Abstract
Hematopoiesis is a regulated multistep process, whereby transcriptional and epigenetic events contribute to progenitor fate determination. miRNAs have emerged as key players in hematopoietic cell development, differentiation and malignant transformation. From embryonic development through to adult life, miRNAs cooperate with, or are regulated, by epigenetic factors. Moreover, recent findings suggest that they contribute to chromatin structural modification, and the functional relevance of this ‘epigenetic–miRNA axis‘ will be discussed in this article. Finally, emerging evidence has highlighted that miRNAs have functional control in human hematopoietic cells, involving targeted recruitment of epigenetic complexes to evolutionarily conserved complementary genomic loci. We propose the existence of epigenetic–miRNA loops that are able to organize the whole gene expression profile in hematopoietic cells.
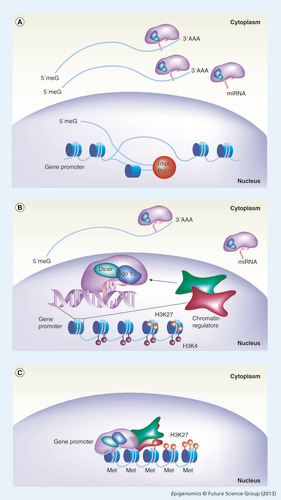
(A) Classical miRNA action on the regulation of gene expression. The miRNA (pink line) embedded into the RNA-induced silencing complex (RISC; purple). Mature miRNA guides the RISC complex to the 3´ untranslated region of a complementary capped (5‘meG) and polyadenilated (3‘AAA) mRNA (blue line), and is exported to the cytoplasm after its transcription into the nucleus by RNA Pol II (red sphere). (B) The action of miRNA on mRNA associates with a new mechanism whereby miRNA and RISC components Ago1/2 (blue) and Dicer (light green), once imported into the nucleus, mediate chromatin remodeling factor (red and dark green squares) recruitment on DNA. (B) A detailed magnified view of the proposed recruitment mechanism. Here a feedback loop is proposed where the same gene is regulated at both (A & B) post-transcriptional and (B) transcriptional levels. A bivalent domain is exemplified by the coexistence of both activating (H3K4me3; dark red circles) and inhibitory (H3K27me3; light red circles) histone marks. (C) This double-step regulation ensures long-term efficient silencing, by extensive chromatin remodeling with resolution of the bivalent domain into heterochromatin via extensive H3K27me3 and Met.
Met: DNA methylation; RNA Pol II: RNA polymerase II.
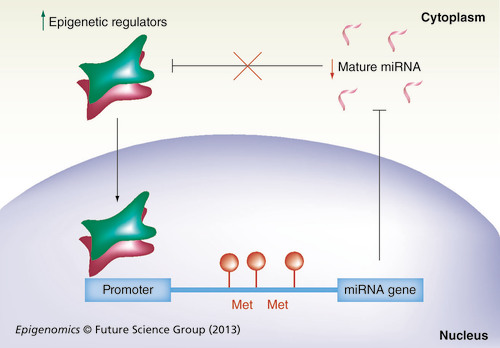
When the miRNA gene promoter is hypermethylated or modified in its chromatin structure (red circles) by the binding of epigenetic regulators (red and green squares), mature miRNA transcription is impaired (black line). Therefore, cytosolic miRNA levels are decreased (red arrow). Mature miRNA impairs the expression of epigenetic regulators (black line). However, decreased miRNA levels allow (red cross on black line) the increased expression of epigenetic regulators (green arrow) that bind to the miRNA gene promoter in the nucleus (black arrow).
Met: DNA methylation.
Epigenetic mechanisms provide a plastic system for regulating gene expression, and are extensively implicated in hematopoiesis and leukemia development and progression Citation[1]. In this framework, a new class of regulators, the miRNAs, have recently emerged as players in the extensive changes responsible for hematopoietic cell development, differentiation and malignant transformation. This class of noncoding RNA, classically linked to transcription factor networks Citation[2,3], is now being integrated into epigenetics Citation[4–6]. miRNAs control gene expression, cooperate with or are regulated by epigenetic factors, including DNA methyltransferases (DNMTs), histone deacetylases (HDACs) and polycomb group (PcG) family proteins Citation[4,5,7,8]. Therefore, miRNAs take part in chromatin structural modifications in a direct or indirect way. miRNAs can affect the expression of epigenetic factors in many systems, providing evidence for the role of these molecules as indirect players in the epigenetic dynamism of the cells.
We will describe the complicated networking of miRNAs and epigenetic pathways, proposing the existence of epigenetic–miRNA loops that are able to organize the whole gene expression profile in every cell. We will then highlight disruptions in this regulatory loop, and how physiological functions are likely to be interfered with, possibly contributing to the establishment of disease Citation[6]. Finally, we will discuss emerging evidence suggesting that in human hematopoietic cells miRNAs functionally control gene transcription by the targeted recruitment of epigenetic complexes to evolutionarily-conserved complementary loci in the genome.
The aim of this review is to encompass the most recent findings underlying the functional relevance of the epigenetic–miRNA axis in normal and malignant hematopoiesis, highlighting the synergistic action of ‘classical‘ epigenetic machineries and miRNAs.
Hematopoiesis
Hematopoiesis is a dynamic and highly complex developmental process that gives rise to a multitude of cell types circulating in the blood of multicellular organisms. Blood cells provide tissues with oxygen and guard against infection. They also prevent bleeding by clotting and mediate inflammatory reactions. As the hematopoietic system plays a central role in human physiology and diseases, such as anemia, infection, bleeding disorders, cancer and autoimmunity, it has been intensely studied for more than a century.
The wide variety of differentiated elements present in animal blood is generated from one unique hematopoietic stem cell (HSC). The hematopoietic system is organized in a hierarchical manner and includes clear-cut nodal progenitor cells that have intermediate levels of lineage potency. The design of this network apparently serves to provide stable substations, perpetually programmed to supply new downstream lineage cells whenever needed. In this scenario, the orchestration of epigenetic events tightly regulates the system Citation[9] and, at the same time, drives the transcriptional activity and expression of genes underlying lineage commitment, whose deregulation can result in cancer onset and progression Citation[10].
The differentiation/maturation of HSCs leads to sequential developmentally restricted states. Initially, they give rise to multipotent progenitors (MPPs), which have lost the ability to self-renew, but still maintain a full-lineage differentiation potential. MPPs can differentiate into lymphoid-primed MPPs, common lymphoid progenitors and common myeloid progenitors. Common lymphoid progenitors give rise to lymphocytes and natural killer cells, whereas common myeloid progenitors differentiate into granulocyte–monocyte and megakaryocyte–erythrocyte progenitors Citation[10,11]. From embryonic development through to adult life, hematopoiesis is tightly regulated by transcriptional and epigenetic events, which prime and commit HSCs and progenitors to their definitive destiny Citation[9,10].
Epigenetics
In the hematopoietic context, epigenetic regulation is required not only for development, but also for tissue homeostasis via the self-renewal and differentiation of somatic stem cells. Accumulating evidence suggests that epigenetic regulators play critical roles in HSCs multipotency and self-renewal ability. Therefore, these properties must be precisely balanced to preserve the multipotent cell pool throughout life Citation[12]. Using microarray technology, the genome-wide DNA methylation pattern has been studied throughout the different stages of hematopoietic differentiation. Promoters of different genes, initially methylated in HSCs or MPPs, are found to be progressively demethylated in a tissue- or lineage-specific way, reflecting a progressive activation of lineage-specific genes. Among these genes, we can recognize those usually considered to be ‘signatures‘ of each hematopoietic cell type, such as myeloperoxidase in granulocyte–monocyte progenitors, globin genes in erythroid precursors, FoxP3 transcription factor in T-regulatory cells differentiation, G-CSF receptor in granulocyte precursors and GP6 during megakaryocyte differentiation Citation[13]. More recently, a potential role for promoter DNA demethylation was investigated by combining genome-wide promoter methylation with gene expression approaches, comparing a variety of somatic tissues. Interestingly, this study revealed that promoter demethylation-associated gene regulation is more frequent in the hematopoietic system than in nonhematopoietic tissues (463 demethylated genes in blood vs an average of 241 demethylated genes in nonhematopoietic samples), supporting the overall strong influence of epigenetic signals in driving the tightly time- and step-controlled cell fate determination during hematopoiesis Citation[13]. The demethylation process is quite complex and involves proteins, such as the TET family of proteins, which are frequently mutated in neoplastic hematopoietic diseases Citation[14].
In parallel, during late commitment of HSCs, other subsets of genes are affected by de novo methylation, allowing a definitive switch to a unique developmental/differentiation program. Similarly, the global methylation status of HSCs undergoes large variations during aging, reflecting the reduced self-renewal and differentiation capability of stem cells Citation[15]. Moreover, de novo methylation has been observed for a subset of genes associated with PcG complexes during aging and tumorigenesis. These events are usually associated with gene silencing and could thus contribute to the reduced phenotypic plasticity and self-renewal of aged stem cells Citation[16]. Besides DNA methylation status, histone tail modifications play a fundamental role in the regulation of chromatin assembly.
Although acetylation of histone 3 (H3) lysine 9 and 14 always correlates with accessible euchromatin and therefore promotes gene transcription, histone methylation correlates with both permissive and nonpermissive chromatin states, and consequently with either transcriptional activation or repression. Indeed, coexistence of H3K27me3 and activating H3K4me3 marks the same DNA region defining the so-called ‘bivalent domains‘, which are usually located at regulatory regions of gene promoters. In the hematopoietic context, these chromatin modifications reflect parallel changes in gene expression and transcriptional competence of developmentally regulated genes Citation[10,17,18]. This bivalency allows cell commitment to be postponed and, contemporarily, progenitor cells to be ‘poised‘ within alternative lineage fates; chromatin resolution into either the active or repressed state represents a definitive cell fate decision Citation[17,18]. Evolutionarily conserved PcGs and trithorax protein groups are responsible for the trimethylation of H3K27 and H3K4, respectively and preserve the epigenetic memory of each cell type. This ensures the correct execution of key developmental programs, including self-renewal and commitment of hematopoietic cells Citation[10,19,20].
A thorough coverage of the abovementioned epigenetic changes can be found in outstanding papers recently reviewed by Dawson and Kouzarides Citation[21].
miRNA biogenesis & function
In 1993, small regulatory RNAs were recognized in Caenorhabditis elegansCitation[22], and later were discovered in plants and mammals Citation[23–25], these RNAs were designated ‘miRNA‘ Citation[26]. The miRNA world has rapidly expanded and now counts 2042 mature human entries registered in the miRBase database Citation[27]. miRNAs are short molecules, 19–25 nucleotides in length, and belong to a class of conserved noncoding transcripts that finely regulate gene expression by an RNA-mediated gene-silencing mechanism, involving translational repression and mRNA degradation Citation[28]. In animals, target recognition occurs mainly through incomplete base-pairing of the miRNA and the target gene 3´ untranslated region. While complementarity at the 5´ end of the miRNA, designated as the ‘seed‘ region Citation[29], appears to be essential for target recognition, a certain flexibility is permitted at the 3´ end for successful binding to the target mRNA Citation[29,30]. These small regulatory RNAs function as part of the effector RNA-induced silencing complex, together with members of the Argonaute (Ago) family of proteins Citation[31].
miRNA genes are found in intergenic regions as well as in introns of protein-coding genes. When located within introns, in some cases, their expression is coregulated with their host genes Citation[32]. The majority of mammalian miRNA genes are transcribed by RNA polymerase II as long 5´-capped and 3´-polyadenilated precursors. Primary miRNA transcripts can be hundreds of bases to several kilobases in length and might contain one or several miRNAs Citation[33]. The biogenesis of miRNAs involves two processing steps whereby the long primary miRNA is first cotranscriptionally cleaved by the nuclear ‘microprocessor‘ complex Citation[34,35], containing Drosha, an RNase III-like enzyme, and its cofactor DGCR8 Citation[36,37]. Then a short stem-looped precursor, the pre-miRNA, is released and exported to the cytoplasm where it is further processed by a second complex containing the RNase III-like enzyme, Dicer. This mechanism liberates the mature miRNA duplexes for loading into the RNA-induced silencing complex Citation[38]. The relatively simple mechanism for target recognition, together with the small portion of the miRNA sequence required for efficient binding to the cognate target mRNA, provides a combinatorial system for gene expression regulation Citation[3].
miRNA-mediated regulation has indeed been described as a central hub in cellular and differentiation pathways and it is extensively involved in hematopoiesis and hematological malignancies Citation[39,40]. The homozygous Dicer deletion in mice is incompatible with a functional HSC state and leads to a marked defect in hematopoietic progenitor competitive repopulation assays Citation[41]. Moreover, haploinsufficiency of Dicer in B cells failed to promote B-cell malignancy or accelerate Myc-induced B-cell lymphomagenesis in mice, thus suggesting a role for Dicer in B-cell lymphoma development and survival Citation[42]. The deletion of Ago2 also leads to severe defects in erythroid and B-cell development Citation[43].
The role of miRNAs in hematopoiesis was largely determined from profiling studies that revealed how several miRNAs are expressed at various stages of hematopoietic development and are specific for each lineage Citation[44]. A recent report encompasses the expression of known hematopoietic miRNAs throughout the mouse hematopoietic system, providing a comprehensive atlas of miRNA expression, from early stem cells through the differentiation stages to mature blood elements Citation[45].
Many miRNAs are also involved in malignant transformation, and sometimes prove to be good diagnostic markers for the different leukemia subtypes. Since comprehensive reviews listing miRNAs involved in myeloid leukemias and myelodysplastic syndromes (MDS) are now available Citation[46,47], we focused on miRNAs connecting the epigenetic myelodysplastic machinery to these diseases. Indeed, miRNAs affect the expression of epigenetic factors in many systems, including hematopoiesis, providing evidence for their role as indirect players in the epigenetic dynamism of the cell.
Epigenetic–miRNA loops in normal & malignant hematopoiesis
Epigenetic regulation of miRNA genes
Epigenetic regulation of miRNA gene transcription appears to be crucial as miRNAs are increasingly defined as critical molecules in normal and aberrant development, cell proliferation and commitment processes.
The primary regulatory mechanism for miRNA expression is probably their transcriptional control Citation[6]. The initial evidence showing that miRNAs undergo epigenetic regulation was obtained in a variety of cancer models. In addition, either pharmacologic or genetic approaches unmasked hypermethylated miRNA genes Citation[48,49]. Interestingly, an extensive analysis of genomic sequences of miRNAs have shown that they are frequently associated with CpG islands Citation[50]. The expression of miRNAs is affected by promoter hypermethylation or global hypomethylation Citation[51].
Although the network complexity underlying aberrant miRNA genes epigenetic regulation in pathophysiological conditions is yet to be determined, single factors, such as cancer-specific fusion proteins or cytokines, have been implicated in driving epigenetic silencing of miRNA genes Citation[52,53]. For instance, the AML1–ETO oncoprotein, resulting from the t(8;21) translocation of AML binds to the miR-223 gene promoter Citation[52], where it recruits an epigenetic silencing complex consisting of HDACs, DNMTs, and methyl-CpG-binding proteins. Through CpG methylation, this complex inhibits pri-miR-223 expression and, in turn, sustains the block in myeloid differentiation underlying the pathogenesis of this acute myeloid leukemia (AML) subtype Citation[52]. Similarly, AML1–ETO triggers the heterochromatic silencing of miR-193a by binding to AML1-binding sites and recruiting chromatin-remodeling enzymes (HDACs and DNMTs) on its gene promoter. Suppression of miR-193a expands the oncogenic activity of the fusion protein AML1–ETO Citation[54].
In a recent report it was shown that in AMLs presenting the t(8;16) translocation, which fuses genes encoding two histone acetyltransferases, MYST3 (or MOZ) on chromosome 8 and CREBBP (or CBP) on chromosome 16, 90 miRNAs are downregulated and among those 29 present CpG islands in their promoter regions. Treatment of myeloid cell lines and primary patient cells with DNMT and HDAC inhibitors (5-Aza-deoxycytidine and Trichostatin A, respectively) rescued the expression of 27 out of 29 of these miRNAs suggesting that their suppression is indeed mediated by epigenetic silencing mechanisms Citation[55]. In addition, an altered regulation of miR-203 was described in T cell lymphoma mouse model and human cells, where its promoter was found hypermethylated Citation[56]. miR-203 also controls the activation of ABL1, a classic oncogene that is extensively characterized in hematopoietic malignancies. miR-203 is epigenetically silenced in chronic myelogenous leukemias carrying the t(9;22) Philadelphia (Ph) translocation; this enhances the t(9;22)-fusion product BCR–ABL1 activation, thus elevating the growth rate of chronic myeloid leukemia cells Citation[56].
Interestingly, in hematopoietic malignancies, 12% of miRNAs are located in fragile genomic regions that only encompass 0.2% of the whole genome Citation[4], and one of these regions hosts miR-203 Citation[4]. miR-203 hypermethylation was also observed in cell lines and primary cells from Ph chromosome-negative myeloproliferative neoplasms (Ph-MPNs), although there was no correlation between miRNA methylation and clinical demographic data or outcome Citation[57]. Moreover, in a large screening (150 primary samples) for methylated miRNAs, miR-203 was found to be methylated in 5% of acute lymphoblastic leukemia (ALL), in 42% of chronic lymphocytic leukemia and 38% of non-Hodgkin lymphoma Citation[58]. Thus, the oncogenic activity of this particular miRNA spreads across different leukemia subtypes (lymphoid and myeloid), but the mechanism leading to miR-203 promoter hypermethylation has not currently been determined.
miR-29b was reported to be a central hub in the gain-of-function mutations of KIT (product of oncogene c-kit) characterizing a specific subset of AMLs (i.e., core binding factor AMLs). In these AMLs, miR-29b-dependent KIT overexpression contributes to leukemia growth. miR-29b participates in a Sp1/NF-κB/HDAC/regulatory network, which mediates c-kit overexpression and that can be successfully targeted by pharmacological disruption of the Sp1/NF-κB/HDAC complex with HDAC inhibitors Citation[59]. In addition, miR-193a was shown to be epigenetically silenced in primary AML blasts and AML-derived cell lines, by a promoter methylation at specific CpG islands. This contributes to c-kit overexpression, with c-kit being one of miR-193a validated targets Citation[60].
One of the features of AML is also the overexpression of CREB factor, which is targeted by miR-34b Citation[61]. This miRNA was shown to be repressed in primary AML samples by promoter hypermethylation, occurring at leukemia onset after a MDS Citation[61]. These results demonstrate that epigenetic deregulation of miRNA genes is relevant in oncogene regulation and may represent a therapeutic target in specific subsets of human leukemias.
Specific miRNAs are downregulated in MDS, a group of hematopoietic malignancies characterized by ineffective hematopoiesis. Among these, miR-124 expression level is inversely correlated with the degree of its promoter methylation Citation[46]. This observation was then supported by data from a mouse model where both miR-124 methylation and silencing trigger an MDS-like disease Citation[62].
Downregulated miRNAs in MDS (e.g., miR-140, -378 and -632) are predominantly intragenic and show a similar expression pattern to their host genes, suggesting mechanisms of coregulation during myeloid maturation. Indeed, the increased methylation status of shared promoters induces the downregulation of several miRNAs and their host genes. Thus, epigenetic regulation in MDS involves both protein-coding genes and miRNAs, unifying the research fields of ‘miRNAs in MDS‘ and ‘epigenetic regulation in MDS‘ Citation[63].
Notably, miR-124 expression was to be found downregulated in ALL patients‘ samples (59% of 353 analyzed samples), where its promoter was hypermethylated Citation[64]. Moreover, functional studies showed upregulation of miR-124 after treatment of ALL cell lines with 5-aza-cytitdine, suggesting a role for CpG methylation in transcriptional silencing of this miRNA in ALL pathogenesis Citation[65].
The epigenetic regulation of miRNAs was extensively investigated in aberrant hematopoiesis, which is a new putative target for therapeutic intervention. Moreover, some studies address its physiological importance in HSCs and differentiating precursors. It has been reported that, upon macrophage differentiation of myeloid progenitors, the transcription factor PU.1, which is essential for myeloid and lymphoid differentiation, induces the expression of Egr2 that, in turn, represses miR-17-92 polycistron. miR-17-92 is a cluster encoding six miRNAs, miR-17, -18a, -19a, -20a, -19b-1 and -92a; the repression of this miRNA cluster is mediated by the histone demethylase Jarid1b, leading to H3K4 demethylation within the CpG island at the miR-17-92 promoter. The reduced expression of the miR-17-92 cluster is a conditio sine qua non for macrophage differentiation Citation[66].
Moreover, several miRNAs are epigenetically repressed by HDACs and G9 histone methyltransferase Citation[67], with a mechanism mediated by zinc-finger repressor Gfi-1, a key intrinsic regulator of HSC self-renewal Citation[68], and its interacting factor PRDM5, a zinc finger protein belonging to the PR (PRD1-BF1 and RIZ homology) domain-containing tumor suppressor protein family, acting in different pathways during hematopoiesis and leukemia Citation[68,69].
miRNA-mediated regulation of epigenetic factors
The epigenetic signature of stem cells has been recently defined Citation[70] and miRNA-mediated regulation has been included in this picture Citation[71–73]. The epigenetic role of miRNAs was globally evaluated in two recent reports where the authors investigated the role for mammalian Dicer and Dicer-dependent small RNAs, particularly miRNAs, in mouse Dicer1-null embryonic stem cells (ESCs). Their findings showed that Dicer1 abrogation results in decreased expression of DNMTs Citation[72,73]. Benetti and colleagues reported that Dicer ablation impairs telomere homeostasis Citation[72]. Thus, miRNAs activity extends to chromosome structure and telomere length maintenance. They observed that in Dicer1-null ESC the decrease in Dnmt levels paralleled the significant increase of Rbl2, a known transcriptional inhibitor of Dnmt expression, and that the substantial downregulation of miR-290 cluster efficiently targets the Rbl2. The authors suggested a mechanism whereby the conserved mammalian miR-290 cluster regulates Rbl2 at the post-transcriptional level, leading to Dnmt3a and Dnmt3b transcriptional repression. This also induces DNA methylation defects within subtelomeric regions, where telomere recombination is increased and aberrant telomere elongation is detectable Citation[72]. These results identify a novel mechanism by which miR-290 regulates Rbl2-dependent Dnmt expression, thus affecting telomere-length homeostasis. Sinkkonen and colleagues also demonstrated that defective DNA methylation can be rescued by ectopic expression of de novo Dnmts or by transfection of miRNAs belonging to the miR-290 cluster into Dicer-/- ESCs, further supporting the notion that miRNAs control de novo DNA methylation in ESCs Citation[73].
The general miRNA-mediated mechanisms described for the control of DNMTs‘ function in pluripotent ESCs could possibly occur in other cellular contexts, such as in HSCs and progenitor cells, where the modulation of DNA methylation during lineage-specific differentiation plays a crucial role Citation[74]. Indeed, So and colleagues reported that inhibition of DNMTs‘ activity or expression in human cord blood-derived multipotent stem cells by 5-azacytidine or RNAis, respectively, led to the induction of cellular senescence, cell cycle arrest and decreased multipotency, accompanied by a decrease in the expression of PcGs, such as EZH2 histone methyltransferase and PcG protein BMI1 Citation[71]. The decrease of PcGs is dependent on miR-214 and -200c, which are known to target EZH2 in skeletal muscle and ESCs, and BMI1 in breast cancer stem cells, respectively Citation[75,76]. Although the same analysis has not currently been performed in the HSC compartment, a central role for both EZH2 and BMI1 in these cells has been extensively described Citation[77–79].
Throughout hematopoietic cell differentiation each transition requires the action of both epigenetic and transcription factors Citation[80,81] as well as miRNAs Citation[82,83]. Terminal erythroid cells differentiation involves a progressive heterochromatin formation and chromatin condensation, leading to enucleation in mammals Citation[74]. Chromatin condensation preceding nucleus expulsion from orthochromatophilic erythroblasts is accompanied by nucleosomal histone modifications Citation[84]. The requirement of HDACs activity in erythroblast chromatin condensation is suggested by the consequences of their inhibition. Treatment of mouse erythroblasts with HDAC inhibitor Trichostatin A, maintains histone acetylation and inhibits chromatin condensation and nuclear extrusion Citation[85]. Recently, miRNAs have been thought to be indirectly involved in the late phases of erythroid differentiation. Downregulation of the developmentally regulated miR-191, identified by RNA deep sequencing performed in terminally differentiating CFU-E erythroid progenitors, is required for erythrocyte precursor chromatin condensation, global gene expression shutdown and enucleation processes Citation[86]. In these events, the involvement of miR-191 is indirect; it targets the erythroid-enriched gene Riok3, which belongs to the RIO family of atypical protein kinases, and Mxi1, a well-known c-Myc antagonist. Riok3 and Mxi1 function in a complex mechanism eventually leading to the downregulation of the histone acetyltransferase Gcn5, which is necessary for proper erythrocyte maturation. Either knockdown of Riok3 and Mxi1, or overexpression of miR-191, blocks Gcn5 downregulation and impairs enucleation Citation[86]. Thus, in mammalian cells, miR-191 repression is essential for erythroid chromatin condensation and enucleation, by allowing upregulation of Riok3, Mxi1 and downregulation of Gcn5.
With regards to physiological differentiation patterns, accumulating data revealed how a combination of genetic and epigenetic abnormalities triggers cancer evolution. The most representative aberrations are CpG island hypermethylation at promoter genes and deacetylation or methylation of histone proteins. The action of miRNAs on the epigenetic network complicates this scenario, as many miRNAs have been found to be deregulated in leukemia Citation[87]. In fact, miR-29b, involved in lung cancer epigenetic abnormalities Citation[88], participates in the global hypomethylation observed in AML Citation[7]. miR-29b hypomethylating role was tested during myeloid leukemogenesis; its expression promotes DNA hypomethylation through the direct targeting of DNMT-3A and -3B. miR-29b also decreased DNMT1 expression indirectly, via downregulation of Sp1, a known DNMT1 transactivating factor. This epigenetic activity of miR-29b was also confirmed in primary leukemic blasts, thus providing the basis for a future use of synthetic miR-29b in a miRNA-based intervention for AML therapy Citation[7]. To date, miR-29b is the only leukemia-related miRNA linked to epigenetic changes by direct targeting of one component of the epigenetic machinery; however, it is likely that other miRNAs are involved in epigenetic factor regulation in solid tumors Citation[89] and they may also have a role in leukemia .
A new direct action of miRNA on chromatin?
Approximately half a century ago, Jacob and Monod proposed that base complementarity would allow specific RNA interaction with other nucleic acid sequences Citation[90]. Besides forming RNA–RNA interactions, RNA can potentially bind to complementary DNA sequences, working as a ‘guide‘ in the control and maintenance of chromatin status Citation[91].
Although, the in vivo existence of RNA–DNA triplexes and their direct action on epigenetic remodeling have not yet been validated, some recent data provided evidence for a role for ncRNA in chromatin-based processes Citation[8,91].
Interestingly, genome-wide analyses revealed an over-representation of putative triple-helix target at human gene promoters, highlighting the potential interaction of ncRNAs with the major groove of the DNA double helix to control gene expression Citation[92,93].
Growing evidence now highlights that miRNAs localize to the nucleus. Despite the canonical miRNA biogenesis that implies Exportin-5-mediated transfer of pre-miRNA from nucleus to cytoplasm, a number of studies indicate that mature miRNAs can be shuttled from the cytoplasm back to the nucleus. This shuttling also involves Ago proteins nuclear import Citation[94]. Moreover, deep-sequencing data revealed a complex subcellular distribution of miRNAs, with 300 of them being identified in both the nucleus and cytoplasm, and 39 having a preferential nuclear localization Citation[95].
Initial evidence on the action of exogenous sequence-specific siRNAs in transcriptional gene silencing Citation[96–100] triggered the idea of a role for endogenous small ncRNAs in evolutionarily conserved epigenetic gene silencing pathways Citation[96,101]. In fact, the existence of endogenous miRNA-directed epigenetic processes in mammals has been reported Citation[102–104].
miR-223 was described as a nucleation center for chromatin remodeling complex recruitment during myeloid lineage determination Citation[8]. A role for miR-223 in driving transcriptional gene silencing via PcG–RNAi complexes was suggested by the presence of two DNA sequences complementary to the miR-223 seed on the promoter region of NFI-A, whose mRNA is also targeted by miR-223 Citation[2]. Our group found that during human granulopoiesis, miR-223 translocates to the nucleus and localizes at its complementary sequences on the NFI-A promoter Citation[8]. The nuclear localization of miR-223 was also observed during metaphase, when RNA synthesis is blocked, suggesting a direct miR-223 binding to DNA Citation[8]. miR-223 is part of a ribonucleoprotein repressive complex formed by PcG proteins YY1 and Suz12, Dicer-1 and transiently Ago1, and the complex‘s localization on the NFI-A promoter is RNA dependent Citation[8]. This complex is recruited on the NFI-A promoter region, marked by an H3K27me3/H3K4me3 bivalent domain, which is then resolved (H3K27me3 increase and H3K4me3 decrease); this induced nearby heterochromatin formation and NFI-A silencing.
An analogous miRNA-dependent complex assembly on DNA has been recently reported for the promoters of the retinoblastoma repressor E2F/Rb1 complex targets, CDC2 (also known as CDK1) and CDCA8. Similarly, a miRNA-complementary sequence was found for let-7f in both promoters, in the antisense and sense strand, respectively Citation[102].
As miR-223 acts on NFI-A mRNA homeostasis and promoter chromatin accessibility, resulting in a more efficient and irreversible repression Citation[2,8], we hypothesize a general double-step control mechanism that would initially ensure fine tuning and later, long-term efficient silencing of developmentally regulated genes during HSC differentiation along each hematopoietic lineage . Therefore, once progenitors are committed, one unique differentiation destiny is ensured, providing the organism with the proper blood elements according to developmental timing, blood cell replacement and environmental signals (e.g., inflammation, growth factors, hormones and wounds).
Conclusion: extending the concept of epigenetics to miRNA-mediated gene regulation
Epigenetic mechanisms play an important role in hematopoiesis since they allow time- and environment-dependent modulation of genes encoding designated key regulatory factors of differentiation, proliferation and function of different hematopoietic cell types Citation[3].
The epigenetic modifications occurring on DNA or nucleosomal histones are not necessarily mutually exclusive processes. They occur by sequential recruitment of different enzymes and, represent dynamic processes affecting the transcriptional status of a specific gene Citation[104].
Since hematopoietic cells can respond to a plethora of stimuli, the use of many different ‘ways‘ to switch genes on and off, gradually or sharply, confers plasticity and a timely response to different signals. An altered balance of this intricate network can be one of the leading mechanisms for pathological conditions such as neoplastic transformation Citation[105].
miRNAs can act as targets as well as effectors of this dynamic regulation Citation[8,89,106], therefore, a monocausal relationship between miRNA action on gene expression and a unique specific function is unlikely Citation[40].
As highlighted in this review, miRNAs can be regulated by epigenetic events such as DNA methylation or histone modifications Citation[8,107] and they can silence members of the epigenetic machinery at post-transcriptional level , therefore, creating ‘epigenetic–miRNA loops‘, as exemplified in . This points to a new level of complexity in chromatin remodeling and epigenetic regulation mediated by miRNAs, which complements the most studied regulatory circuits involving transcription factors in hematopoiesis Citation[2,3,80]. These regulatory circuits are mostly repressing miRNA expression; however, the information regarding the epigenetic mechanisms involving miRNA is still limited.
Future perspective
Hematopoietic differentiation is often depicted as an intricate pattern, the ‘hematopoietic tree‘, with its root in the HSC compartment and the branches in each committed progenitor and mature precursor cell populations. Each transition from one stage to the next during progenitor maturation is governed by the action of epigenetic and transcription factors Citation[80,81] as well as miRNAs Citation[82,83].
Chromatin remodeling factors and miRNAs functions often overlap and cross-regulate each other, making the epigenetic system even more dynamic and intricate.
Moreover, the connection between miRNA nuclear localization and function is remarkable and suggests new pathways for miRNA-mediated gene expression regulation, directly acting on chromatin structure.
Two tightly linked systems, chromatin remodeling factors and miRNA Citation[83], exert a crucial role in HSC self-renewal, commitment and differentiation Citation[108]; now these two main mechanisms of regulation of HSC fate may be brought together, as a direct action of miRNAs on chromatin has been discovered. Therefore, miRNAs can be considered to be part of a multilevel regulatory mechanism, that modulates gene expression from the level of the chromatin structure through to mRNA translation/stability.
The regulatory circuit involving epigenetic mechanisms and miRNAs in hematopoiesis and leukemia has been extensively described Citation[108], but in the years to come, more focus should be put on the HSC compartment, where an interaction is likely, but has yet to be investigated in detail.
Filling in these gaps will help translational research to put new findings into clinical practice, given the high potential of miRNA in regenerative medicine Citation[40]. The possible use of miRNAs as a new class of drug able to control the epigenetic machinery is fascinating Citation[89]. As compared with the general action of chemical compounds, this would give researchers the chance to target specific effector enzymes, inhibiting their activities and affecting the expression of a wide range of epigenetically modulated genes.
Table 1. Examples of epigenetic regulation of miRNAs.
Table 2. miRNA-mediated regulation of epigenetic factors.
Hematopoiesis
Hematopoiesis is a complex and multistep process, whereby the wide variety of differentiated elements present in an animal‘s blood is generated from one unique stem cell. From embryonic development through to adult life, this process is tightly regulated by epigenetic events, which prime and commit progenitors to their definitive destiny.
Epigenetics & miRNA regulation crosstalk
Epigenetic gene regulation has been extensively studied in hematopoiesis, in particular hematopoietic stem cells, and this recently extended to hematopoietic malignancies. In this framework, a new class of regulators, the miRNAs have recently emerged as a player for the extensive changes responsible for hematopoietic cells development, differentiation and malignant transformation.
miRNAs are a species of short noncoding RNA that post-transcriptionally regulate gene expression. This class of noncoding RNA, classically linked to transcription factor networks, is now being integrated into epigenetics.
Some miRNAs control gene expression, cooperate with or are regulated by important epigenetic factors, including DNA methyltransferases, histone deacetylases and polycomb group genes. Therefore, miRNAs take part in chromatin structural modification in a direct or indirect way.
miRNA–epigenetic factors‘ direct cooperation
The complicated network linking miRNAs and epigenetic pathways suggests the existence of an epigenetic–miRNA loop, able to organize the whole gene expression profile in each and every cell.
Disruption of this regulatory loop is likely to interfere with physiological functions, possibly contributing to the establishment of disease processes.
A general double-step control mechanism could be envisioned ensuring fine tuning first and long-term efficient silencing later of key regulatory genes for differentiation along each hematopoietic lineage.
Acknowledgements
The authors apologize to the researchers whose works are not cited here due to space limitations. Some experimental concepts described in this article are based on work conducted in the laboratories of the authors. The authors wish to thank past and present members of their laboratories for their contribution to these experimental studies.
Financial & competing interests disclosure
The authors work is supported by research funding from the Italian Association for Cancer Research (IG-9390 to F Grignani and IG-11949 to C Nervi); University of Roma ‘La Sapienza‘, Ministero dell‘Istruzione, dell‘Università e della Ricerca (PRIN) and Fondazione Roma. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Shih AH , Abdel-WahabO, PatelJP, LevineRL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer12(9) , 599–612 (2012).
- Fazi F , RosaA, FaticaA et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 123(5) , 819–831 (2005).
- Fazi F , NerviC. MicroRNA: basic mechanisms and transcriptional regulatory networks for cell fate determination. Cardiovasc. Res.79(4) , 553–561 (2008).
- Sato F , TsuchiyaS, MeltzerSJ, ShimizuK. MicroRNAs and epigenetics. FEBS J.278(10) , 1598–1609 (2011).
- Kunej T , GodnicI, FerdinJ, HorvatS, DovcP, CalinGA. Epigenetic regulation of microRNAs in cancer: an integrated review of literature. Mutat. Res.717(1–2) , 77–84 (2011).
- Rouhi A , MagerDL, HumphriesRK, KuchenbauerF. MiRNAs, epigenetics, and cancer. Mamm. Genome19(7–8) , 517–525 (2008).
- Garzon R , LiuS, FabbriM et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113(25) , 6411–6418 (2009).
- Zardo G , CiolfiA, VianL et al. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 119(17) , 4034–4046 (2012).
- Cedar H , BergmanY. Epigenetics of haematopoietic cell development. Nat. Rev. Immunol.11(7) , 478–488 (2011).
- Zardo G , CiminoG, NerviC. Epigenetic plasticity of chromatin in embryonic and hematopoietic stem/progenitor cells: therapeutic potential of cell reprogramming. Leukemia22(8) , 1503–1518 (2008).
- Bryder D , RossiDJ, WeissmanIL. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am. J. Pathol.169(2) , 338–346 (2006).
- Sashida G , IwamaA. Epigenetic regulation of hematopoiesis. Int. J. Hematol.96(4) , 405–412 (2012).
- Calvanese V , FernándezAF, UrdinguioRG et al. A promoter DNA demethylation landscape of human hematopoietic differentiation. Nucleic Acids Res. 40(1) , 116–131 (2012).
- Franchini DM , Schmitz K-M, Petersen-Mahrt SK. 5-methylcytosine DNA demethylation: more than losing a methyl group. Annu. Rev. Genet.46 , 419–441 (2012).
- Geiger H , de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol.13(5) , 376–389 (2013).
- Bocker MT , HellwigI, BreilingA, EcksteinV, HoAD, LykoF. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood117(19) , e182–e189 (2011).
- Cui K , ZangC, Roh T-Y et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell4(1) , 80–93 (2009).
- Pietersen AM , van Lohuizen M. Stem cell regulation by polycomb repressors: postponing commitment. Curr. Opin. Cell Biol.20(2) , 201–207 (2008).
- Cao R , ZhangY. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED–EZH2 complex. Mol. Cell15(1) , 57–67 (2004).
- Viré E , BrennerC, DeplusR et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439(7078) , 871–874 (2006).
- Dawson MA , KouzaridesT. Cancer epigenetics: from mechanism to therapy. Cell150(1) , 12–27 (2012).
- Lee RC , FeinbaumRL, AmbrosV. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell75(5) , 843–854 (1993).
- Lagos-Quintana M , RauhutR, LendeckelW, TuschlT. Identification of novel genes coding for small expressed RNAs. Science294(5543) , 853–858 (2001).
- Lau NC , LimLP, WeinsteinEG, BartelDP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science294 , 858–862 (2001).
- Lee RC , AmbrosV. An extensive class of small RNAs in Caenorhabditis elegans. Science294(5543) , 862–864 (2001).
- Ambros V . MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell113(6) , 673–676 (2003).
- Griffiths-Jones S . The microRNA Registry. Nucleic Acids Res.32 , D109–D111 (2004).
- Filipowicz W , BhattacharyyaSN, SonenbergN. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genetics9(2) , 102–114 (2008).
- Lewis BP , ShihI, Jones-RhoadesMW, BartelDP, BurgeCB. Prediction of mammalian microRNA targets. Cell115(7) , 787–798 (2003).
- Lewis BP , BurgeCB, BartelDP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell120(1) , 15–20 (2005).
- Peters L , MeisterG. Argonaute proteins: mediators of RNA silencing. Mol. Cell26 , 611–623 (2007).
- Baskerville S , BartelDP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA11(3) , 241–247 (2005).
- Kim VN . MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol.6 , 376–385 (2005).
- Morlando M , BallarinoM, GromakN, PaganoF, BozzoniI, ProudfootNJ. Primary microRNA transcripts are processed co-transcriptionally. Nat. Struct. Mol. Biol.15(9) , 902–909 (2008).
- Ballarino M , PaganoF, GirardiE et al. Coupled RNA processing and transcription of intergenic primary micrornas coupled RNA processing and transcription of intergenic. Mol. Cell. Biol. 29(20) , 5632–5638 (2009).
- Denli AM , TopsBB, PlasterkRH, KettingRF, HannonGJ. Processing of primary microRNAs by the microprocessor complex. Nature432(7014) , 231–235 (2004).
- Gregory RI , YanKP, AmuthanG et al. The microprocessor complex mediates the genesis of microRNAs. Nature 432(7014) , 235–240 (2004).
- Chendrimada TP , GregoryRI, KumaraswamyE et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436(7051) , 740–744 (2005).
- Alemdehy MF , ErkelandSJ. MicroRNAs: key players of normal and malignant myelopoiesis. Curr. Opin. Hematol.19(4) , 261–267 (2012).
- Bissels U , BosioA, WagnerW. MicroRNAs are shaping the hematopoietic landscape. Haematologica97(2) , 160–167 (2012).
- Guo S , LuJ, SchlangerR et al. MicroRNA miR-125a controls hematopoietic stem cell number. Proc. Natl Acad. Sci. USA 107(32) , 14229–14234 (2010).
- Arrate MP , VincentT, OdvodyJ, KarR, JonesSN, EischenCM. MicroRNA biogenesis is required for Myc-induced B-cell lymphoma development and survival. Cancer Res.70(14) , 6083–6092 (2010).
- O‘Carroll D , MecklenbraukerI, DasPP et al. A Slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev. 21(16) , 1999–2004 (2007).
- Georgantas RW , HildrethR, MorisotS et al. microRNA expression and function: a circuit diagram of differentiation control. Proc. Natl Acad. Sci. USA 104(8) , 2750–2755 (2007).
- Petriv OI , KuchenbauerF, DelaneyAD et al. Comprehensive microRNA expression profiling of the hematopoietic hierarchy. 107(35) , 15443–15448 (2010).
- Rhyasen GW , StarczynowskiDT. Deregulation of microRNAs in myelodysplastic syndrome. Leukemia26(1) , 13–22 (2012).
- Gordon JE , WongJJ, RaskoJE. MicroRNAs in myeloid malignancies. Br. J. Haematol.162(2) , 162–176 (2013).
- Esteller M . Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum. Mol. Genet.16 , R50–59 (2007).
- Jones PA , BaylinSB. The fundamental role of epigenetic events in cancer. Nat. Rev. Genetics3(6) , 415–428 (2002).
- Weber B , StresemannC, BruecknerB, LykoF. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle6(9) , 1001–1005 (2007).
- Bernstein BE , MeissnerA, LanderES. The mammalian epigenome. Cell128(4) , 669–681 (2007).
- Fazi F , RacanicchiS, ZardoG et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer cell 12(5) , 457–466 (2007).
- Meng F , HensonR, Wehbe-JanekH, SmithH, UenoY, PatelT. The microRNA let-7a modulates interleukin-6-dependent STAT-3 survival signaling in malignant human cholangiocytes. J. Biol. Chem.282(11) , 8256–8264 (2007).
- Li Y , GaoL, LuoX et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 121(3) , 499–509 (2013).
- Díaz-Beyá M , NavarroA, FerrerG et al. Acute myeloid leukemia with translocation (8;16)(p11;p13) and MYST3–CREBBP rearrangement harbors a distinctive microRNA signature targeting RET proto-oncogene. Leukemia 27(3) , 595–603 (2013).
- Bueno MJ , Perez de Castro I, Gomez de Cedron M et al. Genetic and epigenetic silencing of MicroRNA-203 enhances ABL1 and BCR–ABL1 oncogene expression. Cancer cell13(6) , 496–506 (2008).
- Chim CS , WanTS, WongKY, FungTK, DrexlerHG, WongKF. Methylation of miR-34a, miR-34b/c, miR-124-121 and miR-203 in Ph-negative myeloproliferative neoplasms. J. Transl. Med.9(1) , 197 (2011).
- Chim CS , WongKY, LeungCY et al. Epigenetic inactivation of the hsa-miR-203 in haematological malignancies. J. Cell. Mol. Med. 15(12) , 2760–2767 (2011).
- Liu S , Wu L-C, Pang J et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer cell17(4) , 333–347 (2010).
- Gao X -N, Lin J, Li Y-H et al. MicroRNA-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia. Oncogene30(31) , 3416–3428 (2011).
- Pigazzi M , ManaraE, BresolinS et al. MicroRNA-34b promoter hypermethylation induces CREB overexpression and contributes to myeloid transformation. Haematologica 98(4) , 602–610 (2013).
- Dickstein J , SenyukV, PremanandK et al. Methylation and silencing of miRNA-124 by EVI1 and self-renewal exhaustion of hematopoietic stem cells in murine myelodysplastic syndrome. Proc. Natl Acad. Sci. USA 107(21) , 9783–9788 (2010).
- Erdogan B , BosompemA, PengD et al. Methylation of promoters of microRNAs and their host genes in myelodysplastic syndromes. Leuk. Lymphoma doi:10.3109/10428194.2013.790542 (2013) (Epub ahead of print)
- Agirre X , Vilas-ZornozaA, Jiménez-VelascoA et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Res. 69(10) , 4443–4453 (2009).
- Roman-Gomez J , AgirreX, Jiménez-VelascoA et al. Epigenetic regulation of microRNAs in acute lymphoblastic leukemia. J. Clin. Oncol. 27(8) , 1316–1322 (2009).
- Pospisil V , VargovaK, KokavecJ et al. Epigenetic silencing of the oncogenic miR-17–92 cluster during PU.1-directed macrophage differentiation. EMBO J. 30(21) , 4450–4464 (2011).
- Duan Z , PersonRE, Lee H-H et al. Epigenetic regulation of protein-coding and microRNA genes by the Gfi1-interacting tumor suppressor PRDM5. Mol. Cell. Biol.27(19) , 6889–6902 (2007).
- Hock H , HamblenMJ, RookeHM, SchindlerJW, SalequeS, FujiwaraY. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature431(7011) , 1002–1007 (2004).
- Duan Z , HorwitzM. Targets of the transcriptional repressor oncoprotein Gfi-1. Proc. Natl Acad. Sci. USA100(10) , 5932–5937 (2003).
- Spivakov M , FisherAG. Epigenetic signatures of stem-cell identity. Nat. Rev. Genetics8(4) , 263–271 (2007).
- So AY , JungJW, LeeS, KimHS, KangKS. DNA methyltransferase controls stem cell aging by regulating BMI1 and EZH2 through microRNAs. PloS One6(5) , e19503 (2011).
- Benetti R , GonzaloS, JacoI et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat. Struct. Mol. Biol. 15(3) , 268–279 (2008).
- Sinkkonen L , HugenschmidtT, BerningerP et al. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 15(3) , 259–267 (2008).
- Ji H , EhrlichLI, SeitaJ et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467(7313) , 338–342 (2010).
- Juan AH , KumarRM, MarxJG, YoungRA. Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol. Cell.36(1) , 61–74 (2009).
- Shimono Y , ZabalaM, ChoRW et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138(3) , 592–603 (2009).
- Oguro H , YuanJ, TanakaS et al. Lethal myelofibrosis induced by Bmi1-deficient hematopoietic cells unveils a tumor suppressor function of the polycomb group genes. J. Exp. Med. 209(3) , 445–454 (2012).
- Kamminga LM , BystrykhLV, de Boer A et al. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood107(5) , 2170–2179 (2006).
- Kent DG , DykstraBJ, CheyneJ, MaE, EavesCJ. Steel factor coordinately regulates the molecular signature and biologic function of hematopoietic stem cells. Blood112(3) , 560–567 (2008).
- Cantor AB , OrkinSH. Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene21(21) , 3368–3376 (1992).
- Rice KL , HormaecheI, LichtJD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene26(47) , 6697–6714 (2007).
- Chen C , LodishHF. MicroRNAs as regulators of mammalian hematopoiesis. Semin. Immunol.17(2) , 155–165 (2005).
- Nervi C , FaziF. Oncoproteins, heterochromatin silencing and microRNAs. Epigenetics3(1) , 1–4 (2008).
- Grigoryev SA , BulynkoYA, PopovaEY. The end adjusts the means: heterochromatin remodelling during terminal cell differentiation. Chromosome Res.14(1) , 53–69 (2006).
- Popova EY , KraussSW, ShortSA et al. Chromatin condensation in terminally differentiating mouse erythroblasts does not involve special architectural proteins but depends on histone deacetylation. Chromosome Res. 17(1) , 47–64 (2009).
- Zhang L , FlygareJ, WongP, LimB, LodishHF. miR-191 regulates mouse erythroblast enucleation by down-regulating Riok3 and Mxi1. Genes Dev.25(2) , 119–124 (2011).
- Fabbri M , CroceCM, CalinGA. MicroRNAs in the ontogeny of leukemias and lymphomas. Leuk. Lymphoma50(2) , 160–170 (2009).
- Fabbri M , GarzonR, CimminoA et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl Sci. USA 104(40) , 15805–15810 (2007).
- Iorio M V, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim. Biophys. Acta.1799(10–12) , 694–701 (2010).
- Jacob F , MonodJ. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol.3 , 318–356 (1961).
- Schmitz KM , MayerC, PostepskaA, GrummtI. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev.24(20) , 2264–2269 (2010).
- Goñi JR , De la Cruz X, Orozco M. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res.32(1) , 354–360 (2004).
- Belotserkovskii BP , De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J. Biol. Chem.282(44) , 32433–32441 (2007).
- Weinmann L , HöckJ, IvacevicT et al. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell 136(3) , 496–507 (2009).
- Liao JY , MaLM, GuoYH et al. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3´ trailers. PLoS One 5(5) , e10563 (2010).
- Zaratiegui M , IrvineDV, MartienssenRA. Noncoding RNAs and gene silencing. Cell128(4) , 763–776 (2007).
- Han J , KimD, MorrisKV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc. Natl Acad. Sci. USA104(30) , 12422–12427 (2007).
- Ting AH , SchuebelKE, HermanJG, BaylinSB. Short dsRNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nat. Genet.37(8) , 906–910 (2005).
- Kim DH , VilleneuveLM, MorrisK V, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol.13(9) , 793–797 (2006).
- Weinberg MS , VilleneuveLM, EhsaniAL et al. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA 12(2) , 256–262 (2006).
- Matzke M , BirchlerJ. RNAi-mediated pathways in the nucleus. Nat. Rev. Genetics6(1) , 24–35 (2005).
- Benhamed M , HerbigU, YeT, DejeanA, BischofO. Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat. Cell Biol.14(3) , 266–275 (2012).
- Kim DH , SætromP, Sn⊘veO, RossiJJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc. Natl Acad. Sci. USA105(42) , 16230–16235 (2008).
- Zardo G , FaziF, TravagliniL, NerviC. Dynamic and reversibility of heterochromatic gene silencing in human disease. Cell Res.15(9) , 679–690 (2005).
- Baer C , ClausR, PlassC. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res.73(2) , 473–477 (2013).
- Duursma AM , KeddeM, SchrierM, le Sage C, Agami R. miR-148 targets human DNMT3b protein coding region. RNA14(5) , 872–877 (2008).
- Zardo G , CiolfiA, VianL et al. Transcriptional targeting by microRNA-polycomb complexes: a novel route in cell fate determination. Cell Cycle 11(19) , 3543–3549 (2012).
- Garzon R , CroceCM. MicroRNAs in normal and malignant hematopoiesis. Curr. Opin. Hematol.15(4) , 19–21 (2008).
- Hsu SD , LinFM, WuWY et al. miRTarBase: a database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 39 , D163–D169 (2011).