Abstract
Caveolae are omega-shaped membrane invaginations present in essentially all cell types of the cardiovascular system, including endothelial cells, smooth muscle cells, macrophages, cardiac myocytes, and fibroblasts. Numerous functions have been ascribed to this omega-shaped structure. Caveolae are enriched with different signaling molecules and ion channel regulatory proteins and function both in protein trafficking and signal transduction in these cell types. Caveolins are the structural proteins that are necessary for the formation of caveola membrane domains. Mechanistically, caveolins interact with a variety of downstream signaling molecules, as, for example, Src-family tyrosine kinase, p42/44 mitogen-activated protein (MAP) kinase, and endothelial nitric oxide synthase (eNOS) and hold the signal transducers in the inactive condition until activated with proper stimulus. Caveolae are gradually acquiring increasing attention as cellular organelles contributing to the pathogenesis of several structural and functional processes including cardiac hypertrophy, atherosclerosis, and heart failure. At present, very little is known about the role of caveolae in cardiac function and dysfunction, although recent studies with caveolin knock-out mouse have shown that caveolae and caveolins play a pivotal role in various human pathobiological conditions. This review will discuss the possible role and mechanism of action of caveolae and caveolins in different cardiac diseases.
Key words::
Introduction
The cell membrane is a heterogeneous mixture of proteins, cholesterol, and lipids including glycerolipid, phospholipid, and sphingolipids. The sphingolipids and cholesterol are associated with one another within the plasma membrane and form lipid microdomains, commonly known as lipid rafts (Citation1,Citation2). Lipid rafts are sphingolipid- and cholesterol-rich domains of the plasma membrane, which contain a variety of signaling and transport proteins. Different subtypes of lipid rafts can be distinguished according to their protein and lipid composition. Caveolae, a subset of lipid rafts, are flask-like invaginations of plasma membrane that contain proteins of the caveolin family (caveolin-1, caveolin-2, and caveolin-3). The organization and function of caveolae are mediated by coat proteins (caveolins) and support or adapter proteins (cavins). In addition, lipid rafts have been implicated in the modulation of multiple different types of ion channel proteins. Various types of lipid rafts have been proposed based on different protein markers, morphological features, and their relative cholesterol-to-sphingolipid content (Citation3,Citation4).
Caveolae and caveolins
Caveolae were first identified in 1953 by Palade using electron microscopy to examine the endothelial cells of rat capillaries (Citation5). Caveolae were named based on their morphological appearance on electron microscopy as ‘little caves’. Typically, caveolae exhibit a flask-shaped invaginated structure of 50–100 nm in diameter which is contiguous with the surface plasmalemma (Citation6,Citation7). Different cell types possess different densities of caveolae in their plasma membrane. For example, approximately 50% of the surface plasmalemma of the adipocyte consists of caveolae (Citation8), while only 5% of fibroblast plasma membrane is made up of caveolae (Citation9). A major component of caveolae is the presence of caveolin proteins (Citation10,Citation11). Cav-1 (also called vesicular integral membrane protein (VIP-) 21) was the first protein to be identified as a prominent resident of caveolae (Citation11,Citation12). Cav-1 and -2 are co-expressed in most cell types, while expression of caveolin-3 is muscle-specific. Thus, endothelial cells and fibroblasts are rich in Cav-1 and -2, while cardiac myocytes and skeletal muscle fibers express cav-1 and caveolin-3. Recent work of Chow et al. provided evidence that cav-1 is found in cardiomyocytes (Citation13,Citation14) and that cav-1 and membrane-bound matrix metalloproteinase-2 (MMP-2) co-localize as another means to keep MMP-2 activity in check, and that caveolin-2 knock-out (KO) mice have enhanced cardiac MMP-2 activity (Citation15). The hearts from young cav-1 KO mice do not necessarily show defects in contractile function (Citation16). In contrast, smooth muscle cells express all three caveolins (Cav-1, -2, and -3) (Citation17).
Caveolins are multiple acylated 22–24 kDa proteins embedded in the cytosolic leaflet of cell membranes, with both N and C termini residing in the cytosol (Citation18,Citation19). Caveolins are first inserted into the membrane of the endoplasmic reticulum; they then transit through the secretory pathway, form homo- and hetero-oligomeric complexes in the Golgi apparatus, and are thought to exit the Golgi for delivery to the plasma membrane as assemblies of around 100–200 caveolin molecules (Citation20,Citation21). Cav-1 expression is necessary for Cav-2 to be exported from the Golgi, and for its stability (Citation22,Citation23). Importantly, no morphological caveolae are found in the Golgi (Citation24).
The recent identification of a family of proteins termed cavins has the potential to lead to significant advances in our understanding of the biology of caveolae. Cavins are localized to caveolae and are important for caveolar biogenesis, caveolin expression, caveola morphology, and have differential tissue distributions.
Caveola regulatory proteins
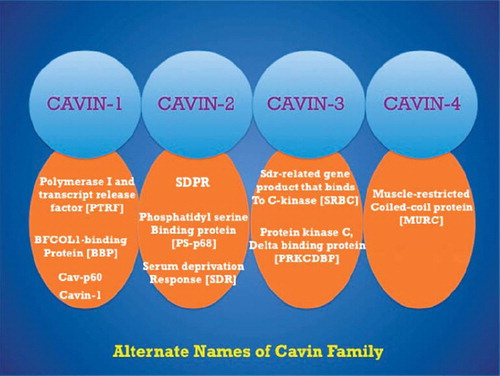
Cavins act a regulator of caveolar function and organization, and each of them has been assigned different roles based on caveola morphologies and cell type. Cavin proteins function primarily as scaffolding proteins and also regulate availability of caveolins. So far, four different cavin proteins have been identified that include cavin-1 (polymerase transcript release factor (PTRF)), cavin-2 (serum deprivation protein response (SDPR)), cavin-3 (Sdr-related gene product that binds to c-kinase (SRBC)), and cavin-4 (muscle-restricted coiled-coil protein (MURC)) ().
Figure 1. Different names of Cavin family protein.
Cavin-1 (PTRF)
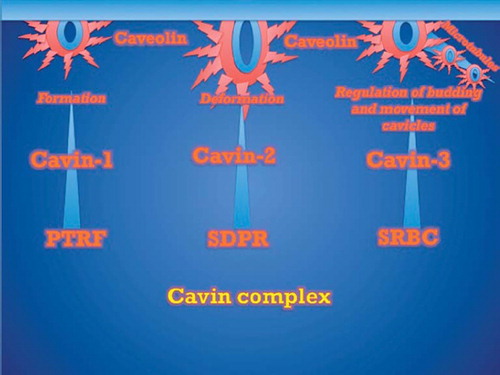
Cavin-1 was initially identified in a yeast two-hybrid screen using transcription termination factor (TTF)-I as the bait (Citation25). TTF-I is involved in the polymerase (Pol) I-mediated transcription of ribosomal RNAs (Citation26,Citation27). Cavin-1 was shown to interact both with TTF-I and Pol I and to function as a trans factor for dissociation of TTF-I-paused Pol I transcription complexes (Citation22,Citation28). The original name for cavin-1 was, therefore, polymerase I and transcript release factor (PTRF) (Citation25). Early studies showed that cavin-1 co-localizes with Cav-1 in adipose tissue and co-distributes with Cav-1 in lipid rafts (Citation29). Regulation of Cav-1 bioavailability by cavin-1 was demonstrated in vitro, as cavin-1 over-expression causes increased levels of Cav-1, and cavin-1 knock-down reduces Cav-1 levels (Citation29). These data are similar to the well appreciated stabilizing effect of Cav-2 with Cav-1 and vice versa (Citation29). In 2008, Liu et al. (Citation30) showed that genetic deletion of cavin-1 resulted in global loss of caveolae through decreased availability of all caveolin proteins, e.g. dyslipidemia, reduced adipose tissue, and glucose intolerance—similar phenotypes to Cav-1/Cav-3 KO mice. In 2008, Hill et al. (Citation31) showed that cavin-1 associates with caveolae at the plasma membrane, where it is required for the formation of caveolae via sequestration of caveolins into caveolae. These authors also demonstrated that the loss of cavin-1 enhances the lateral mobility of Cav-1 and its accelerated lysosomal degradation (Citation27). The function of cavin complexes is shown in .
Figure 2. The function of Cavin complexes.
Cavin-2 (SDPR)
Cavin-2 was first purified as a phosphatidylserine (PS)-binding protein from human platelets and was shown in vitro to be a substrate for protein kinase C (PKC) isoforms (Citation32,Citation33). Mineo et al. also identified a stretch in the middle part of cavin-2 that binds to the regulatory domain of PKC and showed that cavin-2 localizes to caveolae (Citation33). Cavin-2 was also separately identified as a protein with greater expression upon serum deprivation (hence the alternative name serum deprivation protein response (SDPR)) (Citation34,Citation35). In 2009, Hansen et al. (Citation32) showed that cavin-2 directly binds cavin-1 and recruits it to the plasma membrane and that cavin-2 is required for the stable expression levels of both Cav-1 and cavin-1 proteins. Cavin-1/cavin-2 binding results in the formation of complexes containing Cav-1 contributing to stable caveola structures (Citation36). Interestingly, the over-expression of cavin-2 in cultured cells results in the formation of elongated tubular caveolae, implying that it provides an organizational role to generate membrane curvature (Citation36).
Cavin-3 (SRBC)
Cavin-3, or SRBC (Src-related gene product that binds to c-kinase), was initially identified as a PKCδ-binding protein (Citation37,Citation38). In 2009, it was discovered that cavin-3 is localized to caveolae, hence prompting determination of its role in caveolar function. Cavin-3 co-precipitates with Cav-1 and has a similar distribution to Cav-1; either Cav-1 or Cav-3 must be present for cavin-3 localization to the plasma membrane (Citation39). Furthermore, cavin-3 participates in the formation of caveolar vesicles based on two observations: cavin-3 remains associated with caveolae when budding occurs, and the absence of cavin-3 impairs intracellular vesicular/cavicle trafficking (Citation39).
Cavin-4 (MURC)
The most recent addition to the cavin family is cavin-4, MURC (muscle-restricted coiled-coil protein). This cavin was discovered as a cardiac and skeletal muscle-specific cytosolic protein (Citation40–42). In 2009, cavin-4 was characterized as a predominantly muscle-expressed protein component associated with sarcolemmal caveola complexes. On the basis of its expression in muscle and co-localization with Cav-3, it was suggested that cavin-4 plays a predominant role in caveolin-associated muscle disease and disturbs cavin-4 distribution in patients with caveolinopathies (Citation42).
Caveolin knock-down
A vast scientific literature confirmed the roles of caveolae and caveolin in the regulation of many cellular processes in cultured cells, and many investigators considered them as an essential platform of signaling molecules and also as the new therapeutic target. However, in the past few years, development of animal models and usage of genetically altered mice have been instrumental in deciphering their physiological functions in vivo. The most appropriate approach for the study of caveolin is the use of conditional KO mice, tissue-specific KO mice, or a system biology approach. Caveolin KO mice (Cav-1, -2, -3) and Cav-1/3 double KO mice have already been developed. They are viable as well as fertile but display numerous phenotypes. Transgenic over-expression of Cav-1 or Cav-3 in mice or targeted disruption of each of the caveolin gene loci in mice (Cav-1, Cav-2, and Cav-3 genes) has provided significant insight into the roles of caveolin and caveolae (Citation43). The potential role of caveolin in cardiovascular physiology has become apparent by the discovery of Cav-1 and Cav-3 KO mice and double KO mice which have a cardiomyopathic phenotype. Cav-1 KO mice show complete ablation of the presence of the caveolae, cellular organelles, in the endothelium and fat. Similarly, Cav-1 and Cav-3 KO mice lack caveolae in cells that normally express this protein such as skeletal muscle, heart, and diaphragm. Heart tissue is made up of different types of cells. In heart, differentiated cardiomyocytes are surrounded by a network of cardiac fibroblasts and endothelial cells and less abundant vascular smooth muscle cells. There is also a controversy regarding expression of caveolin isoforms in the heart muscle. It is well known that cardiac myocytes express Cav-3, and other cell types in the heart express Cav-1 and Cav-2. But recent studies provided the evidence of the existence of Cav-1 in cardiomyocytes (Citation44). Cav-1 KO mice develop progressive cardiac hypertrophy as demonstrated by transthoracic echocardiography (TTE) and magnetic resonance imaging (MRI) (Citation45). In contrast, Cav-3 KO mice develop cardiomyopathy characterized by hypertrophy, vasodilatation, and reduced contractility as well (Citation46). Cav-1 and Cav-3 double KO mice completely lacking caveolae are deficient in all three caveolin proteins because Cav-2 is degraded in the absence of Cav-1. The double KO mice developed a severe cardiomyopathic phenotype with cardiac hypertrophy and decreased contractility (Citation47). Additionally, Cav-1 KO mice exhibited myocardial hypertrophy, pulmonary hypertension, and alveolar cell hyperproliferation caused by constitutive activation of p42/44 mitogen-activated protein kinase and Akt (Citation48). Interestingly, in Cav-1-reconstituted mice, cardiac hypertrophy and pulmonary hypertension were completely rescued (Citation48). Again, genetic ablation of Cav-1 leads to a striking biventricular hypertrophy and to a sustained eNOS hyperactivation yielding increased systemic NO levels (Citation49). Furthermore, a diminished ATP content and reduced level of cyclic AMP in hearts of KO mice was also reported (Citation49). Taken together, these results indicate that genetic disruption of Cav-1 is sufficient to induce severe biventricular hypertrophy with signs of systolic and diastolic heart failure (Citation49).
Interestingly, Cav-3 KO mice show a number of myopathic changes, consistent with a mild to moderate muscular dystrophy phenotype. However, it remains unknown whether a loss of Cav-3 affects the phenotypic behavior of cardiac myocytes in vivo. Cav-3 KO hearts display significant hypertrophy, dilation, and reduced fractional shortening as revealed by gated cardiac MRI and transthoracic echocardiography. Histological analysis reveals marked cardiac myocyte hypertrophy, with accompanying cellular infiltrates and progressive interstitial/perivascular fibrosis. It has also demonstrated that p42/44 MAPK (ERK1/2) is hyperactivated in heart derived from Cav-3 KO mice, which can lead to cardiac hypertrophy (Citation46) ().
Figure 3. Caveolin and cardiovascular disease.
In the endoplasmic reticulum, Cav-3 initiates the biogenesis of caveola organelles by forming homo-oligomers and hetero-oligomers with Cav-1 (Citation50). At the plasmalemma, Cav-3 interacts with dystrophin and its associated glycoproteins (Citation51,Citation52). Cav-3 and dystrophin competitively bind to the same site of β-dystroglycan, suggesting that Cav-3 may regulate the membrane recruitment of dystrophin and the assembly of the dystrophin glycoprotein complex (DGC) (Citation53). At the cell surface, Cav-3 co-localizes also with signaling molecules such as Gi2α, Gβγ, c-Src, other Src kinases, as well as nitric oxide synthases (neuronal and inducible NOS), indicating that muscle caveolae might be involved in the modulation of these signaling processes (Citation54,Citation55). In addition, Cav-3 localized a glycolytic enzyme in striated muscle and plays a role in the regulation of energy metabolism of muscle cells as it is required for the cell membrane targeting of phosphofructokinase, an enzyme that catalyzes a rate-limiting reaction in glycolysis (Citation56). It is Cav-1 that targets various glycolytic enzymes including phosphofructokinase in smooth muscle, lymphocyte, and astrocyte (Citation56).
In-vitro studies have shown that Cav-3 plays a critical role in myoblast cell differentiation and survival and in myotube formation (Citation57). The relevance of Cav-3 in muscle physiology was further confirmed by the findings that mutations in the CAV3 gene result in distinct neuromuscular and cardiac disorders, such as limb girdle muscular dystrophy (LGMD) 1-C, idiopathic persistent elevation of serum creatine kinase (hyperCKemia), inherited rippling muscle disease (RMD), distal myopathy, and familial hypertrophic cardiomyopathy (HCM) (Citation58–60).
The CAV3 gene (OMIM no. 601253) spans 12 kb of genomic DNA on chromosome 3p25 and contains two exons. At present, 20 different point mutations, 2 base-pair deletions, and 1 novel splice site mutation have been reported (Citation57). More recently, four novel CAV3 mutations have been identified in patients affected by congenital long-QT syndrome (LQTS) in the absence of signs of primary cardiomyopathy, suggesting a possible role for Cav-3 in the regulation of cardiac ion channels (Citation61,Citation62).
Caveolae and cardiac arrhythmia
Modulation of ion channel activity plays a critical role in regulating cardiovascular function. Recently, it has become apparent that the regulation of channel function is not the only means of controlling excitability; the trafficking and localization of ion channels with signaling molecules also play a significant role. The cardiac action potential is generated by the highly orchestrated activity of different of ion channel proteins as well as membrane transporters and exchangers. These transmembrane proteins govern the flux of ions across the sarcolemma of cardiomyocytes generating the ionic currents responsible for excitation. Abnormalities in the function or regulation of the ion channel proteins underlie many different forms of arrhythmias.
Most cells in the cardiovascular system express multiple channel types (e.g. voltage-gated Na+, K+, and Ca2+ channels) and even multiple isoforms of a particular channel, with each channel uniquely contributing to excitability (Citation63,Citation64). Voltage-gated Na+ channels are responsible for the initial depolarization of the cardiac sarcolemma, to permit the opening of voltage-gated L-type Ca2+ channels, resulting in Ca2+ influx and contraction. Membrane repolarization is controlled by K+ channels. Therefore, altering the number of channels and/or their function can have significant impact on both resting membrane potential and the cardiac action potential wave form. Defects in either of these processes can have life-threatening implications (Citation63,Citation64).
Along with the essential scaffolding protein Cav-3, a number of different ion channels and transporters have been localized to caveolae in the heart, including L-type Ca2+ channels (Cav1.2), Na+ channels (Nav1.5), pacemaker channels (HCN4), Na+/Ca2+ exchanger (NCX1), and others. Closely associated with these channels are specific macromolecular signaling complexes that provide highly localized regulation of the channels. Mutations in the Cav-3 gene (CAV3) have been linked with the congenital long QT syndrome (LQTS), and mutations in caveolar localized ion channels may contribute to other inherited arrhythmias. Changes in the caveolar microdomain in acquired heart disease may also lead to dysregulation and dysfunction of ion channels, altering the risk of arrhythmias in conditions such as heart failure (Citation65).
In several cell types, including smooth muscle and endothelial cells, mediators of calcium signaling, such as Ca2+-ATPase, inositol-triphosphate receptor (IP3R), Ca2+ pumps and L-type Ca2+ channels, large conductance Ca2+-activated K+ channel, calmodulin and transient receptor potential (TRP) channels, localize in cholesterol-rich membrane domains. Such localization suggest that membrane rafts and/or caveolae have a role in calcium handling and Ca2+ entry that control excitation-contraction of heart muscle (Citation66,Citation67). TRP channels, in particular TRPC-1, -3, and -4, are enriched in caveolae and Cav-1 and regulate the plasma membrane localization and function of TRP channels (Citation68). Current evidence indicates that caveolae regulate calcium entry, and depletion of cholesterol by methyl-β-cyclodextrin reduces co-localization of Cav-1 and TRPC1, and redistribution of TRPC1, thus preventing Ca2+ influx (Citation69). Moreover, the Na+ pump, Na/K-ATPase, contains two caveolin-binding motifs and resides in caveolae in a number of cells, including smooth muscle cells and cardiomyocytes, thereby helping to maintain the Na+ gradient (Citation70). Voltage-gated K+ channels are also localized in caveolae and play an important role in maintaining cellular excitability. In fibroblast, the Kv 1.5 subunit co-localizes with Cav-1, Kv 2.5 localizes with membrane raft, and depletion of cholesterol with MβCD redistributes and alters the function of K+ channels (Citation71). These findings imply that alteration of caveolae and/or caveolin by any disease or drug treatments can shift the localization of the channels, thereby altering cellular excitability and functional activity.
Caveolae and atherosclerosis
Experimental evidence indicates that caveolae and caveolins have the possibility to influence atherogenesis in many ways. Cav-1 is a cholesterol-binding protein that can transport cholesterol from the endoplasmic reticulum (ER) to the plasma membrane. The major receptors for high-density lipoprotein, SR-B1, and a scavenger receptor for modified forms of LDL, CD36, can also reside in and signal in caveola-type microdomains (Citation72). In addition, oxidized LDL can extract caveola cholesterol, unlocalize eNOS, and impair NO release (Citation73). Conversely, blockade of HMG CoA reductase with statin-based drugs reduces caveolin levels and promotes eNOS activation (Citation74). This concept has been validated in apolipoprotein E-deficient (ApoE−/−) mice where statin treatment decreases Cav-1 expression and promotes NOS function in vivo. However, to date, there are no data showing changes in Cav-1 levels in atherosclerotic lesions from humans (Citation43).
Several lines of evidence now suggest that Cav-1 might play a pro-atherogenic role. In endothelial cells, Cav-1 is up-regulated on LDL exposure (Citation75). Moreover, down-regulation of Cav-1 is associated with reduced uptake of oxidized LDL by endothelial cells (Citation76). This finding is especially important because caveolae are proposed to play a major role in the transcytosis of native and modified LDL.
Cav-1 translocation to the plasma membrane is also enhanced on incubation of endothelial cells with LDL. This movement is accompanied by increased association of Ras with caveolae and results in the activation of Ras, an important upstream activator of the p42/44 MAP kinase pathway (Citation75). Blair et al. (Citation75) have also shown that oxidized LDL can modify the distribution of both Cav-1 and eNOS. This redistribution is accompanied by a reduction in eNOS activation by acetylcholine. This observation might be the result of disruption of the signal transduction complex containing eNOS, Cav-1, and other molecules required for eNOS activation. Recent work by Kincer et al. has shown that CD36, a class B scavenger receptor associated with caveolae, was probably responsible for this effect (Citation77). This observation is important in view of the fact that in hypercholesterolemic patients or animal models impairment of endothelium-derived relaxation is observed (Citation78,Citation79). It is also known that in smooth muscle cells Cav-1 and CD36 interacts and Cav-1 plays a role in the increase in apoptosis and lipotoxicity (Citation80). In agreement with this finding, Feron et al. have shown that exposure of endothelial cells to serum from hypercholesterolemic patients promotes an increase in the Cav-1–eNOS interaction (Citation81).
To verify if Cav-1 influenced lesion progression in mice, Lisanti and his co-workers cross-bred Cav-1−/− mice with ApoE−/− mice that developed atheromas. Interestingly, the loss of Cav-1 in the ApoE−/− mice resulted in a pro-atherogenic lipid profile, similar to that seen in CD36−/− mice bred to an ApoE background (Citation82,Citation83). Surprisingly, despite a pro-atherogenic lipid profile, the loss of Cav-1 reduced the lesion burden by 80%, suggesting Cav-1 regulated LDL-mediated vascular dysfunction, inflammation, and lesion progression. The authors suggested this may be caused by a decrease in stability of the scavenger receptor for oxidized or modified LDL, CD36 in macrophages, and an increase in endothelium-derived NO production, which would reduce vascular inflammation. These remarkable findings unequivocally support the importance of Cav-1/caveolae in the pathogenesis of atherosclerosis (Citation43).
Caveolae and angiogenesis
Angiogenesis, a process of new blood vessel formation, occurs in three clearly distinct phases: initiation, proliferation of vascular cells, and morphogenesis. It has been demonstrated that VEGF stimulates endothelial cell proliferation, induces the expression of proteases and receptors important in cellular invasion and tissue remodeling, modifies endothelial cell permeability by stimulating NO and PGI2 production, and finally prevents endothelial cell apoptosis (Citation84,Citation85). The interaction of VEGF with VEGF receptor-2 (Flk-1/KDR) is required to induce the full spectrum of biological responses involved in angiogenesis (Citation86). It has been recently observed that VEGFR-2 and endothelial NO synthase, activated by VEGF, co-localize with Cav-1 in plasma membrane caveolae of HUVE cells, suggesting the caveolar localization of VEGF signaling machinery in endothelium (Citation87).
It is interesting to note that several important proteins involved in angiogenesis have been localized to caveolae. Some of these macromolecules include the VEGF receptor (VEGFR), the urokinase receptor (uPAR), and eNOS. Recent studies by Labrecque et al. (Citation88) have shown that Cav-1 tonically inhibits VEGFR-2 signaling, but interaction of VEGFR-2 with Cav-1 appears to be required for the proper ligand-induced activation of the receptor within caveolae membranes. Brouet et al. (Citation89) found that eNOS-dependent atorvastatin stabilization of microvascular endothelial cell tube formation was associated with decreased Cav-1 expression, as well as other modifications that enhance eNOS activity. On stimulation of cells with bradykinin, the G protein-coupled bradykinin B2 receptor (B2R) and downstream effectors (Tyk2 and STAT3) are translocated outside the caveolae (Citation90). This finding also has repercussions for eNOS regulation, because B2R can also interact with eNOS and inhibit its activity in a ligand- and calcium-dependent manner (Citation91).
Morphogenesis implicates cell matrix adhesion events dependent on integrins (Citation92) and cell-to-cell adhesion events dependent on interactions between cell surface ligands and receptors, belonging to the family of Eph tyrosine kinase receptors (Citation93). β1-Integrin was found to be associated with Cav-1, and caveolin appears to be a general regulator of β1-integrin function involving Fyn kinase activation (Citation94,Citation95). Moreover, caveolae could function as assembling sites on the cell surface to allow the interaction between ephrin–Eph receptor system and integrins—an event preceding capillary morphogenesis. Data showing that knock-down of Cav-1 disrupts caveolae in endothelial cells and inhibits angiogenesis in vitro and in vivo support a central role of caveolae in angiogenic events (Citation96).
Finally, in support of a role for Cav-1 in the regulation of angiogenesis in vivo, scientists have recently shown, using Matrigel plugs supplemented with basic fibroblast growth factor, that angiogenesis in Cav-1-null (−/−) mice is markedly reduced (Citation97). Similar observations were made regarding tumor angiogenesis that was induced by injecting the B16 melanoma cell line into Cav-1-deficient (−/−) and wild-type animals. In addition, ultrastructural analysis of newly formed capillaries within the exogenous tumors revealed disorganized and incomplete capillary formation in Cav-1-null mice.
Caveolae and hypertrophy
Cardiac hypertrophy is the consequence of an increase in cardiac myocyte size and/or mass. Since cardiac myocytes have no capacity for cellular proliferation, their only means of growth is by cellular enlargement. Given that cardiac failure is the most common result of insufficiency of myocardium, it is not surprising that cardiomyocyte hypertrophy is the dominant cellular response to virtually all forms of hemodynamic overload (Citation98). However, long-term adaptive/compensatory hypertrophy is associated with progressive ventricular dilation. As a consequence of cardiac enlargement and wall thinning, stress on the wall also increases, despite constant intracavitary pressure. This mathematical increase in wall stress generates its own hemodynamic stress on the heart, further stimulating the overloaded hypertrophy signaling pathway and thereby altering the balance from cell growth response to cell death. Once these processes have progressed to this stage (decompensation, loss of cardiac myocytes), irreversible functional deterioration develops, which leads to heart failure and, ultimately, death (Citation99,Citation100).
Over-expression of Cav-3 in neonatal cardiac myocytes decreases the ability of the adrenergic agonist phenylephrine or endothelin-1 to increase cell size (Citation101). A similar kind of effect is seen in cardiac myoblasts (H9C2) in which Cav-3 reduces angiotensin II-promoted hypertrophy (Citation102). Other studies indicate that cardiac hypertrophy results in decreased expression of Cav-3 (Citation103,Citation104) and that right heart (Citation103) and left heart (Citation104) hypertrophy is enhanced in Cav-1 KO and Cav-1/3 double KO mice. Down-regulation of growth signals is the most likely cause of expressed caveolin-induced inhibition of cardiomyocyte growth. Cav-1 and -3 KO mice show hyperactivation of p42/44 MAPK (Citation50) and up-regulation of eNOS activity and nitrosative stress (Citation44,Citation104,Citation50). In contrast, increased caveolin expression down-regulates activity of those entities (Citation102,Citation105). Chronic myocardial hypoxia increases eNOS expression while decreasing the expression of Cav-3, consistent with the idea that the expression and activity of eNOS is dependent on caveolin (Citation106). Alterations in caveolin expression almost certainly change the ability of the hypertrophied heart to respond to a variety of physiologic and pharmacologic agonists/stimuli (Citation44).
Caveolae and ischemic cardiomyopathy
As coronary artery disease is the leading cause of mortality, cardiologists have been attempting for years to identify techniques to minimize the deleterious effects of myocardial ischemia and to diminish the extent of myocardial infarction after coronary occlusion. When the heart is subjected to a transient non-lethal period of ischemia, it quickly adapts itself to become resistant to infarction from a subsequent ischemic insult. This adaptation is called preconditioning (PC). Thus ischemic PC is a protective and adaptive mechanism produced by short periods of ischemic stress, rendering the heart more protective against another similar or greater stress. Although initially it was believed that ‘ischemic PC’ could be induced by short cyclic episodes of ischemia and reperfusion, it soon became apparent that a similar phenotype could be elicited by a splendid array of stimuli. For example, a number of pharmacological agents, agonist of adenosine, bradykinin, adrenergic, muscarinic receptor, nitric oxide (NO) donors, phosphodiesterase inhibitors, and various noxious stimuli (endotoxin, cytokine, reactive oxygen species (ROS), etc.) have all been found to generate a PC-like phenotype (Citation107,Citation108), also known as pharmacological preconditioning.
After the discovery of ‘ischemic PC’ in 1986, the next discovery came in 1993, when it was found that PC has a biphasic pattern: an early phase, which develops very quickly (within few minutes from the exposure to the stimuli) and lasts only 1–2 h, and a late phase, which develops more slowly (needs 6–12 h) but lasts 3–4 days. The early phase develops by rapid post-translational modification of pre-existing proteins through a series of signaling cascades. Protein kinase C (PKC) plays a central role in this signaling cascade, although mitogen-activated protein (MAP) kinase (extracellular kinase, p38 MAP kinase, and c-Jun NH(2)-terminal kinase) is equally involved in PC. The late PC is mediated by cardioprotective gene expression and by the synthesis of new cardioprotective proteins. This mechanism involves redox-sensitive activation of transcription factors through PKC and tyrosine kinase signaling pathways that are in common with the early phase of PC.
Ischemia/reperfusion injury activates p42/44 and p38 MAPK, redistributes Cav-3, and down-regulates expression of Cav-1 (Citation109). Disruption of caveolae using MβCD eliminates the ability of ischemia and pharmacological preconditioning to protect the cardiac myocyte from injury (Citation110). This is supported by the decreased ability of Cav-1 KO mice to undergo pharmacological preconditioning (Citation111). Recent investigations also showed that pro-survival signaling components (e.g. ERK1/2, HO-1, eNOS and p38 MAPKβ) translocate and/or interact with caveolin in the ischemia/reperfusion heart, rendering the heart less susceptible to a pro-survival signal, and induces myocardial injury. Similarly, death signaling components (e.g. p38 MAPKα, JNK, and Src) translocate and/or interact with caveolin in the preconditioned heart, rendering the heart less exposed to death signaling components and more susceptible to pro-survival signaling components (Citation112). Although the detailed mechanism of action of caveolin is not very clear, evidence indicates that proteasomes play a very important role in the interaction between caveolin and signaling components. However, overall observation indicates that caveolin plays a pivotal role in cardioprotection against ischemic injury.
Summary and conclusion
Caveolae and caveolins undoubtedly regulate various aspects of the cardiovascular system. The potential role of caveolin in cardiovascular physiology has become apparent by the discovery of Cav-1 and Cav-3 KO mice and double KO mice which have a cardiomyopathic phenotype. Clearly, loss of Cav-1 has a profound effect on the eNOS pathway, indicating the importance of this interaction, whereas the loss of Cav-3 impacts NOS as well as MAPK activation. Although the detailed mechanisms of actions are not very clear, experimental evidence demonstrates the predominant role of caveolin in cardiac hypertrophy, atherosclerosis, ischemic injury, and different myocardial functions. The most recently discovered proteins, cavins 1–4, are involved in regulation of caveolae and modulate the function of caveolins by promoting membrane remodeling and trafficking. The pathogenic role of caveolins is an emerging area; however, the role of cavins in cardiac disease is just beginning to be explored. Recent investigations are disentangling the complex processes of caveolin and cavin-regulated signaling systems in the myocardium and developing novel approaches, aimed at counteracting heart failure and/or cardiovascular diseases.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Brown CM, Petersen NO. An image correlation analysis of the distribution of clathrin associated adaptor protein (AP-2) at the plasma membrane. J Cell Sci. 1998;111:271–81.
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–72.
- Echarri O, Muriel O, Del Pozo MA. Intracellular trafficking of raft/caveolae domains: insights from integrin signaling. Semin Cell Dev Biol. 2007;18:627–37.
- Hanzal-Bayer MF, Hancock JF. Lipid rafts and membrane traffic. FEBS Lett. 2007;581:2098–104.
- Palade GE. An electron microscope study of the mitochondrial structure. J Histochem Cytochem. 1953;1:188–211.
- Palade GE, Bruns RR. Structural modulations of plasmalemmal vesicles. J Cell Biol. 1968;37:633–49.
- Simionescu M, Simionescu N, Palade GE. Segmental differentiations of cell junctions in the vascular endothelium. The microvasculature. J Cell Biol. 1975;67:863–85.
- Thorn H, Stenkula KG, Karlsson, Ortegren MU, Nystrom FH, Gustavsson J, . Cell surface orifices of caveolae and localization of caveolin to the necks of caveolae in adipocytes. Mol Biol Cell. 2003;14:3967–76.
- Guillot FL, Audus KL, Raub TJ. Fluid-phase endocytosis by primary cultures of bovine brain microvessel endothelial cell monolayers. Microvasc Res. 1990;39:1–14.
- Murata M, Peranen J, Schreiner R, Wieland F, Kurzchalia TV, Simons K. VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci USA. 1995;92:10339–43.
- Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–82.
- Kurzchalia TV, Dupree P, Parton RG, Kellner R, Virta H, . VIP21, a 21-kD membrane protein is an integral component of trans- Golgi-network-derived transport vesicles. J Cell Biol. 1992;118:1003–14.
- Cho WJ, Chow AK, Schulz R, Daniel EE. Matrix metalloproteinase-2, caveolins, focal adhesion kinase and c-Kit in cells of the mouse myocardium. J Cell Mol Med. 2007;11: 1069–86.
- Cho WJ, Chow AK, Schulz R, Daniel EE. Caveolin-1 exists and may function in cardiomyocytes. Can J Physiol Pharmacol. 2010;88:73–6.
- Chow AK, Cena J, El-Yazbi AF, Crawford BD, Holt A, Cho WJ, . Caveolin-1 inhibits matrix metalloproteinase-2 activity in the heart. J Mol Cell Cardiol. 2007;42: 896–901.
- Chow AK, Daniel EE, Schulz R. Cardiac function is not significantly diminished in hearts isolated from young caveolin-1 knockout mice. Am J Physiol. 2010;299: H1183–9.
- Williams TM, Lisanti MP. The caveolin genes: from cell biology to medicine. Ann Med. 2004;36:584–95.
- Monier S, Dietzen DJ, Hastings WR, Lublin DM, Kurzchalia TV, . VIP21-caveolin, a membrane protein constituent of the caveolar coat, oligomerizes in vivo and in vitro. Mol Biol Cell. 1995;6:911–27.
- Tagawa A, Mezzacasa A, Hayer A, Longalti A, Pelkmans L, Helenlus A, . Assembly and trafficking of caveolar domains in the cell: caveolae as stable, cargo-triggered, vesicular transporters. J Cell Biol. 2005;170:769–79.
- Parolini I, Sargiacomo M, Galbiati F, Rizzo G, Grignani F, Engelman JA, . Expression of caveolin-1 is required for the transport of caveolin-2 to the plasma membrane. Retention of caveolin-2 at the level of the Golgi complex. J Biol Chem. 1999;274:25718–25.
- Lee H, Park DS, Razani B, Russell RG, Pestell RG, Lisanti MP, . Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1 (−/−) null mice show mammary epithelial cell hyperplasia. Am J Pathol. 2002;161:1357–69.
- Mogelsvang S, Marsh BJ, Ladinsky MS, Howell KE. Predicting function from structure: 3D structure studies of the mammalian Golgi complex. Traffic. 2004;5:338–45.
- Jansa P, Mason SW, Hoffman RV, Grumnt I, . Cloning and functional characterization of PTRF, a novel protein which induces dissociation of paused ternary transcription complexes. EMBO J. 1998;17:2855–64.
- Leary DJ, Huang S. Regulation of ribosome biogenesis within the nucleolus. FEBS Lett. 2001;509:145–50.
- Grummt I. Regulation of mammalian ribosomal gene transcription by RNA polymerase I. Prog Nucleic Acid Res Mol Biol. 1999;62:109–54.
- Jansa P, Grummt I. Mechanism of transcription termination: PTRF interacts with the largest subunit of RNA polymerase I and dissociates paused transcription complexes from yeast and mouse. Mol Gen Genet. 1999;262: 508–14.
- Liu L, Pilch PF. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem. 2008;283:4314–22.
- Foster LJ, De Hoog CL, Mann M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factor. Proc Natl Acad Sci U S A. 2003;100: 5813–8.
- Scherer PE, Lewis RY, Volonte D, Engelman JA, Galbiati F, Couet J. Cell type and tissue specific expression of cavelin-2. Caveolin-1 and 2 co-localize and form a stable heterooligomeric complex in vivo. J Biol Chem. 1997;272: 29337–46.
- Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH. Deletion of cavin/PTRF causes global loss of caveolae, dyslipidemia and glucose tolerance. Cell Metab. 2008;8:310–7.
- Hill MM, Bastiani M, Luetterforst R, Kirkham M, Nixon SJ. PTRF-cavin, a conserved cystolic protein required for caveolae formation and function. Cell. 2008;132:113–24.
- Burgener R, Wolf M, Ganz T, Baggiolini M. Purification and characterization of a major phosphatidylserine-binding phosphoprotein from human platelets. Biochem J. 1990; 269:729–34.
- Mineo C, Ying YS, Chapline C, Jaken S, Anderson RG, . Targeting of protein kinase C alpha to caveolae. J Cell Biol. 1998;141:601–10.
- Gustincich S, Sand S, Schneider C. Serum deprivation response gene is induced by serum starvation but not by contact inhibition. Cell Growth Differ. 1993;4:753–60.
- Gustincich S, Vatta P, Goruppi S, Wolf M, Saccone S, Della Valle G, . The human serum deprivation response gene (SDPR) maps to 2q32-q33 and codes for a phosphatidylserine-binding protein. Genomics. 1999;57:120–9.
- Hansen CG, Bright NA, Howard G, Nichols BJ. SDPR induces membrane curvature and functions in the formation of caveolae. Nat Cell Biol. 2009;11:807–14.
- Izumi Y, Hirai S, Tamai Y, Fujise-Matsuoka A, Nishimura Y, Ohno S. A protein kinase C delta binding protein SRBC whose expression is induced by serum deprivation. J Biol Chem. 1997;272:7381–9.
- Xu XL, Wu LC, Du F, Davis A, Peyton M, Tomizawa Y. Inactivation of human SRBC, located within the 11p15.5p 15.4 tumor suppressor region, in breast and lung cancer. Cancer Res. 2001;61:7943–9.
- McMahon KA, Zajicek H, Li WP, Peyton MJ, Minna JD, Hernandez VJ. SRBC/cavin-3 is a caveolin adapter protein that regulates caveolae function. EMBO J. 2009;28: 1001–15.
- Ogata T, Ueyama T, Isodono K, Tagawa M, Takehara N, Kawashima T. MURC, a muscle restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol Cell Biol. 2008;28:3424–36.
- Tagawa M, Ueyama T, Ogata T, Takehara N, Nakajima N, Isodono K. MURC, a muscle restricted copiled protein, is involved in the regulation of skeletal myogenesis. Am J Physiol. 2008;295:C490–8.
- Bastiani M, Liu L, Hill MM, Jedrychowski MP, Nixon SJ, Lo HP. MURC/cavin-4 and cavin family members form tissue specific caveolar complex. J Cell Biol. 2009;185: 1259–73.
- Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circ Res. 2004;94:1408–17.
- Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–91.
- Cohen AW, Park DS, Woodman SE, Williams TM, Chandra M, Shirani J, . Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003;284: C457–74.
- Woodman SE, Park DS, Cohen AW, Cheung MW, Chandra M, Shirani J. Caveolin-3 knockout mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem. 2002;277: 38988–97.
- Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M. Caveolin 1/3 double knockout mice are viable but lack both muscle and non-muscle caveolae and develop a severe cardiomyopathic phenotype. Am J Pathol. 2002; 160:2207–17.
- Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, . Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med. 2007;204: 2373–82.
- Wunderlich C, Schober K, Lange SA, Drab M, Braun-dullaens RC, Kesper P, . Disruption of caveolin-1 leads to enhanced nitrosative stress and severe systolic and diastolic heart failure. Biochem Biophys Res Commun. 2006;340:702–8.
- Capozza F, Cohen AW, Cheung MW, Sotgia F, Fchupert W, Battista M, . Muscle-specific interaction of caveolin isoforms: differential complex formation between caveolins in fibroblastic vs. muscle cells. Am J Physiol Cell Physiol. 2005;288:C677–91.
- Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, . Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996; 271:15160–5.
- Crosbie RH, Yamada H, Venzke DP, Lisanti MP, Campbell KP, . Caveolin-3 is not an integral component of the dystrophin glycoprotein complex. FEBS Lett. 1998; 427:279–82.
- Sotgia F, Lee JK, Das K, Bedford M, Petrucci TC, Macioee P, . Caveolin-3 directly interacts with the C-terminal tail of beta-dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem. 2000;275:38048–58.
- Garcia-Cardena G, Fan R, Stern DF, Liu J, Sessa WC. Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J Biol Chem. 1996;271:27237–40.
- Smythe GM, Eby JC, Disatnik MH, Rando T. A caveolin-3 mutant that causes limb girdle muscular dystrophy type 1C disrupts Src localization and activity and induces apoptosis in skeletal myotubes. J Cell Sci. 2003;116:4739–49.
- Sotgia F, Bonuccelli G, Minetti C, Woodman SE, Capozza T, Kemp RG, . Phosphofructokinase muscle-specific isoform requires caveolin-3 expression for plasma membrane recruitment and caveolar targeting: implications for the pathogenesis of caveolin-related muscle diseases. Am J Pathol. 2003;163:2619–34.
- Galbiati F, Volonte D, Engelman JA, Scherer PE, Lisanti MP. Targeted down-regulation of caveolin-3 is sufficient to inhibit myotube formation in differentiating C2C12 myoblasts. Transient activation of p38 mitogen-activated protein kinase is required for induction of caveolin-3 expression and subsequent myotube formation. J Biol Chem. 1999;274: 30315–21.
- Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M, . Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18:365–8.
- Carbone I, Bruno C, Sotgia F, Bado M, Broda P, Masetti E, . Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology. 2000;54: 1373–6.
- Galbiati F, Razani B, Lisanti MP. Caveolae and caveolin-3 in muscular dystrophy. Trends Mol Med. 2001;7:435–41.
- Fulizio L, Nascimbeni AC, Fanin M, Piluso G, Polinato L, Nigro V, . Molecular and muscle pathology in a series of caveolinopathy patients. Hum Mutat. 2005;25:82–9.
- Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, . Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–12.
- O'Connell KM, Martens JR, Tamkun MM. Localization of ion channels to lipid Raft domains within the cardiovascular system. Trends Cardiovasc Med. 2004;14:37–42.
- Maguy A, Hebert TE, Nattel S. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc Res. 2006;69:798–807.
- Wang XL, Ye D, Peterson TE, Cao S, Shah VH, Katusic ZS, . Caveolae targeting and regulation of large conductance Ca(2+)-activated K+ channels in vascular endothelial cells. J Biol Chem. 2005;280:11656–64.
- Fujimoto T, Miyawaki A, Mikoshiba K. Inositol 1,4,5-trisphosphate receptor-like protein in plasmalemma caveolae is linked to actin filaments. J Cell Sci. 1995;108(Pt. 1): 7–15.
- Lohn M, Furstenau M, Sagach V, Elger M, Schulze W, Luft FC, . Ignition of calcium sparks in arterial and cardiac muscle through caveolae. Circ Res. 2000;87:1034–9.
- Kwiatek AM, Minshall RD, Cool DR, Skidgel RA, Malik AB, Tiruppathi C. Caveolin-1 regulates store-operated Ca2+ influx by binding of its scaffolding domain to transient receptor potential channel-1 in endothelial cells. Mol Pharmacol. 2006;70:1174–83.
- Bergdahl A, Gomez MF, Dreja K, Gomez MF, Dreja K, Xu SE, . Cholesterol depletion impairs vascular reactivity to endothelin-1 by reducing store-operated Ca2+ entry dependent on TRPC1. Circ Res. 2003;93: 839–47.
- Fagan KA, Smith KE, Cooper DM. Regulation of the Ca2+-inhibitable adenylyl cyclase type VI by capacitative Ca2+entry requires localization in cholesterol-rich domains. J Biol Chem. 2000;275:26530–7.
- Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1.5 to caveolae. J Biol Chem. 2001;276:8409–14.
- Graf GA, Matveev SV, Smart EJ. Class B scavenger receptors, caveolae and cholesterol homeostasis. Trends Cardiovasc Med. 1999;9:221–5.
- Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J Biol Chem. 1999;274: 32512–9.
- Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–8.
- Pelat M, Dessy C, Massion P, Desager JP, Feron O, Balligand JL. Rosuvastatin decreases caveolin-1 and improves nitric oxide-dependent heart rate and blood pressure variability in apolipoprotein E-/- mice in vivo. Circulation. 2003;107:2480–6.
- Zhu Y, Liao HL, Wang N, Yuan Y, Ma KS, Verna L, . Lipoprotein promotes caveolin-1 and Ras translocation to caveolae: role of cholesterol in endothelial signaling. Arterioscler Thromb Vasc Biol. 2000;20:2465–70.
- Kincer JF, Uittenbogaard A, Dressman J, Guerin TM, Febbraio M, Guo L, . Hypercholesterolemia promotes a CD36-dependent and endothelial nitric-oxide synthase mediated vascular dysfunction. J Biol Chem. 2002;277: 23525–33.
- Seiler C, Hess OM, Buechi M, Suter TM, Krayenbuehl HP. Influence of serum cholesterol and other coronary risk factors on vasomotion of angiographically normal coronary arteries. Circulation. 1993;88:2139–48.
- Reddy KG, Nair RN, Sheehan HM, Hodgson JM. Evidence that selective endothelial dysfunction may occur in the absence of angiographic or ultrasound atherosclerosis in patients with risk factors for atherosclerosis. J Am Coll Cardiol. 1994;23:833–43.
- Dorahy DJ, Lincz LF, Meldrum CJ, Burns GF. Biochemical isolation of a membrane microdomain from resting platelets highly enriched in the plasma membrane glycoprotein CD36. Biochem J. 1996;319:67–72.
- Feron O, Dessy C, Moniotte S, Desager JP, Balligand JL. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J Clin Invest. 1999;103:897–905.
- Febbraio M, Podrez EA, Smith JD, Schmitt D, Silverstein RL, Hazen SL, . Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105: 1049–56.
- Podrez EA, Febbraio M, Sheibani N, Shelbani N, Hoff HF. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. 2000;105:1095–108.
- Ziche M, Morbidelli L, Choudhur RI, Zhang HT, Donnini S, Granger HJ, . Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest. 1997;99:2625–34.
- Gupta K, Kshirsagar S, Li W, Gui L, Ramakrishnan S, Gupta SP, . VEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signaling. Exp Cell Res. 1999;247: 495–504.
- Petrova TV, Makinen T, Alitalo K. Signaling via vascular endothelial growth factor receptors. Exp Cell Res. 1999; 253:117–30.
- Feng Y, Venema VJ, Venema RC, Tsai N, Behzadian MA, Caldwell RB. VEGF-induced permeability increase is mediated by caveolae. Invest Ophthalmol Vis Sci. 1999; 40:157–67.
- Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. 2003;14: 334–47.
- Brouet A, Sonveaux P, Dessy C, Moniotte S, Balligand JL, Feron O. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ Res. 2001;89:866–73.
- Ju H, Venema VJ, Liang H, Harris MB, Zou R, Venema RC. Bradykinin activates the Janus-activated kinase/signal transducers and activators of transcription (JAK/STAT) pathway in vascular endothelial cells: localization of JAK/STAT signalling proteins in plasmalemmal caveolae. Biochem J. 2000;351:257–64.
- Ju H, Venema VJ, Marrero MB, Venema RC. Inhibitory interactions of the bradykinin B2 receptor with endothelial nitric-oxide synthase. J Biol Chem. 1998;273:24025–9.
- Aplin AE, Howe AK, Juliano RL. Cell adhesion molecules, signal transduction and cell growth. Curr Opin Cell Biol. 1999;11:737–44.
- Stein E, Lane AA, Cerretti DP, Schoecklmann HO, Schroff AD, Van Etten RL, . Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev. 1998;12:667–78.
- Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998;94:625–34.
- Wei Y, Yang X, Liu Q, Wilkins JA, Chapman HA. A role for caveolin and the urokinase receptor in integrin-mediated adhesion and signaling. J Cell Biol. 1999;144:1285–94.
- Griffoni C, Spisni E, Santi S, Riccio M, Guarnieri T, Tomasi V. Knockdown of caveolin-1 by antisense oligonucleotides impairs angiogenesis in vitro and in vivo. Biochem Biophys Res Commun. 2000;276:756–61.
- Woodman SE, Ashton AW, Schubert W, Lee H, Williams T, Medina FA, . Caveolin-1 knock-out mice show an impaired angiogenic response to exogenous stimuli. Am J Pathol. 2003;162:2059–68.
- Diwan A, Dorn GW. Decompensation of cardiac hypertrophy: cellular mechanism and novel therapeutic target. Physiology. 2007;22:56–64.
- Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, . Cardiac hypertrophy is not a required compensatory response to short term pressure overload. Circulation. 2000;101:2863–9.
- Sano M, Schneider MD. Still stressed out but doing fine: normalization of wall stress is superfluous to maintain cardiac function in chronic pressure overload. Circulation. 2002;105:8–10.
- Koga A, Oka N, Kikuchi T, Miyazaki H, Kato S, Imaizumi T. Adenovirus-mediated overexpression of caveolin-3 inhibits rat cardiomyocyte hypertrophy. Hypertension. 2003;42:213–9.
- Fujita T, Otsu K, Oshikawa J, Hori H, Kitamura H, Ito T, . Caveolin-3 inhibits growth signal in cardiac myoblasts in a Ca2+-dependent manner. J Cell Mol Med. 2006; 10:216–24.
- De Souza AP, Cohen AW, Park DS, Woodman SE, Tang B, Gutstein DE, . MR imaging of caveolin gene-specific alterations in right ventricular wall thickness. Magn Reson Imaging. 2005;23:61–8.
- Ohsawa Y, Toko H, Katsura M, Morimoto K, Yamada H, Ichikawa Y, . Overexpression of P104L mutant caveolin-3 in mice develops hypertrophic cardiomyopathy with enhanced contractility in association with increased endothelial nitric oxide synthase activity. Hum Mol Genet. 2004;13:151–7.
- Feron O, Balligand JL. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc Res. 2006;69:788–97.
- Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, . Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272:25437–40.
- Bolli R. The late phase of preconditioning. Circ Res. 2000; 87:972–83.
- Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1131–51.
- Ballard-Croft C, Locklar AC, Kristo G, Lasley RD. Regional myocardial ischemia-induced activation of MAPKs is associated with subcellular redistribution of caveolin and cholesterol. Am J Physiol Heart Circ Physiol. 2006;291: H658–67.
- Das M, Gherghiceanu M, Lekli I, Mukherjee S, Popescu LM, Das DK. Essential role of lipid raft in ischemic preconditioning. Cell Physiol Biochem. 2008;21:325–34.
- Patel HH, Tsutsumi YM, Head BP, Niesman IR, Jennings M. Mechanisms of cardiac protection from ischemia/reperfusion injury: a role for caveolae and caveolin-1. FASEB J. 2007;21:1565–74.
- Das M, Cui J, Das DK. Generation of survival signal by differential interaction of p38MAPKalpha and p38MAPKbeta with caveolin-1 and caveolin-3 in the adapted heart. J Mol Cell Cardiol. 2007;42:206–13.