyretin was highly correlated with histopathology in rats exhibiting nephrotoxicity. Additionally, transthyretin accumulated in the proximal tubules of the kidney cortex. Since transthyretin binds megalin at the proximal tubule, binding competition by gentamicin for megalin was proposed as a plausible hypothesis for a mechanism by which transthyretin could be elevated in kidney cortex under gentamicin nephrotoxicity.
Non-Steroidal Anti-Inflammatory Drugs
Non-steroidal anti-inflammatory drugs (NSAIDs) are used chronically in patients for musculoskeletal pain syndromes and headaches and nephrotoxicity results in AKI, interstitial nephritis, and glomerulonephritis. Two studies by the same authors reported proteomic evaluations of second morning urine samples from patients with medication-overuse headache who chronically used NSAIDs.Citation38,Citation39 The methodologies from the studies differed, with the older study using 1-DE and QTOF-LC/MS and the newer study using 2-DE with surface-enhanced laser desorption/ionization (SELDI)-TOF-MS. In the first study,Citation38 seven proteins (including cystatin C) were found to be elevated in urine (). In the second study,Citation39 21 differentially expressed proteins were reported, some of which are included in . While beta-2-microglobulin was included in second study, cystatin C was not found to be differentially expressed in this later analysis. Various cellular processes associated with the toxicity were identified through the proteomics approach and included apoptosis, cytoskeleton, transport, charge selective barrier, renal function and development. Given the high usage of NSAIDs in the general population, proteomic approaches to identify early nephrotoxicity may lead to interventions to prevent DIKD.
Summary
Proteomics is a promising tool for evaluating mechanisms of and biomarkers for DIKD. The studies summarized in this section demonstrate a multitude of potential novel biomarkers that hold promise for detecting kidney injury due to specific nephrotoxins. The majority of studies used in-vitro approaches or animals, with few human studies. The results suggest that future studies of specific biomarker panels for a particular nephrotoxin may be the best pathway forward to evaluating DIKD in humans in a sensitive, specific, and time-dependent manner.
Genomics
The genetic predisposition to DIKD may involve the interplay of genes encoding proteins for drug metabolism and transport, and pathways involved in kidney injury and/or cellular repair (). This section reviews known genomic determinants of common nephrotoxins implicated in DIKD.
Figure 1 Figure depicts factors contributing to the development of kidney injury. Susceptibility of the kidney to toxic injury has been identified in polymorphisms of gap junction proteins and detoxification enzymes. Polymorphisms in drug metabolism or transport can lead to increased exposure of nephrotoxicants. Immune-mediated adverse effects from drugs may be related to genetic polymorphisms in human leukocyte antigens. Lastly, cellular repair mechanisms may be altered increasing the risk of apoptosis and tubular injury from nephrotoxicants.
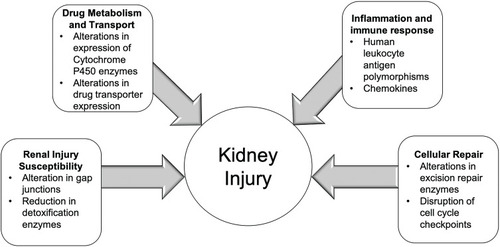
Metabolism and Transport
Several studies have investigated the role of genetic alterations in drug metabolism and transport pathways on nephrotoxicity. Nephrotoxicity due to calcineurin inhibitors can lead to kidney injury in transplant recipients and limit kidney allograft survival. Kuypers and colleagues found that kidney transplant recipients who expressed of at least one CYP3A5*1 allele (intermediate metabolizer) in their blood had higher dose requirements and a significantly increased risk of biopsy-proven calcineurin inhibitor nephrotoxicity (HR 2.39 [95% CI 1.15–4.92, p=0.01]).Citation40 Additionally, Joy and colleagues found that reduced CYP3A5 protein expression in kidney tissue of solid organ transplant recipients resulted in a higher rate of biopsy-confirmed nephrotoxicity.Citation41
P-glycoprotein (P-gp) is an identified xenobiotic transporter responsible for efflux of calcineurin inhibitors from proximal tubule cells. Kidney donors with reduced P-gp expression have an increased risk of calcineurin inhibitor-associated nephrotoxicity (OR 9.2 [95% CI 1.1–79.4, p=0.03]).Citation42 A previous study also documented reduced P-gp expression in the kidney tissue of solid organ transplant recipients with calcineurin nephrotoxicity.Citation43 Naesens and colleagues demonstrated that recipient-donor homozygosity for P-gp/ABCB1 variant C3435T resulted in reduced P-gp expression in both recipients and donors and was significantly associated with chronic damage.Citation44 In a study of approximately 4500 renal transplant recipients, homozygous variant donor genotype of the ABCB1 C3435T polymorphism (representing increased PGP expression in the kidney) was associated with lower rates of death-censored graft survival.Citation45 The authors postulated that increased P-gp expression may be promoting renal injury through anti-apoptotic effects in fibroblasts and cholesterol ester–mediated vascular damage.Citation45
Cisplatin has been shown to be transported into kidney proximal tubule cells by organic cation transporter 2 (SLC22A2, OCT2) and undergoes efflux by multidrug and toxin extrusion protein 1 and 2 (SLC47A1/MATE1 and SLC47A2/MATE2) and multidrug resistance-associated protein 2 (ABCC2/MRP2).Citation46–Citation48 Patients with a reduced function variant in SLC22A2 (G808T) have been shown to have lower risks of nephrotoxicity.Citation46,Citation49 One previous study reported that MRP2 knockout confers increased risk for kidney injury with exposure to cisplatin.Citation15 A recent study evaluated changes in glomerular filtration rate and urinary protein biomarkers by various polymorphisms in SLC22A2/OCT2, SLC31A1/CTR1, SLC47A1/MATE1, ABCC2/MRP2, and GSTP1 in cancer patients receiving cisplatin.Citation50 Several polymorphisms in uptake transporters, rs596881 (SLC22A2/OCT2), rs12686377, and rs7851395 (SLC31A1/CTR1), were associated with maintenance of estimated glomerular filtration rate (eGFR). While one previous study did not find associations between polymorphisms in SLC47A1 and nephrotoxicity in cancer patients receiving cisplatin,Citation49 a recent study reported associations between polymorphisms in SLC47A1/MATE1, as well as SLC22A2/OCT2, SLC31A1/CTRI, ABCC2/MRP2, and GSTP1 with increases in the urinary proteins KIM-1, TFF3, monocyte chemoattractant protein 1 (MCP1), NGAL, clusterin, cystatin C, and calbindin.Citation50 These data suggest the potential for genomics to predict both clinical and sub-clinical kidney injury in cancer patients receiving cisplatin.
Tenofovir is a nucleotide inhibitor of human immunodeficiency virus (HIV) and hepatitis B virus reverse transcriptases and used in the prevention and treatment of these diseases. Tenofovir is eliminated by the kidneys through glomerular filtration and tubular secretion. The drug has been associated with tubule injury resulting in Fanconi syndrome and diabetes insipidus. Tubular secretion of tenofovir involves the organic anion transporter 1 (SLC22A6/OAT1) for influx into proximal tubular cells and ABCC2/MRP2 and ABCC4/MRP4 transporters for efflux into the urinary ultrafiltrate. Studies have demonstrated that reduced efflux of tenofovir due to the ABCC2 G1249A polymorphism is associated with tubular injury (OR 6.11, 95% CI 1.2–31.15, p=0.02).Citation51 Rodriguez-Novoa and colleagues demonstrated a significant association between another ABCC2 variant C-24C and tubular dysfunction.Citation52 However, contrary to these two studies, a genome-wide association study (GWAS) of approximately 500 tenofovir-treated patients failed to demonstrate the association of genetic polymorphisms with decline in kidney function over long-term exposures.Citation53
Kidney Injury
The mechanism for ifosfamide-induced kidney injury is thought to be related to intracellular glutathione depletion secondary to exposures from the chloroacetaldehyde metabolite. Glutathione s-transferase (GST) enzymes are Phase 2 detoxification enzymes, which catalyze the conjugation of glutathione to electrophiles. Zielinska and colleagues investigated the role of GST enzymes and nephrotoxicity in pediatric oncology patients receiving ifosfamide.Citation54 Children with polymorphic locus +313 A-G (+/Val) of the GSTP1 gene compared with homozygous GSTP1a alleles (Ile/Ile) had a decreased creatinine clearance (81.1 mL/min/1.73 m2 vs 105.0 mL/min/1.73 m2, p=0.03) with ifosfamide treatment.Citation54
Vancomycin, a glycopeptide antibiotic used in the treatment of methicillin-resistant Staphylococcus aureus infections, undergoes predominant clearance through the kidney. Kidney injury from vancomycin manifests as acute tubular necrosis and/or immune-mediated acute interstitial nephritis. In a multi-center, observational cohort study, the incidence of vancomycin nephrotoxicity was found to be 19% in the entire cohort and 29.6% in patients with trough concentrations > 15 mg/L.Citation55 A GWAS of vancomycin associated AKI documented a significant association (p=1.1x10−7) with a single nucleotide polymorphism (rs2789047) on chromosome 6 in patients of Northern European ancestry.Citation56 This polymorphism was in the region of gene GJA1, which encodes for connexin43, a gap junction protein in renal tubules.Citation56 Gap junctions have been shown to contribute to tubular injury associated with aminoglycosides; however, their exact role in enhancing injury has not been fully elucidated and may include transporting toxicants to surrounding cells and transmitting damage signals.Citation57
Clofarabine undergoes significant renal filtration, with over 60% of administered drug excreted unchanged in the urine and causes kidney toxicity. Using human embryonic kidney cells (HEK293), Eadon and colleagues demonstrated that SET binding protein 1 (SETBP1) knockout increased HEK293 cell sensitivity to clofarabine toxicity.Citation58 While the role of SETBP1 binding to SET is unclear, SETBP1 variants have been implicated in the development of Schinzel-Giedion Syndrome with renal manifestations including hydronephrosis or calyceal hypertrophy.Citation59
Cellular Repair
Many older chemotherapeutic agents exhibit a mechanism of action by interrupting DNA synthesis and repair and subsequently leading to cell death. Cell cycle progression and arrest processes have been shown to mediate kidney recovery and fibrosis following injury.Citation60 Consequently, genetic alterations in cellular repair mechanisms may increase susceptibility to nephrotoxicity.
Polymorphisms in DNA repair genes have been associated with development of cisplatin nephrotoxicity. Excision repair cross-complementation (ERCC) genes regulate proteins required for repair of DNA damage caused by cisplatin. Patients who were heterozygous for ERCC1 C8092A and T19007C had increased risk of developing nephrotoxicity, OR 3.29 (95% CI 1.4–7.73, p=0.009) and OR 2.51 (95% CI 1.09–5.57, p=0.037), respectively.Citation61 In patients with advanced gastric cancers, the ERCC2 haplotype Asn312/751 increased the risk of cisplatin nephrotoxicity (OR 2.27, 95% CI 1.28–4.02, p=0.005).Citation62 These polymorphisms likely represent suboptimal DNA repair capacity in response to cisplatin injury.
Summary
Studies to date have identified the contribution of genetic polymorphisms in drug metabolism and transport, injury, and detoxification pathways in addition to cell cycle repair to the development of AKI and DIKD. These preliminary genetic findings enhanced by transcriptomic and proteomic evaluations will improve our knowledge of the pathophysiology of AKI and DIKD and improve strategies to identify at-risk patients and treatment of this serious condition.
Transcriptomics
Transcriptional profiling has demonstrated molecular markers and potential regulatory pathways of kidney repair and progression that are altered and persist in AKI.Citation63,Citation64 An understanding of transcriptome changes will help in the identification of biomarkers for detection of AKI and potentially novel therapeutics to mitigate this adverse effect of drug therapy. This section will review changes in mRNA and miRNA induced by exposures to known nephrotoxins. A summary of these changes are provided in Citation65–Citation68 and .Citation69–Citation78
Table 2 Genes Exhibiting ≥2-Fold Expression Changes in Acute Kidney Injury Models versus Respective Controls
Table 3 Micro-RNAs (miRNAs) Evaluated in Acute Kidney Injury Studies with ≥2-Fold Expression Changes Compared to Controls in Tissue, Blood or Urine
Acetaminophen
In a study of acetaminophen (APAP) overdose, AKI was detected in 43 out of 70 patients.Citation74 Urinary KIM-1, miR-21, miR-200c, and miR-423 were significantly increased in these patients who developed AKI compared to healthy volunteersCitation74 and higher urinary concentrations of each biomarker were associated with increased odds of AKI. Wolenski et al dosed male Sprague-Dawley rats with a single dose of APAP (400 or 1250 mg/kg) and found significant changes in 6 miRNAs in urine and 74 miRNAs in plasma 24 h later.Citation77
Antibiotics
Aminoglycosides can cause acute tubular necrosis, with higher risk of toxicity with longer duration of therapy, high trough concentrations, more frequent dosing and concurrent nephrotoxin exposures.Citation79 Qiu and colleagues conducted a time-series gene expression study after administering gentamicin to rats and confirming toxicity by measuring KIM-1 concentrations.Citation80 The authors found an increased expression of CXCL1 which encodes for chemokines and is involved in inflammation and as a chemoattractant for neutrophils.Citation80
Quantitative gene expression changes in kidney tissue of female cynomolgus monkeys after exposure to gentamicin (10 mg/kg), everninomicin (30 mg/kg or 60 mg/kg), or gentamicin (10 mg/kg) + everninomicin (30 mg/kg) for 1 or 7 days was evaluated by Davis et al.Citation67 Increased expression of MMP6 was observed irrespective of treatment and dose. Everninomicin and gentamicin + everninomicin-treated groups demonstrated significantly increased expression of MMP6, Vimentin, waf-1, Tβ10, and GJβ2) and decreased expression of EGF and OATK1.Citation67 Predictors of nephrotoxicity were defined as genes that showed significant expression changes before overt detection of injury as detected by histology. Clusterin and OPN were significantly increased on Day 1 in all groups [except gentamicin (10 mg/kg) and everninomicin (30 mg/kg)], and further increased expression was seen on Day 7.Citation67 This study suggests that clusterin and OPN expression may be predictive biomarkers of nephrotoxicity.
Female Sprague Dawley rats were administered nephrotoxicants [cyclophosphamide (70 mg/kg), amphotericin B (50 mg/kg), gentamicin (100 mg/kg), and cisplatin (2 mg/kg)] and gene expression changes in kidney and blood were evaluated.Citation66 All nephrotoxicants resulted in increased expression of Mpo, Spp1, and Tnf, and decreased expression of Gstp1 and Gstt1 in kidney tissue.Citation66 Mpo encodes for myeloperoxidase, which is a lysosomal enzyme involved in oxidative stress-mediated kidney injury. Spp1 encodes the glycoprotein osteopontin which acts as a proinflammatory cytokine and activator of T lymphocytes. Tumor necrosis factor cytokines are involved in inflammation and regulation of immune cells. Glutathione transferases play a role in detoxification by catalyzing the conjugation of many compounds with glutathione. In blood, Spp1 was decreased and Gstp1 did not correspond to expression in kidney tissue.Citation66 The study demonstrated the divergent gene expression patterns in kidney vs blood in animals exposed to nephrotoxicants and suggests that blood may not inform kidney injury for some potential transcriptomic biomarkers.
In collaboration with the European InnoMed PredTox project, transcriptomic data in kidney and blood of male Wister rats administered gentamicin (25 mg/kg/day to 75 mg/kg/day) for 1, 2, or 14 days were examined.Citation36 Kidney tissue demonstrated global expression of 463 genes at day 14 (303 upregulated and 160 downregulated) with high-dose gentamicin (75 mg/kg/day). In blood, 3241 genes were expressed (919 upregulated and 2322 downregulated) at 14 days with high-dose gentamicin (75 mg/kg/day). No significant gene expression changes in kidney tissue or blood were observed at earlier time points or with lower doses of gentamicin. Gene ontology mapped to biological processes demonstrated activation of NF-kB cascade, induction of protein synthesis, induction of RNA or protein catabolism, induction of inflammatory and immune response, mobilization of reproductive processes, activation of apoptosis, and mobilization of acetyl-CoA catabolic processes.Citation36 The results suggest there are dose- and time-dependent effects on gene expression as well as induction of inflammatory responses with gentamicin treatment.
A global miRNA expression analysis was performed in urine of male Wister rats treated with daily gentamicin exposure of 60 mg/kg (low-dose) or 120 mg/kg (high-dose) for 10 days.Citation78 High-dose gentamicin treatment resulted in significant changes in miRNAs with 34 increased and 88 decreased on day 4. With low-dose gentamicin, 14 miRNAs were increased and 66 miRNAs were decreased. A total of 64 miRNAs overlapped between the low-dose and high-dose gentamicin groups.Citation78 Of the miRNAs screened, 13 were identified as biomarker candidates based on time- and dose-dependent increases (). Analyses demonstrated that miR-140-3p (AUC = 0.964) and miR-138-5p (AUC = 0.946) had high accuracy and sensitivity and were superior to BUN as demonstrated by ROC.Citation78 These miRNAs offer the potential to serve as biomarkers for detection of DIKD from gentamicin.
Saikumar et al performed global miRNA expression profiling in male Sprague Dawley rats that received high doses of gentamicin (200 mg/kg or 300 mg/kg) for 3 days.Citation75 Upregulation of miR-21 was demonstrated in kidney tissue with 300 mg/kg gentamicin. While miR-155 expression was not dose dependent, it was higher in the kidney cortex versus medulla. miR-18a did not show significant changes with gentamicin doses or differences between the cortex and medulla locations.Citation75 miR-21 was significantly decreased in the blood, whereas, only a modest decrease in blood was observed for miR-155 and miR-18a.Citation75 Both miR-21 and miR-155 were significantly decreased in the urine, while miR-18a was undetectable.Citation75 These results suggest a discrepancy between miRNA expression patterns in kidney tissue versus urine and suggest that kidney tissue is best for assessment of miRNAs for kidney injury. These data suggest that urinary data likely lags behind events and expression in tissues.
Colistin is an antibiotic used to treat gram negative infections. Eadon and colleagues used a murine model of colistin nephrotoxicity to identify differentially expressed transcripts in kidney tissue.Citation81 Colistin induced kidney damage resulted in increased expression of LGALS3 (galectin-3), CCNB-1 (cyclin-B1), CDKN1a (p21).Citation81 Galectin-3 is a lectin involved in cell cycle regulation that contributes to regulation of both the G1 to S and G2 to M cell cycle checkpoints via activation of p21. CCNB-1 and CDKN1a genes encode for the cell cycle markers cyclin-B1 and p21, respectively, with CDKN1a arresting progression of the cell cycle from G2 to M phase.Citation81 Cell cycle arrest appears to be a predominant mechanism allowing cells to repair after injury from colistin.
Cisplatin
Changes in kidney expression of genes were evaluated in male Sprague Dawley rats administered a single dose of cisplatin (0.1 mg/kg to 5 mg/kg).Citation65 At 24 h and 144 h, high doses of cisplatin (5 mg/kg) were associated with severe proximal tubule toxicity. Hierarchical clustering was performed and revealed two distinct separations that correlated with severity of toxicity. Cisplatin treatment induced a ≥2-fold increase in Sgp2 and osteopontin (OPN),Citation65 both associated with clearance of cellular debris, apoptosis, and inflammatory processes.
Pavkovic et al measured miRNA biomarkers in the urine of male Han Wister rats 3 days prior to and 3, 5, 8, 15, and 28 days after a single injection of 1 mg/kg or 3 mg/kg cisplatin.Citation73 Maximal changes in miRNA expression (≥2-fold) were detected at 5 days after cisplatin, with levels declining towards baseline by 26 days. Of the 136 miRNAs detected, 18 were chosen as candidate biomarkers for nephrotoxicity (). Fourteen of the selected miRNAs demonstrated increased expression at 3 days and maximal expression at 5 days after cisplatin 3 mg/kg.Citation73 A statistically significant increase in 4 miRNAs (miR-15, miR-16, miR-20a, and miR-192) was also noted in the 1 mg/kg cisplatin dose group at 5 days.Citation73 ROC analyses demonstrated that miR-192 most strongly predicted proximal tubular necrosis after cisplatin exposure (AUC = 0.9203). The 18 candidate miRNAs were also analyzed in kidney tissue. The miRNAs were modestly decreased, with the exception of miR-34a, miR-184, miR-21, and miR-327. Gene expression profiling demonstrated peak increases (162 genes) and decreases (112 genes) in mRNA expression at 8 days after cisplatin. mRNA expression patterns were similar across cisplatin doses, but with smaller fold-changes for the 1 mg/kg dose. Functionally, the genes that were upregulated encoded proteins related to DNA damage response, apoptosis, cell cycle, stress response, and regeneration.Citation73 The mRNAs that were decreased were related to kidney function. Additional analyses revealed that 21 mRNAS were regulated by 11 miRNAs. The pathways affected included p53 and PI3K/AKT.Citation73
Kanki et al collected urine and kidney 1, 3, 5, and 7 days after a single injection of 1, 3, 6 mg/kg cisplatin to male Sprague Dawley rats for the identification of miRNA biomarkers.Citation71 There were 322 miRNAs detected in urine, with 30 chosen for further analysis based on ≥2-fold changes. Two of the miRNAs (miR-196c-5p and miR-1959) were decreased more than 2-fold.Citation71 Increased expression of miRNAs in urine was associated with decreased expression in the kidney. The expression of miRNAs peaked at day 5 and decreased by day 7. No changes in miRNA expression were observed at the 1 mg/kg dose, whereas 8 miRNAs showed a dose-dependent increase with exposure to 3 mg/kg and 6 mg/kg cisplatin doses.Citation71
miRNA and/or mRNA expression in kidney tissue was evaluated in C57BL/6 mice administered a single injection of cisplatin (15 mg/kg).Citation72 Seven miRNAs demonstrated significant changes in expression after cisplatin treatment, with two upregulated (miR-34a and miRlet-7g) and five downregulated (miR-122, miR-10b, miR30e, miR193, miR-26a).Citation72 Time course studies revealed a downregulation of miR-122 on day 1 after cisplatin treatment, with expression remaining low until day 3, with a modest increase at day 5. In contrast, miR-34a was significantly increased at day 3, with further induction at day 5. miR-34a and miR-122 can inhibit Foxo3 translation, a protein found in the p53 pathway.Citation72 Thirty-six mRNAs were found to be differentially expressed by cisplatin treatment. These results suggest that miRNAs play a crucial role in renal injury through regulation of gene transcripts and later through protein translation.
Wolenski et al dosed male Sprague-Dawley rats with a single dose of 2 mg/kg or 5 mg/kg cisplatin and changes in miRNA expression in urine, plasma, and kidney tissue were assessed.Citation77 There were 832 miRNAs detected in all samples. At 72 h post-cisplatin, there were significant changes in 6 miRNAs in urine, 14 in plasma, and 5 in kidney tissue. Of the miRNAs elevated in the urine, 5 of them (miR-378a, miR-140, miR-26b, miR-1839, and miRlet-7g) were validated in a second rat model of cisplatin toxicity. A 6-fold elevation of urinary miR-378a was observed and increased expression of miR-34a in kidney tissue was consistent with previous studies. A total of 4 miRs (miR-34c, miR-128, miR-34a, and miR-130b) had ≥2-fold expression changes in the plasma 72 h post-cisplatin administration.Citation77 From these data, miR-34a was the only miRNA that was significantly changed in both kidney tissue and plasma, but not in urine.
A recently published study evaluated the potential of miRNA biomarkers for predicting cisplatin nephrotoxicity in humans as assessed by KIM-1.Citation74 The authors reported significantly increased miR-21, miR-200c, and miR-423 after cisplatin treatment as compared to baseline. However, urinary miRNA concentrations were not associated with increased odds of AKI development. These data suggest that urinary KIM-1 and miRNAs can be non-invasive methods for the detection of cisplatin-induced AKI in cancer patients.Citation74
Summary
DIKD is associated with changes in the expression of mRNAs and miRNAs that may contribute to the pathogenesis or progression of the disease or may serve protective roles. The expression of the transcriptome can be modulated by many different pathways including transcriptional initiation, RNA processing, and post-translational modification, all of which can initiate changes in expression (including upregulation and downregulation) of genes involved in DIKD pathological or recovery processes. miRNAs contribute to DIKD by changing expression of downstream target genes which may afford renoprotection or contribute to the disease process. Understanding the interplay between transcriptomic and miRNA changes can potentially facilitate early diagnosis, enable the development of novel therapeutics, and assess prognosis in patients with DIKD.
Metabolomics
Metabolomics allows for rapid and reproducible analyses that are directly reflective of biological events and require limited sample material such as plasma and urine.Citation82 Metabolomic biomarkers have the potential to detect early DIKD. Metabolomic changes in urine and kidney tissue of rats were evaluated after treatment with known proximal tubule nephrotoxins including cisplatin, gentamicin, and tobramycin.Citation83 Sprague-Dawley rats were treated with gentamicin 40 mg/kg, cisplatin 0.5 mg/kg, or tobramycin 40 mg/kg. Urine and kidney samples were collected at 1, 5, and 28 days after dosing. GC/MS and LC/MS detected 547 metabolites in kidney tissue and 657 metabolites in urine.Citation83 It was observed that the majority of metabolites increased in urine with drug exposures, while the majority of the metabolites decreased in kidney tissue after drug exposures. A finding of particular interest was the increase in excretion of several polyamines including cadaverine, putrescine, agmatine, and spermidine, which were found to be most pronounced on day 1 after drug exposures with reported fold changes of 1.86, 2.4, 4.37, and 2.35, respectively, for the previous polyamines (). Excretion of polyamines in urine may be of interest as a metabolic biomarker for detection of early nephrotoxicity due to gentamicin, cisplatin, and/or tobramycin.Citation83
Table 4 Summary of Selected Metabolite Biomarkers in Urine and Kidney in Nephrotoxicity Models. Metabolites in Urine and Kidney Showed a Day 1 Response in the Three Nephrotoxicity Models in Terms of Fold Change in Comparison to the Vehicle Sample Through GC-MS (GC) or LC-MS (LC) AnalysisCitation83
Cisplatin
The effects of dose and time-dependent changes on metabolite levels and nephrotoxicity after exposure to cisplatin were evaluated using MS-based metabolomics integrated with principle component-based area calculation.Citation84 In this study, cisplatin was administered to Sprague-Dawley rats in low-dose (LD) 2.5 mg/kg, middle-dose (MD) 5.0 mg/kg, and high-dose (HD) 10 mg/kg, and samples were collected for 7 days. A control group received saline. Significant differences (p < 0.0001) in BUN and SCr were observed between the control and MD groups at days 3 and 5 and between the control and HD groups at days 3, 5, and 7. The LD group showed no difference in BUN and SCr when compared to the control group. Moderate histological damage was observed in the MD group, while the HD group exhibited more severe histological damage.Citation84 Based on GC-MS and LC-MS data, fold-change area under the curve (FCAUC) was calculated for each group to quantitate trends in metabolic changes, with absolute values >1.50 deemed to be significant.Citation84 LD did not result in any significant metabolic changes. MD was found to have induced significant metabolic changes at day 5 with an FCAUC of −1.94 and −1.82 for GC-MS and LC-MS data, respectively, and a time-dependent effect was observed, with FCAUC values decreasing over time to −2.27 for GC-MS and −2.03 for LC-MS at day 7.Citation84 The HD group saw an even greater decrease in FCAUC values (−2.44 for GC-MS and −2.69 for LC-MS at day 5 to −3.72 for GC-MS and −3.50 for LC-MS at day 7), indicating even more significant metabolic changes.Citation84 LC-MS was found to have a higher sensitivity than GC-MS for detecting metabolites, with MD and HD groups having significant FCAUC values at day 3 (−1.57 and −1.83, respectively), where significant metabolic change occurred at day 5.Citation84 This study not only identified potential metabolic biomarkers, but it also reported a high sensitivity method for quantitatively analyzing metabolic changes to reveal dose- and time-dependent-induced nephrotoxicity secondary to cisplatin.
Gentamicin
In the human neonatal population, sensitivity and specificity limitations for detection of kidney injury using SCr and BUN are complicated by initial postnatal SCr reflecting the maternal values. Additionally, changes in creatinine clearance occur as growth and renal development ensues.Citation85 As a result, kidney injury in newborns often goes undetected until a substantial reduction in function occurs, signifying the need for early, sensitive, and specific biomarkers.Citation86 Rat kidneys continue to develop until approximately 3 weeks after birth, similar to the pattern of postnatal renal development that occurs in human preterm neonates.Citation87 In the study by Hanna et al, newborn Sprague Dawley rats were treated for 7 days with gentamicin, receiving either a low dose (10 mg/kg/day) or high dose (20 mg/kg/day),Citation85 corresponding to two and four times the recommended dose for human newborn infants. BUN and SCr did not exhibit a significant change in either treatment group 3 days after injection, but a significant increase was seen in both treatment groups 7 days after injection (p < 0.05). Histopathology of kidneys 7 days after low-dose injection showed minimal focal tubular necrosis. High-dose treatment showed more distinguishable changes, which included proximal convoluted tubules with loss of brush border on the surface, damaged mitochondria, and tubular casts.Citation85 Altered metabolite levels, as measured by GC/MS and HPLC/MS were observed at days 3 and 7 for both dosing groups.Citation85 Multivariate data analysis found 10 metabolites that contributed to significant perturbations of kidney injury by gentamicin (). Significant changes in the abundance of metabolites after treatment with gentamicin suggest that several biochemical metabolic pathways are altered and these could serve as superior biomarkers to BUN and SCr for detection of kidney injury in the neonatal population.Citation85
Table 5 Metabolite Changes in Urine After Low or High Dose of Gentamicin. Metabolites Identified in Urine After Treatment with a Low or High Dose of Gentamicin in Comparison to the Control Group That Showed Significant Fold Changes (p < 0.05) and Showed Good Fitness and High Predictability with High Statistical Values of R2 and Q2, Respectively, Through PLS-DA Analysis.Citation85
Bioinformatics in Combination with Metabolomics
A large reference database of metabolic profiles was compared to the metabolic profiles obtained from animals treated with nephrotoxic compounds as a potential method to predict toxic events post exposures.Citation88 This method could be of particular importance to drug development, as 30% of new drug candidates are halted in development because of toxicity profiles and side effects in clinical studies.Citation89 In this study, Wistar rats were treated for 28 days with either a low dose or high dose, respectively, of phenytoin (600 ppm/day or 2400 ppm/day; negative control), doxorubicin (0.5 mg/kg or 2 mg/kg biweekly), cyclosporin A (20 mg/kg or 45 mg/kg biweekly), lisinopril (40 mg/kg or 400 mg/kg biweekly) or captopril (20 mg/kg or 200 mg/kg biweekly). Blood samples were obtained at 7, 14, and 28 days post-treatment, and metabolite profiling analysis of the samples was performed by GC-MS, LC-MS/MS, and solid phase extraction (SPE)-LC-MS/MS. The MetaMap Tox system analyzes metabolite profiles based on the strength of the change and the biochemical importance for a certain mode of action.Citation88 This study found that metabolite profile analysis with the MetaMap Tox system was successfully able to predict nephrotoxicity to treatments known to induce this adverse event, whereas nephrotoxicity was not predicted with negative controls.Citation88 Incorporating metabolomics database like MetaMap Tox into the drug development process can be a useful tool providing the ability of identifying toxicities, including nephrotoxicity, in response to drug treatments. The prospect of identifying potential nephrotoxicity prior to starting clinical studies would be highly beneficial to human patients and would significantly decrease drug development costs.
Summary
Metabolomics has good potential for the early and sensitive detection of DIKD. Ability to detect DIKD through metabolic profiles would aid in early interventions including removal of offending agents, reduction in dosages, treatment changes, and mitigation therapies. Understanding the patterns of metabolic changes associated with DIKD would also contribute to drug safety in preclinical and clinical drug development.
Disclosure
The authors report no conflicts of interest for this work.
References
- Group K. KDIGO Clinical Practice Guidelines for Acute Kidney Injury. Kidney Int. 2012;2:1–138.
- Hoste EAJ, Bagshaw SM, Bellomo R, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Medicine. 2015;41(8):1411–1423. doi:10.1007/s00134-015-3934-726162677
- Mehta RL, Pascual MT, Soroko S, et al. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int. 2004;66(4):1613–1621. doi:10.1111/j.1523-1755.2004.00927.x15458458
- Meyboom RHB, Egberts ACG, Edwards IR, Hekster YA, de Koning FHP, Gribnau FWJ. Principles of signal detection in pharmacovigilance. Drug Saf. 1997;16(6):355–365. doi:10.2165/00002018-199716060-000029241490
- Mehta RL, Awdishu L, Davenport A, et al. Phenotype standardization for drug-induced kidney disease. Kidney Int. 2015;88(2):226–234. doi:10.1038/ki.2015.11525853333
- Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129–1136. doi:10.1056/NEJMoa070733018337603
- Cortazar FB, Marrone KA, Troxell ML, et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016;90(3):638–647.27282937
- Perazella MA. Checkmate: kidney injury associated with targeted cancer immunotherapy. Kidney Int. 2016;90(3):474–476. doi:10.1016/j.kint.2016.05.02427521108
- Jaimes EA. Renal Toxicity of Systemic Therapy for Renal Cell Carcinoma. Semin Nephrol. 2020;40(1):49–58.32130966
- Lefebvre J, Glezerman IG. Kidney Toxicities Associated With Novel Cancer Therapies. Adv Chronic Kidney Dis. 2017;24(4):233–240. doi:10.1053/j.ackd.2017.05.00628778363
- Malyszko J, Kozlowska K, Kozlowski L. Nephrotoxicity of anticancer treatment. Nephrol Dial Transplant. 2017;32(6):924–936.28339935
- Murakami N, Motwani S, Riella LV. Renal complications of immune checkpoint blockade. Curr Probl Cancer. 2017;41(2):100–110. doi:10.1016/j.currproblcancer.2016.12.00428189263
- Nussbaum EZ, Perazella MA. Update on the nephrotoxicity of novel anticancer agents. Clin Nephrol. 2018;89(3):149–165. doi:10.5414/CN10937129350175
- Wanchoo R, Abudayyeh A, Doshi M, et al. Renal Toxicities of Novel Agents Used for Treatment of Multiple Myeloma. Clin J Am Soc Nephrol. 2017;12(1):176–189. doi:10.2215/CJN.0610061627654928
- Lu H, Han Y-J, Xu J-D, Xing W-M, Chen J, Zhang C. Proteomic characterization of acyclovir-induced nephrotoxicity in a mouse model. PLoS One. 2014;9(7):e103185. doi:10.1371/journal.pone.010318525055032
- Rocha PN, Macedo MN, Kobayashi CD, et al. Role of urine neutrophil gelatinase-associated lipocalin in the early diagnosis of amphotericin B-induced acute kidney injury. Antimicrob Agents Chemother. 2015;59(11):6913–6921. doi:10.1128/AAC.01079-1526303800
- Perez JD, Colucci JA, Sakata MM, Cunha TS, Arita DY, Casarini DE. Proteomic approaches in understanding a detected relationship between chemotherapy-induced nephrotoxicity and cell respiration in HK-2 cells. Nephron Physiol. 2011;119(1):pp1–10. doi:10.1159/000327575
- Wilmes A, Bielow C, Ranninger C, et al. Mechanism of cisplatin proximal tubule toxicity revealed by integrating transcriptomics, proteomics, metabolomics and biokinetics. Toxicol in Vitro. 2015;30(1):117–127. doi:10.1016/j.tiv.2014.10.00625450742
- Montoro-Molina S, Quesada A, Zafra-Ruiz PV, et al. Immunological detection of glutamyl aminopeptidase in urine samples from cisplatin-treated rats. Proteomics Clin Appl. 2015;9(5–6):630–635. doi:10.1002/prca.20140009625470983
- Zhang W, Zhang L, Chen Y-X, et al. Identification of nestin as a urinary biomarker for acute kidney injury. Am J Nephrol. 2014;39(2):110–121. doi:10.1159/00035826024503548
- Bulacio RP, Torres AM. Organic anion transporter 5 (Oat5) renal expression and urinary excretion in rats treated with cisplatin: a potential biomarker of cisplatin-induced nephrotoxicity. Arch Toxicol. 2013;87(11):1953–1962. doi:10.1007/s00204-013-1062-023649842
- Furukawa K, Yamane M, Tatsukawa H, Hitomi K. Early response as shown by enhancement of transglutaminase 1 expression after cisplatin-induced acute kidney injury. Arch Biochem Biophys. 2015;586(586):27–32. doi:10.1016/j.abb.2015.09.02126427353
- George B, Wen X, Mercke N, et al. Profiling of Kidney Injury Biomarkers in Patients Receiving Cisplatin: time-dependent Changes in the Absence of Clinical Nephrotoxicity. Clin Pharmacol Ther. 2017;101(4):510–518. doi:10.1002/cpt.60628002630
- George B, Wen X, Mercke N, et al. Time-dependent changes in kidney injury biomarkers in patients receiving multiple cycles of cisplatin chemotherapy. Toxicol Rep. 2020;7:571–576. doi:10.1016/j.toxrep.2020.04.00332382514
- Lamoureux F, Gastinel LN, Mestre E, Marquet P, Essig M. Mapping cyclosporine-induced changes in protein secretion by renal cells using stable isotope labeling with amino acids in cell culture (SILAC). J Proteomics. 2012;75(12):3674–3687. doi:10.1016/j.jprot.2012.04.02422564819
- Klawitter J, Klawitter J, Kushner E, et al. Association of immunosuppressant-induced protein changes in the rat kidney with changes in urine metabolite patterns: a proteo-metabonomic study. J Proteome Res. 2010;9(2):865–875. doi:10.1021/pr900761m19994912
- Klawitter J, Klawitter J, Schmitz V, et al. Low-salt diet and cyclosporine nephrotoxicity: changes in kidney cell metabolism. J Proteome Res. 2012;11(11):5135–5144. doi:10.1021/pr300260e23057591
- Betton GR, Kenne K, Somers R, Marr A, Dixit R. Protein biomarkers of nephrotoxicity; a review and findings with cyclosporin A, a signal transduction kinase inhibitor and N-phenylanthranilic acid. Cancer Biomarkers. 2005;1(1):59–67. doi:10.3233/CBM-2005-110717192032
- Shu Z, Pu X, Xiong X, Li Q, Wang Y, Zhai S. Differential expression of plasma proteins in cyclosporine A-induced rat acute nephrotoxicity. Biosci Biotechnol Biochem. 2009;73(3):592–598. doi:10.1271/bbb.8067719270397
- Ferreira L, Quiros Y, Sancho-Martinez SM, et al. Urinary levels of regenerating islet-derived protein III β and gelsolin differentiate gentamicin from cisplatin-induced acute kidney injury in rats. Kidney Int. 2011;79(5):518–528. doi:10.1038/ki.2010.43920980976
- Rouse R, Siwy J, Mullen W, Mischak H, Metzger J, Hanig J. Proteomic candidate biomarkers of drug-induced nephrotoxicity in the rat. PLoS One. 2012;7(4):e34606. doi:10.1371/journal.pone.003460622509332
- Vicente-Vicente L, Ferreira L, Gonzalez-Buitrago JM, Lopez-Hernandez FJ, Lopez-Novoa JM, Morales AI. Increased urinary excretion of albumin, hemopexin, transferrin and VDBP correlates with chronic sensitization to gentamicin nephrotoxicity in rats. Toxicology. 2013;304(304):83–91. doi:10.1016/j.tox.2012.12.00623261757
- Zhou Y, Vaidya VS, Brown RP, et al. Comparison of kidney injury molecule-1 and other nephrotoxicity biomarkers in urine and kidney following acute exposure to gentamicin, mercury, and chromium. Toxicol Sci. 2008;101(1):159–170. doi:10.1093/toxsci/kfm26017934191
- Gautier J-C, Zhou X, Yang Y, et al. Evaluation of novel biomarkers of nephrotoxicity in Cynomolgus monkeys treated with gentamicin. Toxicol Appl Pharmacol. 2016;303(303):1–10. doi:10.1016/j.taap.2016.04.01227105553
- Zhou X, Ma B, Lin Z, et al. Evaluation of the usefulness of novel biomarkers for drug-induced acute kidney injury in beagle dogs. Toxicol Appl Pharmacol. 2014;280(1):30–35. doi:10.1016/j.taap.2014.07.00225034533
- Com E, Boitier E, Marchandeau J-P, et al. Integrated transcriptomic and proteomic evaluation of gentamicin nephrotoxicity in rats. Toxicol Appl Pharmacol. 2012;258(1):124–133. doi:10.1016/j.taap.2011.10.01522061828
- Meistermann H, Norris JL, Aerni H-R, et al. Biomarker discovery by imaging mass spectrometry: transthyretin is a biomarker for gentamicin-induced nephrotoxicity in rat. Mol Cell Proteomics. 2006;5(10):1876–1886. doi:10.1074/mcp.M500399-MCP20016705188
- Bellei E, Cuoghi A, Monari E, et al. Proteomic analysis of urine in medication-overuse headache patients: possible relation with renal damages. J Headache Pain. 2012;13(1):45–52. doi:10.1007/s10194-011-0390-921997203
- Bellei E, Monari E, Cuoghi A, et al. Discovery by a proteomic approach of possible early biomarkers of drug-induced nephrotoxicity in medication-overuse headache. J Headache Pain. 2013;14(1):6. doi:10.1186/1129-2377-14-623565828
- Kuypers DRJ, Naesens M, de Jonge H, Lerut E, Verbeke K, Vanrenterghem Y. Tacrolimus dose requirements and CYP3A5 genotype and the development of calcineurin inhibitor-associated nephrotoxicity in renal allograft recipients. Ther Drug Monit. 2010;32(4):394–404. doi:10.1097/FTD.0b013e3181e0681820526235
- Joy MS, Hogan SL, Thompson BD, Finn WF, Nickeleit V. Cytochrome P450 3A5 expression in the kidneys of patients with calcineurin inhibitor nephrotoxicity. Nephrol Dial Transplant. 2007;22(7):1963–1968. doi:10.1093/ndt/gfm13317395652
- Hauser IA, Schaeffeler E, Gauer S, et al. ABCB1 genotype of the donor but not of the recipient is a major risk factor for cyclosporine-related nephrotoxicity after renal transplantation. J Am Soc Nephrol. 2005;16(5):1501–1511. doi:10.1681/ASN.200410088215772250
- Joy MS, Nickeleit V, Hogan SL, Thompson BD, Finn WF. Calcineurin Inhibitor–Induced Nephrotoxicity and Renal Expression of P-glycoprotein. Pharmacotherapy. 2005;25(6):779–789. doi:10.1592/phco.2005.25.6.77915927895
- Naesens M, Lerut E, de Jonge H, Van Damme B, Vanrenterghem Y, Kuypers DRJ. Donor age and renal P-glycoprotein expression associate with chronic histological damage in renal allografts. J Am Soc Nephrol. 2009;20(11):2468–2480. doi:10.1681/ASN.200902019219762492
- Moore J, McKnight AJ, Dohler B, et al. Donor ABCB1 Variant Associates with Increased Risk for Kidney Allograft Failure. J Am Soc Nephrol. 2012;23(11):1891–1899. doi:10.1681/ASN.201203026023064017
- Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86(4):396–402. doi:10.1038/clpt.2009.13919625999
- Nakamura T, Yonezawa A, Hashimoto S, Katsura T, Inui K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem Pharmacol. 2010;80(11):1762–1767. doi:10.1016/j.bcp.2010.08.01920813096
- Wen X, Buckley B, McCandlish E, et al. Transgenic expression of the human MRP2 transporter reduces cisplatin accumulation and nephrotoxicity in Mrp2-null mice. Am J Pathol. 2014;184(5):1299–1308. doi:10.1016/j.ajpath.2014.01.02524641901
- Iwata K, Aizawa K, Kamitsu S, et al. Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin Exp Nephrol. 2012;16(6):843–851. doi:10.1007/s10157-012-0638-y22569819
- Chang C, Hu Y, Hogan SL, et al. Pharmacogenomic Variants May Influence the Urinary Excretion of Novel Kidney Injury Biomarkers in Patients Receiving Cisplatin. Int J Mol Sci. 2017;18(7):7. doi:10.3390/ijms18071333
- Izzedine H, Hulot J-S, Villard E, et al. Association between ABCC2 Gene Haplotypes and Tenofovir-Induced Proximal Tubulopathy. J Infect Dis. 2006;194(11):1481–1491. doi:10.1086/50854617083032
- Rodríguez‐Nóvoa S, Labarga P, Soriano V, et al. Predictors of kidney tubular dysfunction in HIV-infected patients treated with tenofovir: a pharmacogenetic study. Clin Infect Dis. 2009;48(11):e108–16. doi:10.1086/59850719400747
- Wanga V, Venuto C, Morse GD, et al. Genomewide association study of tenofovir pharmacokinetics and creatinine clearance in AIDS Clinical Trials Group protocol A5202. Pharmacogenet Genomics. 2015;25(9):450–461. doi:10.1097/FPC.000000000000015626148204
- E, Zubowska M, Misiura K. Role of GSTM1, GSTP1, and GSTT1 gene polymorphism in ifosfamide metabolism affecting neurotoxicity and nephrotoxicity in children. J Pediatr Hematol Oncol. 2005;27(11):582–589. doi:10.1097/01.mph.0000187429.52616.8a16282887
- Bosso JA, Nappi J, Rudisill C, et al. Relationship between vancomycin trough concentrations and nephrotoxicity: a prospective multicenter trial. Antimicrob Agents Chemother. 2011;55(12):5475–5479. doi:10.1128/AAC.00168-1121947388
- Van Driest SL, McGregor TL, Velez Edwards DR, et al. Genome-Wide Association Study of Serum Creatinine Levels during Vancomycin Therapy. PLoS One. 2015;10(6):e0127791.26030142
- Yao J, Huang T, Fang X, et al. Disruption of gap junctions attenuates aminoglycoside-elicited renal tubular cell injury. Br J Pharmacol. 2010;160(8):2055–2068. doi:10.1111/j.1476-5381.2010.00860.x20649601
- Eadon MT, Wheeler HE, Stark AL, et al. Genetic and epigenetic variants contributing to clofarabine cytotoxicity. Hum Mol Genet. 2013;22(19):4007–4020. doi:10.1093/hmg/ddt24023720496
- Albano LMJ, Sakae PPO, Mataloun MMGB, Leone CR, Bertola DR, Kim CA. Hydronephrosis in Schinzel-Giedion syndrome: an important clue for the diagnosis. Rev Hosp Clin Fac Med Sao Paulo. 2004;59(2):89–92. doi:10.1590/S0041-8781200400020000815122424
- Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int. 2009;76(6):604–613. doi:10.1038/ki.2009.22419536080
- Khrunin AV, Moisseev A, Gorbunova V, Limborska S. Genetic polymorphisms and the efficacy and toxicity of cisplatin-based chemotherapy in ovarian cancer patients. Pharmacogenomics J. 2010;10(1):54–61. doi:10.1038/tpj.2009.4519786980
- Goekkurt E, Al-Batran S-E, Hartmann JT, et al. Pharmacogenetic analyses of a Phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil and leucovorin plus either oxaliplatin or cisplatin: a study of the arbeitsgemeinschaft internistische onkologie. J Clin Oncol. 2009;27(17):2863–2873. doi:10.1200/JCO.2008.19.171819332728
- Supavekin S, Zhang W, Kucherlapati R, Kaskel FJ, Moore LC, Devarajan P. Differential gene expression following early renal ischemia/reperfusion. Kidney Int. 2003;63(5):1714–1724. doi:10.1046/j.1523-1755.2003.00928.x12675847
- Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol. 2008;19(3):547–558. doi:10.1681/ASN.200704046918235097
- Amin RP, Vickers AE, Sistare F, et al. Identification of putative gene based markers of renal toxicity. Environ Health Perspect. 2004;112(4):465–479. doi:10.1289/ehp.668315033597
- Dadarkar SS, Fonseca LC, Mishra PB, et al. Phenotypic and genotypic assessment of concomitant drug-induced toxic effects in liver, kidney and blood. J Appl Toxicol. 2011;31(2):117–130.20623750
- Davis JW, Goodsaid FM, Bral CM, et al. Quantitative gene expression analysis in a nonhuman primate model of antibiotic-induced nephrotoxicity. Toxicol Appl Pharmacol. 2004;200(1):16–26. doi:10.1016/j.taap.2004.02.00115451304
- Hung Y-C, Huang GS, Lin L-W, Hong M-Y, Se P-S. Thea sinensis melanin prevents cisplatin-induced nephrotoxicity in mice. Food Chem Toxicol. 2007;45(7):1123–1130. doi:10.1016/j.fct.2006.12.01717303299
- Aguado-Fraile E, Ramos E, Conde E, et al. A Pilot Study Identifying a Set of microRNAs As Precise Diagnostic Biomarkers of Acute Kidney Injury. PLoS One. 2015;10(6):e0127175. doi:10.1371/journal.pone.012717526079930
- Gutierrez-Escolano A, Santacruz-Vazquez E, Gomez-Perez F. Dysregulated microRNAs involved in contrast-induced acute kidney injury in rat and human. Ren Fail. 2015;37(9):1498–1506. doi:10.3109/0886022X.2015.107732226337190
- Kanki M, Moriguchi A, Sasaki D, et al. Identification of urinary miRNA biomarkers for detecting cisplatin-induced proximal tubular injury in rats. Toxicology. 2014;324(324):158–168. doi:10.1016/j.tox.2014.05.00424863737
- Lee CG, Kim JG, Kim HJ, et al. Discovery of an integrative network of microRNAs and transcriptomics changes for acute kidney injury. Kidney Int. 2014;86(5):943–953. doi:10.1038/ki.2014.11724759152
- Pavkovic M, Riefke B, Ellinger-Ziegelbauer H. Urinary microRNA profiling for identification of biomarkers after cisplatin-induced kidney injury. Toxicology. 2014;324(324):147–157. doi:10.1016/j.tox.2014.05.00524880025
- Pavkovic M, Robinson-Cohen C, Chua AS, et al. Detection of Drug-Induced Acute Kidney Injury in Humans Using Urinary KIM-1, miR-21, −200c, and −423. Toxicological Sciences. 2016;152(1):205–213. doi:10.1093/toxsci/kfw07727122240
- Saikumar J, Hoffmann D, Kim T-M, et al. Expression, circulation, and excretion profile of microRNA-21, −155, and −18a following acute kidney injury. Toxicol Sci. 2012;129(2):256–267. doi:10.1093/toxsci/kfs21022705808
- Sun S, Zhang T, Ding D, et al. Circulating microRNA-188, −30a, and −30e a early biomarkers for contrast-induced acute kidney injury. J Am Heart Assoc. 2016;116:1–11.
- Wolenski FS, Shah P, Sano T, et al. Identification of microRNA biomarker candidates in urine and plasma from rats with kidney or liver damage. J Appl Toxicol. 2016;11.
- Zhou XB, Qu Z, Zhu C, et al. Identification of urinary microRNA biomarkers for detection of gentamicin-induced acute kidney injury in rats. Regulatory Toxicology Pharmacology. 2016;78:78–84. doi:10.1016/j.yrtph.2016.04.00127074385
- Oliveira JF, Silva CA, Barbieri CD, Oliveira GM, Zanetta DM, Burdmann EA. Prevalence and risk factors for aminoglycoside nephrotoxicity in intensive care units. Antimicrob Agents Chemother. 2009;53(7):2887–2891.19364846
- Qiu Y, Hong M, Li H, et al. Time-series pattern of gene expression profile in gentamycin-induced nephrotoxicity. Toxicol Mech Methods. 2014;24(2):142–150. doi:10.3109/15376516.2013.86978024274596
- Eadon MT, Hack BK, Alexander JJ, Xu C, Dolan ME, Cunningham PN. Cell cycle arrest in a model of colistin nephrotoxicity. Physiol Genomics. 2013;45(19):877–888. doi:10.1152/physiolgenomics.00076.201323922129
- Uehara T, Horinouchi A, Morikawa Y, et al. Identification of metabolomic biomarkers for drug-induced acute kidney injury in rats. J Appl Toxicol. 2014;34(10):1087–1095.24114878
- Boudonck KJ, Mitchell MW, Nemet L, et al. Discovery of metabolomics biomarkers for early detection of nephrotoxicity. Toxicol Pathol. 2009;37(3):280–292.19380839
- Zhang P, Chen J, Wang Y, et al. Discovery of Potential Biomarkers with Dose- and Time-Dependence in Cisplatin-Induced Nephrotoxicity Using Metabolomics Integrated with a Principal Component-Based Area Calculation Strategy. Chem Res Toxicol. 2016;29(5):776–783.27030963
- Hanna MH, Segar JL, Teesch LM, Kasper DC, Schaefer FS, Brophy PD. Urinary metabolomic markers of aminoglycoside nephrotoxicity in newborn rats. Pediatr Res. 2013;73(5):585–591.23411940
- Andreoli SP. Acute renal failure in the newborn. Semin Perinatol. 2004;28(2):112–123.15200250
- Marquez MG, Cabrera I, Serrano DJ, Sterin-Speziale N. Cell proliferation and morphometric changes in the rat kidney during postnatal development. Anat Embryol (Berl). 2002;205(5–6):431–440.12382146
- Mattes WB, Kamp HG, Fabian E, et al. Prediction of clinically relevant safety signals of nephrotoxicity through plasma metabolite profiling. Biomed Res Int. 2013;2013:202497.23762827
- Thukral SK, Nordone PJ, Hu R, et al. Prediction of nephrotoxicant action and identification of candidate toxicity-related biomarkers. Toxicol Pathol. 2005;33(3):343–355.15805072