Abstract
Raltegravir has recently been licensed for the treatment of HIV-1 infection. Currently its use is limited to treatment-experienced patients and subjects with resistant virus. In addition to its activity in the setting of resistance and treatment failure, it appears to have great potential for first-line therapy and as a switch option for subjects with intolerance to other agents, as well. Overall tolerability in clinical trials was excellent, and the toxicity profile is non-overlapping with other agents, with no clear neuropsychiatric, gastrointestinal, or metabolic toxicity. Its metabolization occurs mainly via UGT1A1 rather than by the CYP450 system, resulting in a relatively unproblematic drug interaction profile. The independence of the compound from “boosting” of drug levels with ritonavir is an attractive feature for many patients suffering from ritonavir-associated side effects. However, it has to be dosed twice daily.
The unique effect of raltegravir on the establishment of viral latency makes it a logical component of treatment attempts aiming at reducing and controlling this viral sanctuary.
This review summarizes the clinical view on the role of this novel compound in HIV therapy.
Until 2007, highly active antiretroviral therapy (HAART) was based on combinations of nucleoside (NRTI), nucleotide (NtRTI), non-nucleoside reverse transcriptase inhibitors (NNRTI), and protease inhibitors (PI), as well as the fusion inhibitor enfuvirtide. In late 2007 and early 2008, the CCR5 chemokine receptor blocker maraviroc and the first integrase inhibitor raltegravir were licensed in most countries, representing two new classes of drugs with novel modes of action.
Raltegravir as the first drug in its class targets HIV-1 integrase, an enzyme in the viral replication cycle that is essential for inserting HIV-1 proviral DNA into the host cell genome. In contrast to drugs from all other classes, this mode of action affects viral latency directly.
Mechanism of action
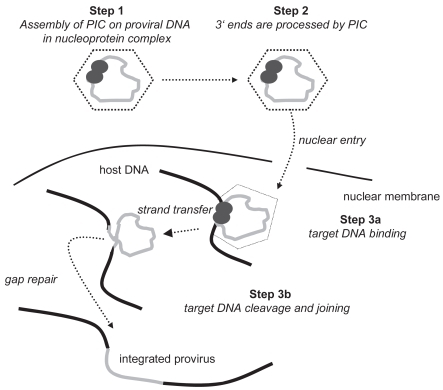
The complete viral life cycle requires integration of HIV-1 proviral DNA into the host cell genome. It occurs in three steps (CitationCraigie 2001; CitationHazuda et al 2000; CitationLafemina et al 1992). The double-stranded DNA copy generated by reverse transcription of the HIV-1 viral RNA genome is associated with a “preintegration complex”, containing both cellular and viral proteins, including integrase. In a first step, two nucleotides are removed from the 3′ ends of the proviral DNA (see ). In a second step, the proviral DNA strands are inserted into the host DNA and joined with it. This process is termed “strand transfer”. Subsequently, gaps in DNA are repaired by cellular enzymes by removing the two unpaired nucleotides at the 5′ end of the proviral DNA. HIV integrase catalyses the first two steps.
Figure 1 Steps of viral integration. Adapted with permission from: New Classes of Antiretrovirals: The Potential Clinical Role of Integrase Inhibitors and Entry Inhibitors. Clinical Care Options; Reston, Virginia; Slideset accessed at clinicaloptions.com/hiv.
With the discovery of selective inhibitors of strand transfer, which share a β-diketo acid moiety, orally bioavailable agents came within the reach (CitationHazuda et al 1999, Citation2004; CitationEspeseth et al 2000; CitationPais et al 2002; CitationEmbrey et al 2005). Chemical optimisation of these compounds led to naphthyridine derivatives and L-900612 or MK-0518 (raltegravir) as a promising candidate compound.
Pharmacological properties, metabolism
Oral absorption of raltegravir is rapid, and oral bioavailability approaches 32%. Plasma protein binding reaches 83%. Raltegravir has a rapid initial half-life of ~1 hour and a terminal half-life of ~7 hours (CitationMerck and Co. 2007), not supporting once daily administration. Steady state pharmacokinetics are reached within 2 days after the first dose (CitationIwamoto et al 2008b).
Although a high fat meal postpones the maximum plasma concentration by approximately 7.5 hours and decreases it by 34%, this effect is outweighed by an augmentation of exposure over time, as assessed by an increase of the area under the curve (AUC) by 19%. As a consequence, no specific restrictions are recommended with regard to food.
In contrast to most other antiretroviral drugs, raltegravir is metabolized by glucuronidation via UGT1A1 (CitationKassahun et al 2006, Citation2007; CitationMerck and Co. 2007, Citation2008). Excretion in feces (51%) and in urine (31%) accounts for most of the elimination. No dose adjustment is required for gender, age, hepatic or renal function, or body mass index.
Drug interactions
Raltegravir neither induces nor inhibits cytochrome P450 enzymes, nor is it a substrate (CitationIwamoto et al 2008a). Therefore, few interactions have to be expected with other drugs metabolized via the P450 enzyme system, such as protease inhibitors (PI), non-nucleoside reverse transcriptase inhibitors (NNRTI), maraviroc or frequent concomitant medication (eg, anticonvulsants, lipid-lowering drugs).
Of note, coadministration of raltegravir with tipranavir/ritonavir led to a 55% decrease in raltegravir C12 h trough levels (CitationWenning et al 2006b), while reducing maximum concentrations (Cmax) and the AUC0–12 h less markedly (24 and 18%).
Indinavir and Atazanavir inhibit UGT1A1. Because Indinavir is used rarely due to its specific toxicity, atazanavir is a likely combination partner for raltegravir. Pharmacokinetic studies with atazanavir, with or without ritonavir-“boosting”, showed a 30%–70% increase in raltegravir AUC (CitationMistry et al 2007). In view of its good tolerability (see below), this is probably not clinically relevant, and no dose adjustment is required for either drug. No relevant interaction was observed with efavirenz, etravirine, or tenofovir, either (CitationIwamoto et al 2006b; CitationWenning et al 2006a). Rifampicin, however, reduces raltegravir concentrations at 12 hours by 61%(CitationIwamoto et al 2006a), which appears clinically relevant. Combinations of raltegravir with rifampicin or similarly potent inducers should therefore be avoided where possible (CitationKassahun et al 2006, Citation2007). Importantly, there appears to be no relevant interaction with contraceptives (CitationAnderson et al 2007).
Antiviral activity in vitro
Marked and broad in vitro antiviral activity was observed against HIV-1 variants sensitive or resistant to NRTIs, NNRTIs, and PIs, as well as against SIV (Miller et al 2006). Despite considerable (40%) heterogeneity between HIV-1 and HIV-2 integrase genes, clinical HIV-2 isolates also show a high sensitivity to raltegravir (CitationRoquebert et al 2008).
Clinical trials in HIV-infected patients
For a list of relevant trials and a brief summary of their results, see .
Table 1 Clinical trial results with Raltegravir (MK-0518)
Phase II
In protocol 004 (CitationMarkowitz et al 2007), doses of 100, 200, 400, and 600 mg of raltegravir twice daily were compared to placebo (part 1) or efavirenz 600 mg (part 2) in antiretroviral naïve subjects in a randomized, double-blind manner, stratified for initial HIV plasma viremia (< or > 50000 copies/mL). Minimum CD4 cell count at screening was 100/μL, minimum viral load 5000 copies/mL. Thirty-five subjects entered part 1 and received a 10-day course of raltegravir monotherapy at the doses described above or placebo. Baseline characteristics were not significantly different between the study arms. The mean decrease at the different raltegravir doses after 10 days of monotherapy was 1.66 to 2.16 log10 copies/mL, as compared with placebo (−0.17 log10 copies/mL), with no significant difference between the raltegravir dosage arms.
Of these trial participants, 30 entered the second part, together with 198 additional subjects. They were randomized to receive either raltegravir at the doses mentioned above, or efavirenz (EFV) 600 mg, all in conjunction with tenofovir (TDF) and lamivudine (3TC). Baseline characteristics were comparable between the groups, with plasma viremia ranging between 4.6 and 4.8 log10 copies/mL and CD4+ T-cells from 271 to 314 cells/mm3. None of the study subjects had primary resistance to TDF, 3TC, or EFV. Viral load reduction to less than 50 copies of HIV RNA per mL was more rapid and significantly more frequent at weeks 4 and 8 in all raltegravir arms, but response was not different at any time-point thereafter up to and including week 48. CD4+ T-cells increased to a comparable extent in all arms. Adverse events were infrequent, of mild to moderate intensity, with no obvious difference between the treatment arms. Continued follow-up until week 96 confirmed the sustained efficacy of raltegravir in this study (CitationMarkowitz et al 2008).
Protocol 005 (CitationGrinsztejn et al 2007) was a multicenter, randomized, double-blind, placebo-controlled dose-ranging trial in treatment-experienced patients. 178 patients with greater than 5000 HIV RNA copies per mL, a minimum of 50 CD4+ T-cells on 3 months of stable antiretroviral therapy, and resistance to at least 3 drug classes were included and analyzed. Investigators selected an optimal background regimen according to all available resistance test results. Randomization was performed in a 1:1:1:1 fashion to raltegravir doses of 200, 400, and 600 mg twice daily, or placebo, stratified by PI resistance at baseline and prior enfuvirtide use. Primary end-points were week 24 response and toxicity.
Virological response was significantly better for all raltegravir arms as compared to placebo, with no significant difference between the raltegravir arms. This difference in favour of raltegravir was observed with any extent of resistance to the compounds of the background regimen, as assessed by genotypic or phenotypic sensitivity scores (GSS or PSS), ie, the number of drugs a patient received, which were predicted to be active at baseline. Use of additional enfuvirtide was also associated with a better virological outcome. There was no obvious difference of toxicity profile for any raltegravir dose in comparison to placebo.
Phase III
Based on the results of the aforementioned trials, 400 mg twice daily was selected as a dose for the phase III studies.
MK-0518-018 and MK-0518-019 (BENCHMRK-1 and BENCHMRK-2 (CitationCooper et al 2007; CitationSteigbigel et al 2007) were two parallel, double-blind, placebo-controlled studies. In both trials, an investigator-selected, resistance analysis-based optimal regimen was combined with either raltegravir or placebo. Randomization was performed in a 2:1 manner. Patients were required to have triple-class resistant virus at baseline and HIV plasma viremia greater than 1000 copies per mL. BENCHMRK-1 was conducted in Europe, Asia, the Pacific, and Peru, BENCHMRK-2 in the Americas. The primary end-point was viral suppression to < 400 copies per mL at week 16, with virus suppression to < 50 copies per mL and change from baseline viral load and CD4+ T-cell count evaluated as secondary end-points.
Baseline demographic variables and patient characteristics were comparable between the study arms and the studies.
The raltegravir arms in both trials were superior over placebo with regard to all end-points. The difference was maintained irrespective of the extent of baseline resistance (as assessed by GSS and PSS), baseline HIV RNA levels, and baseline CD4+ T-cell count. There was no significant difference with respect to toxicity between the study arms.
A superior response was sustained until week 24 in a combined analysis of both trials. The difference in favour of raltegravir persisted regardless of the background regimen. A repeat analysis after 48 weeks of follow-up confirmed the week 24 results: The response rate (HIV RNA < 50 copies per mL) in the raltegravir group at week 48 was 89% in combination with enfuvirtide and darunavir, when both were used for the first time (CitationCooper et al 2008). This approaches response rates in previously untreated patients and therefore represents a marked improvement for subjects with resistant virus.
Toxicity
Study 005 and the BENCHMRK studies are more difficult to interpret with respect to toxicity, since all participants received complex regimens with a high potential of untoward reactions, eg, gastrointestinal side effects associated with ritonavir-boosted protease inhibitors. However, side effects and laboratory abnormalities were balanced between the groups in both trials, with no significant difference between the study arms. In this regard, MK-0518-004 provides a clearer picture, because there was a 10-day run-in mono-therapy phase. Moreover, subsequently all subjects received a background regimen comprising of only tenofovir and lamivudine. From the 004 and 005 studies (CitationGrinsztejn et al 2007; CitationMarkowitz et al 2007), diarrhea, nausea, fatigue, and muscle aches were reported. Laboratory abnormalities include increases in pancreatic amylase and hepatic trans-aminase levels. Of note, no lipid abnormalities were reported from the 004 study (CitationMarkowitz et al 2007).
When analyzing the phase II and III trials together, only a few patients on raltegravir or placebo discontinued for adverse events. Adverse events were mostly mild to moderate. Headache, nausea, and diarrhea were the most common treatment related adverse events occurring in >10% of subjects. They were evenly distributed between the arms.
Fatigue (7.9% vs 4.6%), herpes zoster (4.1% vs 0.7%), nasopharyngitis (6.1% vs 3.9%), and mild to moderate rash (5.3% vs 2.5%) not requiring treatment discontinuation, as well as elevated creatine phosphokinase (CPK, 3.7% vs 1.1%) were observed more frequently on raltegravir (> 2% difference). Myopathy and rhabdomyolysis were also reported, but the causal role of raltegravir in these instances is unclear. It is therefore recommended that raltegravir be used with caution in combination with other drugs associated with muscle toxicity.
Overall, more hepatic laboratory events were observed on raltegravir. However, no clear difference was noted with respect to other hepatic adverse events in the total study population or within the subgroup of hepatitis B or C coinfected subjects.
In the pooled analysis, a higher rate of malignancies was observed in patients on raltegravir (2.5% vs 1.5% for placebo). After adjustment for duration of exposure, however, the adjusted risk of malignancy per 100 patient-years was not significantly different with a rate of 2.32 for raltegravir and 1.92 for the comparator groups (relative risk 1.209, 95% confidence interval 0.44–4.14). Moreover, the malignancies occurred in subjects with prior AIDS and were very heterogeneous, with 8/19 in the raltegravir groups representing recurrent disease and the others occurring within three months of enrolment. This indicates no causal relationship to raltegravir (CitationMerck and Co 2007).
A recent report of four cases suggests that raltegravir might lead to exacerbation of depression in subjects on psychotropic medication for mental disorders (CitationHarris et al 2008a), possibly due to drug-drug interactions. This observation, however, remains to be substantiated.
Raltegravir is currently classified as FDA pregnancy category C, mainly due to lack of data on reproductive toxicity.
Resistance
Knowledge about resistance selection patterns for raltegravir is rapidly expanding, and they are yet understood incompletely. Several characteristic mutations leading to typical amino acid exchanges were characterized in cell culture studies and confirmed in clinical samples from trial participants with virological failure on raltegravir (CitationMerck and Co 2007). The Q148K mutation was typically followed by an amino acid substitution at other positions (E138A, G140A, and V54I), with additional mutations developing at higher drug concentrations. Insertional mutagenesis confirmed the role of these amino acid changes. The Q148K, E138A/Q148K, and E138A/G140A/Q148K resulted in a substantial fold-shift (46- to 508-fold) in raltegravir IC50. Another study identified the E92Q, G140S Q148H, N155H, and E157Q as key mutations (CitationMalet et al 2008). Furthermore, virus carrying the Q148K as well as the E138A, G140A, and V54I changes was highly cross-resistant to elvitegravir and had a significantly reduced sensitivity against other integrase inhibitors in development (CitationGoethals et al 2008). In another study, mutated integrase enzymes displayed an impairment of enzyme function (CitationMalet et al 2008), indicating reduced replicative fitness. The clinical relevance of this observation, however, remains to be demonstrated. In vitro and in vivo data correspond very well, as the majority of patients in the BENCHMRK trials with virological failure on raltegravir, in whom genotyping was performed, exhibited mutations in integrase known to be associated with raltegravir resistance (CitationCooper et al 2008). This reflects the strong selective pressure of the drug and its barrier to resistance, which appears lower than for ritonavir-boosted protease inhibitors, but somewhat higher than for first-generation NNRTIs.
Subjects failing on raltegravir in clinical trials exhibited two pathways to resistance. The virus appears to reach a moderate level of resistance (10- to 25-fold) by acquiring a change at position Q148 (H,K, or R) or the N155H. Other mutations, which by themselves have only limited impact on resistance (L74M, E92Q, T97A, E138K, G140S, V151I, G163G/R, and D232D/N), appear to decrease susceptibility further in conjunction with either of the two (CitationHazuda et al 2007).
The observation of at least partial in vitro cross-resistance between raltegravir and elvitegravir, another integrase inhibitor in phase III studies, was substantiated by clinical case studies. Two participants of an elvitegravir phase II trial switched to raltegravir upon failure to the study drug. No virological response was noted within one week. One subject harboured virus carrying the Q148R key raltegravir mutation in conjunction with others; the integrase sequence of the other one was not amplifiable (CitationDeJesus et al 2007).
With respect to activity against different HIV clades, a recent study found no impact of clade-specific integrase polymorphisms on raltegravir sensitivity (Citationvan Baelen et al 2008).
Therapeutic potential
Raltegravir as the first of its class has demonstrated high antiviral activity in a series of clinical trials, which have led to its approval for treatment-experienced subjects. Its optimal use in this setting is becoming increasingly clear: a subgroup analysis of the pooled BENCHMRK study data shows that response rates at week 48 were highest when the regimen comprised two other components for which sensitivity was predicted (CitationCooper et al 2008). Of note, response rates were not higher when more than two active components were combined with raltegravir. Therefore, three active components (which include raltegravir) achieve high response rates in treatment-experienced patients, which in fact are similar to those in previously untreated subjects.
Raltegravir also represents an important option for patients who do not tolerate an active agent in their combination, while exhibiting optimal response. In a series of 35 subjects responding to a regimen comprising enfuvirtide, enfuvirtide was replaced by raltegravir, while the rest of the regimen was left unchanged. Virological suppression was sustained until a median of 7 months of follow-up (CitationHarris et al 2008b). The strategy of replacing active components by raltegravir is currently being investigated in several trials.
A large phase III trial currently addresses the activity of raltegravir in first-line therapy. It is compared with the current standard-of-care efavirenz, both in combination with tenofovir plus emtricitabine (trial 021). Results are expected for the end of 2008.
It is unclear if the more rapid increase in response rates in the raltegravir arms in study 004 (CitationMurray et al 2007) in comparison with the efavirenz arm indicates higher antiviral potency. Mathematical modelling actually suggests that calculations of viral clearance rates during early therapy depend on the mode of action of the drugs employed, ie on the stage of the viral life-cycle at which they interfere, and that this appears to make a difference only early in treatment (CitationSedaghat et al 2008). This actually challenges previous pathophysiological interpretations of studies on viral clearance rates. Should recently infected cells with unintegrated proviral DNA, which proceed to integration and virus production in the early phase of HAART, contribute a lot to plasma viral RNA in this phase, raltegravir might help a lower number of infected cells to be reached before later phases of viral decay are entered. That might not make a difference for viral RNA suppression in these later phases, but may reduce the viral depository of quasi-species and the basis for re-activation of virus from the latent pool. This view, however, is speculative and remains hypothetical at present.
Another very positive feature of the drug is its independence from pharmacological “boosting” with ritonavir. This makes it a very attractive option for patients with gastrointestinal side effects on ritonavir-boosted PIs and/or intolerance to efavirenz or nevirapine, as well as for patients in whom lipid abnormalities cannot be treated effectively during PI therapy. Several switch studies investigating improvement of neuropsychiatric toxicity of efavirenz or lipid and gastrointestinal toxicity on a ritonavir-boosted PI are ongoing.
Of the few relevant pharmacokinetic interactions, the inhibition of the metabolising enzyme UGT1A1 by atazanavir might even be exploited therapeutically. The concept of “boosting” raltegravir levels in order to circumvent twice daily dosing as one of the few disadvantageous features of the drug will be investigated in a clinical trial.
The recent approval of raltegravir and other compounds active against triple-class resistant virus has been a breakthrough for patients with uncontrolled replication due to resistance. The term “paradigm shift” is frequently employed for this change in therapeutic perspectives. The rapid appearance of resistance with the associated mutational changes in case of failure on raltegravir, however, demonstrates that suboptimal use of this novel compound (ie, in the context of too few active drugs) could ultimately lead to a similar rescue therapy situation. The BENCHMRK studies suggest that two other active drugs provide the necessary background for achieving the optimal effect of raltegravir.
For HIV-2, the number of treatment options unfortunately is far more limited than for HIV-1. In contrast to NNRTIs and some PIs, raltegravir appears to have comparable activity against HIV-2. Controlled clinical data, however, are still lacking. The same applies to pediatric application.
Ongoing or planned clinical trials
Clinical trials are underway to investigate a switch from enfuvirtide or a ritonavir-boosted protease inhibitor, a replacement of nucleoside analogues in a PI-based combination, alternative first-line combinations of lopinavir/ritonavir and raltegravir or raltegravir and tenofovir/emtricitabine, as well as abacavir/lamivudine and raltegravir.
The unique effect of raltegravir on viral integration prompted clinical studies of its impact on viral latency by adding raltegravir to a fully suppressive combination or combining it with the histone deacetylase inhibitor valproic acid. Furthermore, it is being investigated in combination with tenofovir/emtricitabine for non-occupational post-exposure prophylaxis. Other studies address children, African patients and acutely infected subjects, as well as a multitude of pharmacokinetic questions.
Raltegravir provides an important improvement of the therapeutic armamentarium against HIV-1 and probably HIV-2. Future studies will show if its high antiviral activity and its effect on the establishment of viral latency represent the desired step forward towards life-long control of HIV.
Disclosures
The author has served as a scientific advisor to Merck.
References
- AndersonMSWenningLAMoreauA2007Effect of raltegravir (RAL) on the pharmacokinetics (PK) of oral contraceptives [abstract]47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC)17–20 September 2007Chicago, IL, USA abstract A-1425. 2007
- CooperDAGatellJRockstrohJ2007Results of BENCHMRK-1, a Phase III study evaluating the efficacy and safety of MK-0518, a Novel HIV-1 integrase inhibitor, in patients with triple-class resistant virus [abstract]14th Conf Retro Opportun InfectLos Angeles abstract 105aLB
- CooperDASteigbigelRTGatellJM2008Subgroup and resistance analyses of raltegravir for resistant HIV-1 infectionN Engl J Med3593556518650513
- CraigieR2001HIV integrase, a brief overview from chemistry to therapeuticsJ Biol Chem276232131611346660
- DeJesusECohenCElionR2007First report of raltegravir (RAL, MK-0518) use after virologic rebound on elvitegravir (EVT, GS 9137) [abstract]4th IAS Conference on HIV Pathogenesis, Treatment and PreventionSydney, Australia22–25, July 2007 Abstract TUPEB0322007
- EmbreyMWWaiJSFunkTW2005A series of 5-(5,6)-dihydrouracil substituted 8-hydroxy-[1,6]naphthyridine-7-carboxylic acid 4-fluoro-benzylamide inhibitors of HIV-1 integrase and viral replication in cellsBioorg Med Chem Lett154550416102965
- EspesethASFelockPWolfeA2000HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integraseProc Natl Acad Sci U S A9711244911016953
- GoethalsOClaytonRWagemansE2008Resistance mutations in HIV-1 integrase selected with raltegravir or elvitegravir confer reduced susceptibility to a diverse panel of integrase inhibitors [abstract]XVII International HIV Drug Resistance WorkshopJune 10–14, 2008Sitges Spain abstract no. 2008
- GrinsztejnBNguyenBYKatlamaC2007Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II ran-domised controlled trialLancet3691261917434401
- HarrisMLarsenGMontanerJS2008aExacerbation of depression associated with starting raltegravir: a report of four casesAIDS221890218753871
- HarrisMLarsenGMontanerJS2008bOutcomes of multidrug-resistant patients switched from enfuvirtide to raltegravir within a virologically suppressive regimenAids221224618525270
- HazudaDBlauCUFelockP1999Isolation and characterization of novel human immunodeficiency virus integrase inhibitors from fungal metabolitesAntivir Chem Chemother10637010335400
- HazudaDMillerMDNguyenBY2007Resistance to the HIV-integrase inhibitor raltegravir: analysis of Protocol 005, a Phase 2 study in patients with triple-class resistant HIV-1 infection [abstract]Antivir Ther abstract no. 8[12]S10
- HazudaDJAnthonyNJGomezRP2004A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integraseProc Natl Acad Sci U S A10111233815277684
- HazudaDJFelockPWitmerM2000Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cellsScience2876465010649997
- IwamotoMKassahunKTroyerMD2008aLack of a pharmacokinetic effect of raltegravir on midazolam: in vitro/in vivo correlationJ Clin Pharmacol482091418077730
- IwamotoMWenningLALiouSY2006aRifampin (RIF) modestly reduces plasma levels of MK-0518 [abstract]8th international congress on drug therapy in HIV infectionGlasgow, United KingdomNovember 12–16, 2006 abstract P299
- IwamotoMWenningLAPetryAS2006bMinimal effect of ritonavir (RTV) and efavirenz (EFV) on the pharmacokinetics (PK) of MK-0518 [abstract]46th Interscience Conference on Antimicrobial Agents and ChemotherapySeptember 27–30, 2006San Francisco, USA abstract A-373
- IwamotoMWenningLAPetryAS2008bSafety, tolerability, and pharmacokinetics of raltegravir after single and multiple doses in healthy subjectsClin Pharmacol Ther832939917713476
- KassahunKMcIntoshICuiD2007Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzymeDrug Metab Dispos3516576317591678
- KassahunKMcIntoshIHreniukDAbsorption, metabolism and excretion of MK-0518, a potent HIV-1 integrase inhibitor, in healthy male volunteers [abstract]46th Interscience Conference on 1 Agents and ChemotherapySeptember 27–30, 2006San Francisco, USA abstract A-372
- LafeminaRLSchneiderCLRobbinsHL1992Requirement of active human immunodeficiency virus type 1 integrase enzyme for productive infection of human T-lymphoid cellsJ Virol667414191433523
- MaletIDelelisOValantinMA2008Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitroAntimicrob Agents Chemother521351818227187
- MarkowitzMNguyenBYGotuzzoE2008Sustained antiretroviral efficacy of raltegravir as part of combination ART in treatment-naive HIV-1 infected patients: 96-week data [abstract]17th International AIDS ConferenceAugust 3–8, 2008Mexico City, Mexico Abstract TUAB0102
- MarkowitzMNguyenBYGotuzzoE2007Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled studyJ Acquir Immune Defic Syndr461253317721395
- Merck and Co., W. S. N. O. UIsentress™ FDA Briefing DocumentFood And Drug Administration Homepage2007 www.fda.gov/ohmrs/dockets/ac/07/briefing/2007-4314b1-01-Merck.pdf
- Merck and Co., W. S. N. O. UIsentress™ Prescribing InformationUS Food and Drug Administration Homepage2008 www.fda.gov/cder/foi/label/2007/0221451b1.pdf
- MistryGWenningLPetryA2007Atazanavir modestly increases plasma leves of MK-0518 [abstract]4th IAS Conference on HIV Pathogenesis, Treatment and PreventionSydney, Australia22–25, July 2007 abstract 1985
- MurrayJMEmerySKelleherAD2007Antiretroviral therapy with the integrase inhibitor raltegravir alters decay kinetics of HIV, significantly reducing the second phaseAids2123152118090280
- PaisGCZhangXMarchandC2002Structure activity of 3-aryl-1,3-diketo-containing compounds as HIV-1 integrase inhibitorsJ Med Chem4531849412109903
- RoquebertBDamondFCollinGon behalf of the French ANRS HIV- 2008HIV-2 integrase gene polymorphism and phenotypic susceptibility of HIV-2 clinical isolates to the integrase inhibitors raltegravir and elvite-gravir in vitroJ Antimicrob Chemother10.1093/jac/dkn335
- SedaghatARDinosoJBShenL2008Decay dynamics of HIV-1 depend on the inhibited stages of the viral life cycleProc Natl Acad Sci U S A1054832718362342
- SteigbigelRKumarPEronJ2007Results of BENCHMRK-2, a Phase III study evaluating the efficacy and safety of MK-0518, a novel HIV-1 integrase inhibitor, in patients with triple-class resistant virus [abstract]14th Conf Retro Opportun InfectLos Angeles abstract 105bLB
- van BaelenKvan EygenVRondelezE2008Clade-specific HIV-1 integrase polymorphisms do not reduce raltegravir and elvitegravir phenotypic susceptibilityAids Epub ahead of print
- WenningLAFriedmanEKostJT2006aLack of a significant drug interaction between MK-0518 and tenofovir disoproxil fumarate (TDF) [abstract]46th Interscience Conference on Antimicrobial Agents and ChemotherapySeptember 27–30, 2006San Francisco, USA abstract A-375
- WenningLAHanleyHStoneJ2006bEffect of tipranavir + ritonavir (TPV + RTV) on pharmacokinetics of MK-0518 [abstract]46th Interscience Conference on Antimicrobial Agents and ChemotherapySeptember 27–30, 2006San Francisco, USA abstract A-3742006b