
Abstract
Malignant peripheral nerve sheath tumors (MPNSTs) are rare and aggressive soft-tissue sarcomas with dismal prognosis. Complete resection, which is the only known definitive therapy, is not feasible with every tumor, and local recurrence after surgery is another challenge to successful treatment. Treatments used with other sarcoma types have not proven beneficial to MPNST patients. Targeted therapies blocking several signaling pathways known to drive MPNST pathogenesis have also not improved patient outcomes in clinical trials. This review discusses existing therapies and targeted chemotherapeutic options currently being tested clinically, and potential therapeutic avenues identified in preclinical studies that include targeting signaling pathways such as the HIPPO-YAP pathway and epigenetic mechanisms as well as multi-agent strategies.
Epidemiology & pathology of malignant peripheral nerve sheath tumor
Malignant peripheral nerve sheath tumors (MPNSTs) are highly aggressive peripheral nerve-associated tumors of Schwann cell origin that compose 5–10% of all soft-tissue sarcomas. The tumors occur primarily in adults between 20 and 60 years of age [Citation1]. Approximately 20% of cases are diagnosed in the pediatric population, and MPNSTs are one of the most frequent non-rhabdomyosarcoma tumors in children [Citation2]. The occurrence of MPNST is rare with an estimated incidence of 1.46 per 1,000,000 individuals [Citation3]. MPNSTs either occur independently or in association with neurofibromatosis type 1 (NF1) [Citation4]. Between 8 and 13% of individuals with NF1 mutations develop MPNST, and these patients constitute nearly 50% of all MPNST cases. Approximately 45% of MPNSTs occur sporadically with unidentified genetic anomalies, and the remaining 10% of cases are iatrogenically associated with radiotherapy [Citation5–7]. The prognosis of patients with MPNSTs is dismal with a 5-year survival rate of between 16 and 52%. Disease management poses significant challenges, as surgical resection remains the primary treatment. Surgical resection is often impeded by the large size of tumors, their proximity to complex nerve networks and a low rate of negative resection margins [Citation8]. In addition, MPNSTs usually exhibit strong metastatic proclivity, and patients have a high propensity for relapse following tumor resection and poor responses to cytotoxic chemotherapy and irradiation, resulting in high mortality [Citation4,Citation9] ().
The mechanisms of action of potential small-molecule inhibitors targeting each of the above-mentioned machinery are denoted.
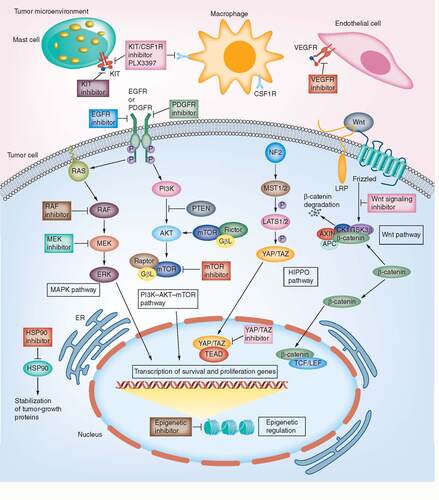
MPNSTs arise in nerve roots and bundles in the extremities and pelvis, in particular, large peripheral nerves such as the sciatic nerve, brachial plexus and sacral plexus. Patients present with rapidly enlarging mass, pain and neurological symptoms, and pulmonary metastasis is observed in up to half of the cases. The majority of MPNSTs are classified as intermediate to high grade at diagnosis. Heterologous differentiation to mesenchymal lineages, such as skeletal muscle, bone, cartilage and blood vessels is seen in approximately 15% of tumors and may be predictive of poorer prognosis [Citation4]. The molecular and signaling events responsible for benign neurofibroma-to-MPNST transformation in NF1 patients, and those involved in tumor development and progression of sporadic MPNSTs, are still incompletely understood.
Genetics & genomics of NF1-associated & sporadic MPNST
In NF1-associated MPNST, malignant transformation of pre-existing benign plexiform neurofibromas, a hallmark of NF1, is believed to contribute to MPNST; MPNSTs are the leading cause of death in neurofibromatosis patients [Citation10]. NF1 is an autosomal-dominant tumor predisposition syndrome caused by a mutation in the NF1 tumor suppressor gene. NF1 is located in the chromosome band 17q11 and codes for a 280-kDa protein called neurofibromin that accelerates GTP hydrolysis on Ras proteins and facilitates the conversion of active Ras-GTP to inactive Ras-GDP in various cell types, thereby inhibiting the p21ras signaling pathway [Citation11]. The loss of NF1 protein leads to activation of the Ras signaling pathway. Progression toward MPNST normally requires biallelic loss of the NF1 gene. NF1 deficiency by itself is insufficient to induce MPNST given that only 8–13% of all NF1 patients develop MPNST. The acquisition of additional genetic aberrations, including the amplification of oncogenes PDGFRA, EGFR or MET or the loss of tumor suppressor genes CDKN2A, RB1, TP53 or the genes encoding epigenetic PRC2 complex components SUZ12 or EED, has been associated with MPNST formation [Citation12]. TP53 point mutations or deletions are observed in approximately 75% of patients with MPNSTs. The loss of TP53 in one allele is sufficient to drive MPNST [Citation13]. Deletions of the CDKN2A tumor suppressor gene, which encodes two cell-cycle inhibitors p16INK4A and p14ARF, occur in about 50% of neoplasms [Citation12]. Moreover, genomic inactivation of CDKN2A can be found in a major subset of atypical neurofibromas, which eventually progress into malignancy. Loss of CDKN2A expression contributes to neurofibroma-to-MPNST transformation [Citation14,Citation15].
Sporadic MPNSTs are caused by unidentified genetic or epigenetic anomalies, although nearly 40% also exhibit somatic mutations of NF1 [Citation16]. TP53 mutations are more frequently detected in sporadic MPNST, whereas Raf/PI3K/AKT activation and EGFR overexpression are more associated with NF1-associated MPNST [Citation4]. Despite so, recurrent mutations of NF1, TP53, CDKN2A, SUZ12 and EED have been reported across MPNST subtypes from multiple next-generation sequencing studies [Citation17], suggesting that distinct subtypes of MPNST can be driven by similar genomic or epigenetic mechanism.
Standard treatment paradigm for MPNST
The conventional treatment for MPNST involves surgical resection with a wide negative margin, which is defined as the resection margin lacking identifiable tumor within 1 mm from an inked surface of tissue. Surgical resection has significantly prolonged overall survival in patients with NF1. Efficacy of surgery depends on tumor size, location and metastasis. The local recurrence rate after surgical resection is between 32 and 65% [Citation18]. Adjuvant radiation therapy is generally recommended for patients with large high-grade lesions (>5 cm) or with positive resected tumor margins as a post-operative measure to reduce local recurrence and to control symptoms such as pain. The use of radiotherapy, however, has not demonstrated an advantage in terms of overall survival [Citation19].
MPNST has been treated with chemotherapy regimens similar to those used in treatment of soft-tissue sarcomas with agents such as doxorubicin, dactinomycin, cyclophosphamide and vincristine. Chemotherapy is largely reserved for unresectable tumors and metastatic MPNSTs, however. NF1 patients respond less well to chemotherapy than do sporadic MPNST patients [Citation4]. The efficacy of chemotherapy is still not clearly defined as current chemotherapy regimens have provided only marginal survival benefit [Citation4].
Despite current aggressive multimodal therapy, the prognosis of MPNST patients remains dismal. There is an urgent need for improved effective therapeutic modalities for MPNST. Identifying key pathways that are vital to MPNST growth is essential for overcoming this challenge. Suboptimal clinical outcomes with conventional chemotherapeutic agents have prompted interest in the development of rationally targeted therapeutics in MPNST, and recent molecular and genomics studies that have identified signaling networks and epigenetic regulators underlying MPNST pathogenesis have potential to guide this development.
Therapeutics targeting activated signaling pathways in MPNST
Receptor tyrosine kinase signaling
Receptor tyrosine kinases (RTKs) are a family of high-affinity cell-surface receptors for a wide array of growth factors, hormones, cytokines, neurotrophic factors and other extracellular signaling molecules. Multiple soluble or membrane-bound growth factors bind and activate RTKs including NGFR, PDGFR, FGFR, EGFR, VEGFR, MET/HGFR, IR and IGFR, and the proto-oncogene c-KIT. These growth factors are key regulators of normal cellular processes including survival, proliferation, differentiation and migration [Citation20,Citation21].
Aberrant regulation of RTK signaling due to acquisition of genetic and epigenetic alterations is a common cause of cell transformation, cancer development and metastasis, and RTKs play critical roles in regulation of cell stemness and proliferation [Citation22,Citation23]. Genetic alterations that activate RTKs or components of downstream pathways such as MAPK, PI3K/AKT and JAK/STAT have been identified in a multitude of malignancies [Citation22]. Small-molecule inhibitors and antibodies that target RTKs and the MAP kinase and PI3K/AKT pathways show moderate efficacy against MPNST [Citation24]. Tyrosine kinase inhibitors imatinib and dasatinib, which target the PDGFR pathway and c-KIT, respectively, both upregulated in MPNST [Citation25], have been explored in clinical trials but no significant responses were observed [Citation26,Citation27].
Gene amplification events in the EGFR pathway and dysregulated EGFR expression are frequent in MPNST [Citation28,Citation29]. The EGFR ligands transforming growth factor alpha and EGF are expressed at higher levels in MPNSTs than in neurofibromas [Citation30], and both promote malignant transformation of neurofibroma in a mouse model [Citation31]. Despite this, a Phase II trial of erlotinib, an EGFR inhibitor, in metastatic or unresectable MPNST did not show obvious benefit [Citation32]. Intriguingly, EGFR may mediate its effects through the STAT3 pathway and inhibition of the EGFR-STAT3 axis efficiently impedes MPNST transformation and tumorigenesis in xenograft models [Citation31]. A specific JAK2 inhibitor FLLL32, which blocks JAK2-mediated phosphorylation of STAT3, delays MPNST growth in an MPNST xenograft model, suggesting that combination therapies targeting JAK/STAT3 might be a rational choice for therapeutics. In summary, targeting RTK signaling pathways is a rational but not yet successful approach for treatment of MPNSTs ().
Table 1. Targeted therapies of past and ongoing clinical trials for malignant peripheral nerve sheath tumor and potential therapeutics tested in preclinical studies.
RAS/RAF/MEK pathway
The RAS/RAF/MEK/ERK cascade integrates inputs from a variety of extracellular cues into intrinsic transcriptional programs that drive a broad spectrum of physiological and pathological cellular processes such as growth, proliferation, differentiation, migration and apoptosis [Citation33]. The tumor suppressor gene NF1 encodes neurofibromin, a GTPase-activating protein that accelerates GTP hydrolysis on Ras proteins, thus negatively regulating RAS/RAF/MEK signaling. NF1 loss-of-function mutations result in an increase of the GTP-bound form of Ras [Citation34]. GTP-bound Ras recruits and activates the serine/threonine protein kinase RAF; RAF subsequently activates MEK1/2 and ERK1/2. Activated ERK1 and ERK2 phosphorylate and stimulate downstream effectors and regulate different transcription factors, leading to expression of genes that stimulate proliferation.
Germline or somatic mutations of NF1 are frequently detected in both NF1-associated and sporadic MPNSTs, suggesting that the RAS-RAF-MEK pathway should be targeted to treat MPNSTs. However, a multicenter Phase II trial exploring responses to sorafenib, a small-molecule orally given RAF kinase and tyrosine kinase inhibitor revealed no significant responses, with overall survival of treated MPNST patients similar to survival of controls [Citation35].
MEK inhibitors show promising antitumor effects in MPNST cells grown in culture [Citation36]. Moreover, a MEK inhibitor, selumetinib, reversed plexiform neurofibroma tumor burden in a recent clinical trial [Citation37]. An ongoing Phase II trial (SARC031; NCT03433183) aims to test the efficiency of selumetinib in combination with the dual mTOR kinase inhibitor vistusertib in patients with unresectable or metastatic NF1-associated or sporadic MPNST.
PI3K/AKT/mTOR pathway
Activation of the mTOR pathway is prevalent in MPNST tumor samples and likely plays a key role in the pathogenesis of MPNST. Alterations in expression or activity of PI3K/mTOR pathway components such as PTEN, p-AKT, p-mTOR and p-S6RP portend a poorer prognosis in MPNST patients [Citation38]. Preclinical studies of mTOR inhibitors in vitro and in xenografts have demonstrated tumor growth suppression [Citation38,Citation39]. The combination of mTOR inhibitors with other agents has also been explored in animal models, and combinations have been identified that surpass the cytostatic effects of mTOR inhibition alone. In an MPNST mouse model, dual inhibition of the mTOR pathway and the molecular chaperone hsp90 effectively abrogated the cellular stress response machinery tumor cells employ to survive proteotoxic stress and eventually caused cell death and tumor regression [Citation40]. A multi-institutional Phase I/II trial (SARC023; NCT02008877) of ganetespib, an Hsp90 inhibitor, in combination with the mTOR inhibitor sirolimus in patients with refractory sarcoma including MPNST was recently completed. The investigators hypothesize that these agents that work on separate and potentially synergistic pathways may be useful for treatment of refractory soft-tissue sarcomas. The outcome analysis is yet to be released. Tests of combination therapies involving mTOR inhibitors with agents targeting additional pathways, such as MEK and VEGFR pathways, are ongoing.
VEGF pathway
VEGF signaling promotes vasculogenesis and angiogenesis and is critical to tumor-induced new vasculature formation, which is essential for tumor growth and metastasis [Citation41]. Anti-angiogenic therapy has been a vital strategy for cancer therapy; however, pharmacological inhibition of the VEGF pathway in Phase II studies did not benefit MPNST patients [Citation42]. Therefore, rationally designed combination therapies are currently being explored. A Phase II trial testing the combination of anti-angiogenic VEGF inhibitor bevacizumab and mTOR inhibitor everolimus in 25 patients with NF1-associated and sporadic MPNSTs has been completed (NCT01661283). The therapy outcome was dismal, with a clinical benefit rate of 12% [Citation43].
Emerging potential therapeutic options
Despite a prevalent association with NF1 genetic aberrations in most MPNST cases, the genomic complexity of the disease renders therapy development challenging. Moreover, the focus of most clinical trials aimed at treating MPNSTs has been on reducing RAS signaling or downstream effector expression using inhibitors of RTK, RAS, MEK, AKT and mTOR signaling pathways. Unfortunately, none of these trials have yet shown efficacy against MPNSTs. Therefore, it is imperative to elucidate additional key molecular drivers of MPNST.
Wnt signaling
Largaespada and colleagues have developed an unbiased in vivo forward genetic screen to identify pathways critical for MPNST tumorigenesis. Using their Sleeping Beauty transposon-based somatic mutagenesis system in mice, in which a mutagenic transposable element tags insertional mutagenesis events that drive cancer in the Schwann cell lineage, they identified Wnt signaling pathway genes as potential drivers of benign neurofibromas and MPNSTs [Citation44]. Evidence suggests that the canonical WNT signaling pathway crosstalks with other pathways to drive MPNST pathogenesis [Citation45,Citation46]. Targeting the Wnt pathway in conjunction with mTOR inhibition synergistically induced cell death in MPNST cell lines [Citation47]. The same Sleeping Beauty screening platform also enabled identification of new proto-oncogenes key to MPNST maintenance such as FOXR2, further validating the potential use of this tool for discovery of cancer-driving genes and therapeutic design [Citation44].
Epigenetic mechanisms
Epigenetic-based therapies have gained tremendous attention in cancer research in recent years. In 70–90% of all MPNST subtypes, loss-of-function mutations in SUZ12 or EED, which encode polycomb repressive complex 2 (PRC2) constituents, have been identified [Citation48]. These mutations are associated with malignant progression from benign plexiform neurofibroma to MPNST [Citation49,Citation50]. SUZ12 or EED mutations cause loss of the repressive mark, trimethylation of histone 3 on K27 (H3K27), resulting in de-repression of genes that drive cell growth. Upon inactivation of PRC2 and loss of H3K27 methylation, H3K27 may become acetylated. The bromodomain and extra terminal domain (BET) family proteins (e.g., BRD protein family) bind to these acetylated loci and recruit transcription factor complexes thereby modulating transcription elongation of specific genes, such as oncogenic Ras signaling genes [Citation51]. A potential strategy is to reverse the loss of PRC2 activity with bromodomain inhibitors such as JQ1. JQ1 potently inhibits bromodomain protein activities, thus suppressing the genes normally repressed by PRC2. Furthermore, JQ1 in conjunction with a MEK inhibitor promotes cell death and tumor regression in a mouse model [Citation49].
Another study finds that the chromatin regulator, BRD4, a member of the BET family, is upregulated in a mouse MPNST model. Inhibition of BRD4 robustly suppresses MPNST growth and tumorigenesis via induction of apoptosis through proapoptotic Bim activation [Citation52]. A Phase II clinical trial is ongoing with the use of CPI-0610, a selective inhibitor of BET family proteins (NCT02986919).
Additionally, tumors with elevated Ras signaling, including MPNSTs, are sensitive to histone deacetylase inhibitors, which induce autophagy in MPNSTs and might be a useful therapeutic option [Citation53].
HIPPO signaling pathway
Despite the rarity of genetic mutations of HIPPO core components, persistent activation of HIPPO signaling effectors YAP and TAZ has been reported in a multitude of cancer types. A few inactivating mutations of the HIPPO pathway components have also been identified in cancers [Citation54], including mutations in NF2, encoding an upstream HIPPO component Merlin, in benign schwannoma [Citation55] and occasionally high-grade tumors [Citation56], as well as LATS2 and SAV1 in malignant mesothelioma [Citation57]. Constitutive activation of YAP/TAZ oncoproteins is observed in a wide variety of sarcoma types including MPNST [Citation58]. YAP/TAZ hyperactivity bestows tumor cells with stem cell properties and resistance to chemotherapeutic agents [Citation59]. Importantly, a recent study shows that the HIPPO/YAP gene signature is activated in both NF1-associated and sporadic MPNSTs regardless of NF1 mutation status [Citation60]. Pharmacological manipulation of the HIPPO pathway may be a potential approach to treat tumors with high YAP and/or TAZ activity.
Our recent study showed that deficiencies in HIPPO signaling kinases LATS1/2 in Schwann cells induces transformation to malignant nerve sheath tumors with full penetrance, suggesting that HIPPO signaling dysregulation is critical for MPNST pathogenesis. Tumors that arise are highly invasive and differentiated, resembling the characteristics of human MPNST. Oncogenic activities of YAP and TAZ are dependent on their association with the TEADs transcription factors. Analysis of TEAD1 genome occupancy and transcript levels demonstrated that YAP/TAZ directly regulate multiple oncogenic pathways including those involved in cell cycle progression, regulation of cell migration, extracellular matrix organization and, in particular, PDGFR signaling. Pharmacologically targeting HIPPO/YAP/TAZ and PDGFR signaling was explored in allograft models as well as in human MPNST cell lines in vitro. Combined attenuation of YAP/TAZ/TEAD1 and PDGFR/RAF1 signaling decelerates tumor cell proliferation in LATS1/2-deficient mice and human MPNST cells. Although single-agent therapy with PDGFR/RAF1 inhibitors alone (imatinib or sorafenib) was not effective in treatment of MPNST patients in a Phase I/II trial [Citation26,Citation35], the combination treatment paradigm has potential.
The YAP/TAZ inhibitor verteporfin is an FDA-approved porphyrin-family drug clinically used as a photosensitizer in photodynamic therapy that has been shown to disrupt the interaction of YAP/TAZ with TEAD [Citation61]. Verteporfin effectively inhibits YAP expression and sensitizes esophageal cancer cells [Citation62] and lung cancer cells [Citation63] in culture to cytotoxic drugs. Although a Phase I/II trial of verteporfin photodynamic therapy in locally advanced pancreatic cancer indicates therapy feasibility [Citation64], its toxicity to non-malignant cells and its low aqueous solubility pose limitations to clinical use.
The interactions between TEAD and YAP/TAZ can also be inhibited by peptides and small molecules. Super-TDU, designed to mimic the tandem TDU domain of VGLL4, which directly competes with YAP for binding to TEADs, robustly inhibits gastric cancer growth in vitro and in xenografts [Citation65]. Intriguingly, its anti-tumor activity appears to be specific for tumors with an elevated ratio of YAP to VGLL4. Another peptide engineered to target the YAP-TEAD interaction is the cyclic peptide 17; the peptide specifically targets the TEAD-binding interface on YAP. In vitro assays and xenograft models show that it potently impairs tumor growth. CA3, a small-molecule inhibitor of TEAD/YAP1 transcriptional activity, inhibits proliferation and tumor sphere formation, induces apoptosis and reduces the proportion of cancer stem cells in esophageal adenocarcinoma xenografts without apparent toxicity [Citation66]. In summary, targeting oncogenic proteins YAP/TAZ is promising for cancer therapeutics [Citation59]. The animal model of MPNST with LATS deficiency will facilitate identification and development of combinatorial strategies coupling inhibition of the HIPPO pathway and other oncogenic pathways.
Targeting cancer-associated cells in the tumor microenvironment
One of the hallmarks of cancer is the evolution of a tumor microenvironment whereby malignant cells recruit, co-exist and communicate with nonmalignant cells such as fibroblasts, macrophages and other immune infiltrates to establish a growth-permissive milieu. The myriads of cellular interactions between cancer cells and cancer-associated cells in the tumor microenvironment can be growth-promoting and growth-inhibitory, which determines whether a given host environment favors tumor proliferation, invasiveness, metastatic potential and stemness maintenance and confer drug resistance [Citation67]. A complete understanding of these cellular interactions may therefore shed light on novel anti-tumor therapies aimed at increasing the resistance of the host environment.
MPNST is composed of a mixture of Schwann cells, fibroblasts and perineurial cells, all of which contribute to the establishment of a tumor niche. Indeed, mouse model studies reveal that Nf1 heterozygosity in the tumor environment promotes neurofibroma formation [Citation68]. In a conditional Nf1 mouse model, in which a floxed Nf1 allele is deleted by a Cre transgene under control of the Schwann cell-specific promoter, Krox-20 (Nf1flox/- ;Krox20-cre; containing NF1 -/- Schwann cells and NF1 +/- mast cells), the Nf1flox/- ;Krox20-cre mice develop plexiform neurofibromas and massive mast cell infiltration, whereas Nf1flox/flox ;Krox20-cre mice (containing NF1 -/- Schwann cells and normal mast cells) only developed small, infrequent hyperplastic lesions in the cranial nerves with reduced mast cell infiltration. This suggests an important role of the heterozygous environment in neurofibroma development. However, subsequent studies by other groups suggests that ablation of Nf1 or codeletion of Nf1 and Pten solely in Schwann cell precursors give rise to neurofibroma and MPNST, respectively [Citation47,Citation69].
Macrophage depletion
Macrophage infiltration is prevalent in neurofibromas and MPNSTs, accounting for nearly 50% of cells within a tumor [Citation70]. While evaluating the efficacy of PLX3397, a selective RTK/c-KIT and colony stimulating factor 1 receptor (CSF1R) inhibitor against MPNST cells, Schwartz and colleagues demonstrated that PLX3397 efficiently blocks c-KIT, PDGFR-β and CSF1R activity in MPNST cells in vitro and in xenografts, impairing tumor growth and inducing macrophage depletion. Combining PLX3397 and an mTOR inhibitor, rapamycin further enhances macrophage depletion and tumor growth inhibition [Citation71]. This strategy is currently being explored in a Phase I/II trial (NCT02584647) of the orally dosed combination of PLX3397 and an mTOR inhibitor, sirolimus.
Immune checkpoint blockade
Tumor cells evade immune surveillance and T-cell mediated clearance by expression of inhibitory immune molecules, including programmed death-ligand 1 (PD-L1). Manipulation of immune checkpoints such as CTLA4 or PD-1 with targeted antibodies has emerged as an effective anticancer strategy in various malignancies. Since PD-L1 expression is more prevalent in MPNST than normal nerves, benign neurofibromas and schwannomas, immune checkpoint blockade immunotherapy may be a potential therapy for MPNST [Citation72,Citation73]. A Phase II clinical trial is ongoing using a checkpoint inhibitor to PD-1, pembrolizumab (MK-3475) in the patients with metastatic or locally advanced/unresectable MPNST (NCT02691026). Pembrolizumab is a humanized antibody that binds to PD-1, expressed on the surface of activated T cells, and blocks the interaction and activation of PD-1 by its ligand PD-L1 on tumor cells, which results in the activation of T-cell-mediated killing of tumor cells.
Oncolytic virus vaccine therapy
There has been growing interest in using oncolytic viruses as therapy due to their ability to kill cancer cells, via direct lytic activity of the virus life cycle or activation of the innate immune response against tumor cells. Oncolytic herpes simplex viruses selectively infect MPNST cells expressing high levels of RAS in culture and inhibit the growth of MPNST cells in immunodeficient mice [Citation74,Citation75]. A Phase I trial for investigating the side effects and the suitable dose of vaccine therapy in the patients with recurrent or unresectable MPNST is ongoing (NCT02700230).
Conclusion & future perspective
The poor prognosis, generally dismal drug response rate, and high rates of drug resistance of patients with MPNST present a pressing need for a better understanding of MPNST biology and development of effective treatment paradigms. The rarity of MPNST poses a challenge for patient recruitment and testing of potential therapies. In the past two decades, the outcome of MPNST has not improved significantly. However, a more thorough understanding of the genomic, epigenetic, signaling pathway and microenvironment changes during plexiform neurofibroma-to-MPNST malignant transformation has offered hope for the development of more effective diagnostic and therapeutic strategies for MPNST. In the next years, developing novel animal models that recapitulate human MPNST transformation from atypical plexiform neurofibroma is necessary to enable dissection of the molecular mechanisms underlying MPNST initiation and progression. Identification of the biomarkers of MPNST and their validation in preclinical models are critically important to the success of clinical trials. In addition, targeting individual steps in a druggable pathway may induce negative feedback responses resulting in treatment failure. Understanding all steps in multiple signaling pathways activated in MPNST and how different pathways crosstalk within tumor cells or with tumor microenvironment will facilitate design of rational combinations of targeted therapies to increase treatment efficacy and reduce resistance mechanisms.
Furthermore, a comprehensive database including genomic, transcriptomic, phenotypic, metabolomic data, drug treatment responses and tumor microenvironment niches is requisite for accelerating our understanding of the molecular basis of this challenging disease. These important resources must be shared to inform therapeutic strategies in MPNST. Coordinated efforts between preclinical and clinical research teams will help establish criteria for rational selection of combination therapies for clinical trial evaluation in MPNST patients.
Epidemiology & pathology of malignant peripheral nerve sheath tumor
The major subtypes of malignant peripheral nerve sheath tumors (MPNSTs) include NF1-associated, sporadic and radiotherapy-associated MPNST. These highly aggressive peripheral nerve-associated tumors of Schwann cell origin compose 5–10% of all soft-tissue sarcomas.
The clinical outcome of MPNSTs is poor with a dismal 5-year survival rate. Surgical resection remains the primary treatment. In addition, MPNSTs are strongly metastatic and resistant to cytotoxic chemotherapy.
Genetics & genomics of NF1-associated & sporadic MPNST
Biallelic loss of the NF1 gene alone is insufficient to induce MPNST.
The acquisition of additional genetic aberrations, including the amplification of oncogenes PDGFRA, EGFR or MET or the loss of tumor suppressor genes CDKN2A, RB1, TP53 or the genes encoding epigenetic PRC2 complex components SUZ12 or EED, has been associated with MPNST formation.
Standard treatment paradigm for MPNST
The conventional treatment for MPNST involves surgical resection.
Adjuvant radiation therapy is generally recommended for patients with large high-grade lesions (>5 cm) or with positive resected tumor margins.
Chemotherapy is largely reserved for unresectable tumors and metastatic MPNSTs, and is similar to treatment paradigms for other soft tissue sarcomas. However, these regimen are considered ineffective.
Therapeutics targeting activated signaling pathways in MPNST
Receptor tyrosine kinase signaling
RAS/RAF/MEK pathway
NF1 loss-of-function mutations result in Ras activation.
MEK inhibitors show promising anti-tumor effects in MPNST cells in vitro.
A clinical trial is ongoing using a MEK inhibitor in combination with other oncogenic pathway inhibition.
PI3K/AKT/mTOR pathway
Preclinical studies of mTOR inhibitors in vitro and in xenografts have demonstrated tumor growth suppression.
Several ongoing clinical trials use combination of mTOR inhibitors and other agents.
VEGF pathway
Pharmacological inhibition of the VEGF pathway in Phase II studies did not benefit MPNST patients.
Emerging potential therapeutic options
Wnt signaling
In vivo forward genetic screen identified Wnt signaling pathway genes as potential drivers of benign neurofibromas and MPNST.
Targeting the Wnt pathway in conjunction with mTOR inhibition synergistically induced cell death in MPNST cell lines.
Epigenetic mechanisms
Loss-of-function mutations of PRC2 components are associated with malignant progression from benign plexiform neurofibroma to MPNST.
BRD4 inhibition suppresses MPNST tumor growth.
HDAC inhibition induces autophagy in MPNST.
HIPPO signaling pathway
HIPPO/YAP gene signature is activated in both NF1-associated and sporadic MPNSTs regardless of NF1 mutation status.
Pharmacological manipulation of the HIPPO pathway may be a potential approach to treat tumors with high YAP and/or TAZ activity.
Targeting cancer-associated cells in the tumor microenvironment
Macrophage depletion
Macrophage infiltration is prevalent in neurofibromas and MPNSTs, accounting for nearly 50% of cells within a tumor.
PLX3397 efficiently blocks c-KIT, PDGFRβ and CSF1R activity in MPNST cells in vitro and in xenografts.
Immune checkpoint blockade
PD-L1 expression is more prevalent in MPNST than normal nerves, benign neurofibromas and schwannomas.
A phase II clinical trial is ongoing using a checkpoint inhibitor to PD-1 in MPNST.
Oncolytic virus vaccine therapy
Future perspective & conclusion
Identification of the biomarkers of MPNST and their validation in preclinical models are critically important to the success of clinical trials.
A comprehensive database including genomic, transcriptomic, phenotypic, metabolomic data, drug treatment responses and tumor microenvironment niches is requisite for accelerating our understanding of the molecular basis of this challenging disease.
Financial & competing interests disclosure
This study was funded in part by grants from NIH R37 NS096359 to QR Lu and Children's Tumor Foundation Young Investigator Award to LM Wu, supported by the NF Research Initiative at Boston Children's Hospital made possible by an anonymous gift. The authors would like to thank Dr Xianyao Zhou for figure drawing. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Widemann BC. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr. Oncol. Rep. 11(4), 322–328 (2009).
- Carli M, Ferrari A, Mattke A et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J. Clin. Oncol. 23(33), 8422–8430 (2005).
- Bates JE, Peterson CR, Dhakal S, Giampoli EJ, Constine LS. Malignant peripheral nerve sheath tumors (MPNST): a SEER analysis of incidence across the age spectrum and therapeutic interventions in the pediatric population. Pediatr. Blood Cancer 61(11), 1955–1960 (2014).
- Farid M, Demicco EG, Garcia R et al. Malignant peripheral nerve sheath tumors. Oncologist 19(2), 193–201 (2014).
- Rodriguez FJ. Peripheral nerve sheath tumors: the elegant chapter in surgical neuropathology. Acta Neuropathol. 123(3), 293–294 (2012).
- Foley KM, Woodruff JM, Ellis FT, Posner JB. Radiation-induced malignant and atypical peripheral nerve sheath tumors. Ann. Neurol. 7(4), 311–318 (1980).
- Lafemina J, Qin LX, Moraco NH et al. Oncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumors. Ann. Surg. Oncol. 20(1), 66–72 (2013).
- Gupta G, Mammis A, Maniker A. Malignant peripheral nerve sheath tumors. Neurosurg. Clin. North Am. 19(4), 533–543; v (2008).
- Stemmer-Rachamimov AO, Louis DN, Nielsen GP et al. Comparative pathology of nerve sheath tumors in mouse models and humans. Cancer Res. 64(10), 3718–3724 (2004).
- Kim A, Stewart DR, Reilly KM, Viskochil D, Miettinen MM, Widemann BC. Malignant peripheral nerve sheath tumors state of the science: leveraging clinical and biological insights into effective therapies. Sarcoma 2017, 7429697 (2017).
- Jouhilahti EM, Peltonen S, Heape AM, Peltonen J. The pathoetiology of neurofibromatosis 1. Am. J. Pathol. 178(5), 1932–1939 (2011).
- Carroll SL. The challenge of cancer genomics in rare nervous system neoplasms: malignant peripheral nerve sheath tumors as a paradigm for cross-species comparative oncogenomics. Am. J. Pathol. 186(3), 464–477 (2016).
- Lothe RA, Smith-Sorensen B, Hektoen M et al. Biallelic inactivation of TP53 rarely contributes to the development of malignant peripheral nerve sheath tumors. Genes Chromosom. Cancer 30(2), 202–206 (2001).
- Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE, Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am. J. Pathol. 155(6), 1879–1884 (1999).
- Beert E, Brems H, Daniels B et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosom. cancer 50(12), 1021–1032 (2011).
- Bottillo I, Ahlquist T, Brekke H et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J. Pathol. 217(5), 693–701 (2009).
- Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci. Rep. 7(1), 14992 (2017).
- Bradford D, Kim A. Current treatment options for malignant peripheral nerve sheath tumors. Curr. Treat. Options Oncol. 16(3), 328 (2015).
- Kahn J, Gillespie A, Tsokos M et al. Radiation therapy in management of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Front. Oncol. 4, 324 (2014).
- Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr. Opin. Cell Biol. 19(2), 117–123 (2007).
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 141(7), 1117–1134 (2010).
- Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, Kundu GC. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol. Cancer 17(1), 34 (2018).
- Regad T. Targeting RTK signaling pathways in cancer. Cancers 7(3), 1758–1784 (2015).
- Reilly KM. Extending the convergence of canonical WNT signaling and classic cancer pathways for treatment of malignant peripheral nerve sheath tumors. Cancer Discov. 3(6), 610–612 (2013).
- Holtkamp N, Okuducu AF, Mucha J et al. Mutation and expression of PDGFRA and KIT in malignant peripheral nerve sheath tumors, and its implications for imatinib sensitivity. Carcinogenesis 27(3), 664–671 (2006).
- Chugh R, Wathen JK, Maki RG et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J. Clin. Oncol. 27(19), 3148–3153 (2009).
- Schuetze SM, Wathen JK, Lucas DR et al. SARC009: Phase 2 study of dasatinib in patients with previously treated, high-grade, advanced sarcoma. Cancer 122(6), 868–874 (2016).
- Du X, Yang J, Ylipaa A, Zhu Z. Genomic amplification and high expression of EGFR are key targetable oncogenic events in malignant peripheral nerve sheath tumor. J. Hematol. Oncol. 6, 93 (2013).
- Tabone-Eglinger S, Bahleda R, Cote JF et al. Frequent EGFR positivity and overexpression in high-grade areas of human MPNSTs. Sarcoma 2008, 849156 (2008).
- Holtkamp N, Malzer E, Zietsch J et al. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro-Oncology 10(6), 946–957 (2008).
- Wu J, Patmore DM, Jousma E et al. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene 33(2), 173–180 (2014).
- Albritton KH, Rankin C, Coffin CM et al. Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST). J. Clin. Oncol. 24(18_Suppl), 9518–9518 (2006).
- Morrison DK. MAP kinase pathways. Cold Spring Harbor Perspect. Biol. 4(11), (2012).
- Carroll SL. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 123(3), 321–348 (2012).
- Maki RG, D'adamo DR, Keohan ML et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J. Clin. Oncol. 27(19), 3133–3140 (2009).
- Jessen WJ, Miller SJ, Jousma E et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Invest. 123(1), 340–347 (2013).
- Dombi E, Baldwin A, Marcus LJ et al. Activity of selumetinib in neurofibromatosis Type 1-related plexiform neurofibromas. New Engl. J. Med. 375(26), 2550–2560 (2016).
- Endo M, Yamamoto H, Setsu N et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin. Cancer Res. 19(2), 450–461 (2013).
- Johansson G, Mahller YY, Collins MH et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol. Cancer Ther. 7(5), 1237–1245 (2008).
- De Raedt T, Walton Z, Yecies JL et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 20(3), 400–413 (2011).
- Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harbor Perspect. Med. 2(7), a006502 (2012).
- D'adamo DR, Anderson SE, Albritton K et al. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. J. Clin. Oncol. 23(28), 7135–7142 (2005).
- Widemann BC, Meyer CF, Cote GM et al. SARC016: Phase II study of everolimus in combination with bevacizumab in sporadic and neurofibromatosis type 1 (NF1) related refractory malignant peripheral nerve sheath tumors (MPNST). J. Clin. Oncol. 34(15_suppl), 11053–11053 (2016).
- Rahrmann EP, Watson AL, Keng VW et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat. Genet. 45(7), 756–766 (2013).
- Hu T, Li C. Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Mol. Cancer 9, 236 (2010).
- Mo W, Chen J, Patel A et al. CXCR4/CXCL12 mediate autocrine cell- cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell 152(5), 1077–1090 (2013).
- Watson AL, Rahrmann EP, Moriarity BS et al. Canonical Wnt/beta-catenin signaling drives human Schwann cell transformation, progression, and tumor maintenance. Cancer Discov 3(6), 674–689 (2013).
- Lee W, Teckie S, Wiesner T et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 46(11), 1227–1232 (2014).
- De Raedt T, Beert E, Pasmant E et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514(7521), 247–251 (2014).
- Zhang M, Wang Y, Jones S et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Gen. 46(11), 1170–1172 (2014).
- Pasini D, Malatesta M, Jung HR et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 38(15), 4958–4969 (2010).
- Patel AJ, Liao CP, Chen Z, Liu C, Wang Y, Le LQ. BET bromodomain inhibition triggers apoptosis of NF1-associated malignant peripheral nerve sheath tumors through Bim induction. Cell Rep. 6(1), 81–92 (2014).
- Lopez G, Torres K, Liu J et al. Autophagic survival in resistance to histone deacetylase inhibitors: novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 71(1), 185–196 (2011).
- Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat. Rev. Cancer 13(4), 246–257 (2013).
- Jacoby LB, Maccollin M, Barone R, Ramesh V, Gusella JF. Frequency and distribution of NF2 mutations in schwannomas. Genes, Chromosom. Cancer 17(1), 45–55 (1996).
- Gehlhausen JR, Park SJ, Hickox AE et al. A murine model of neurofibromatosis type 2 that accurately phenocopies human Schwannoma formation. Hum. Mol. Genet. 24(1), 1–8 (2015).
- Murakami H, Mizuno T, Taniguchi T et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res. 71(3), 873–883 (2011).
- Fullenkamp CA, Hall SL, Jaber OI et al. TAZ and YAP are frequently activated oncoproteins in sarcomas. Oncotarget 7(21), 30094–30108 (2016).
- Zanconato F, Battilana G, Cordenonsi M, Piccolo S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 29, 26–33 (2016).
- Wu LM, Deng Y, Wang J et al. Programming of Schwann Cells by Lats1/2-TAZ/YAP signaling drives malignant peripheral nerve sheath tumorigenesis. Cancer Cell 33 (2018).
- Liu-Chittenden Y, Huang B, Shim JS et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26(12), 1300–1305 (2012).
- Song S, Honjo S, Jin J et al. The Hippo coactivator YAP1 mediates EGFR overexpression and confers chemoresistance in esophageal cancer. Clin. Cancer Res. 21(11), 2580–2590 (2015).
- Cheng H, Zhang Z, Rodriguez-Barrueco R et al. Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget 7(20), 28976–28988 (2016).
- Huggett MT, Jermyn M, Gillams A et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 110(7), 1698–1704 (2014).
- Jiao S, Wang H, Shi Z et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25(2), 166–180 (2014).
- Song S, Xie M, Scott AW et al. A novel YAP1 inhibitor targets CSC-enriched radiation-resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol. Cancer Ther. 17(2), 443–454 (2018).
- Bhavsar C, Momin M, Khan T, Omri A. Targeting tumor microenvironment to curb chemoresistance via novel drug delivery strategies. Exp. Opin. Drug Deliv. 15(7), 641–663 (2018).
- Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 296(5569), 920–922 (2002).
- Wu J, Williams JP, Rizvi TA et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell 13(2), 105–116 (2008).
- Prada CE, Jousma E, Rizvi TA et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol. 125(1), 159–168 (2013).
- Patwardhan PP, Surriga O, Beckman MJ et al. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin. Cancer Res. 20(12), 3146–3158 (2014).
- Shurell E, Singh AS, Crompton JG et al. Characterizing the immune microenvironment of malignant peripheral nerve sheath tumor by PD-L1 expression and presence of CD8+ tumor infiltrating lymphocytes. Oncotarget 7(39), 64300–64308 (2016).
- Budczies J, Mechtersheimer G, Denkert C et al. PD-L1 (CD274) copy number gain, expression, and immune cell infiltration as candidate predictors for response to immune checkpoint inhibitors in soft-tissue sarcoma. Oncoimmunology 6(3), e1279777 (2017).
- Antoszczyk S, Spyra M, Mautner VF et al. Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro-Oncology 16(8), 1057–1066 (2014).
- Mahller YY, Vaikunth SS, Currier MA et al. Oncolytic HSV and erlotinib inhibit tumor growth and angiogenesis in a novel malignant peripheral nerve sheath tumor xenograft model. Mol. Ther. 15(2), 279–286 (2007).