
Abstract
The constantly evolving science of risk assessment is currently faced with many challenges, not only from the interpretation of the volume of data being generated with new innovative technologies, but also in attempting to quantitatively incorporate this information into understanding potential risk of adverse events in human populations. The objective of the case study described was to use the more recent data for di-(2-ethylhexyl)phthalate (DEHP) to investigate the impact of innovative quantitative approaches on the risk assessment of a compound, specifically as it can be used to move towards the new vision of risk assessment involving the integration of the available toxicological data to understand underlying biological processes. What emerged were several outcomes that demonstrated clearly the importance of the integration of the toxicological data, specifically to understand the biological processes being impacted, because standard statistical modeling approaches may not be adequate to describe the dose–response relationships observed. Alternative approaches demonstrate that a definitive mode of action is not needed to justify the shape of the low-dose region or a threshold, when the integration of the available data assist risk assessors in understanding the shape of the dose–response curve for both noncancer and cancer endpoints. Many of the challenges described as part of this case study would likely be encountered with compounds other than DEHP, especially other receptor-mediated compounds or compounds that “perturb” biological pathways, such as endocrine disruptors. This case study also highlights the importance of communication between risk assessors and the research community to focus on the generation of data most relevant for assessing the potential for chemicals to impact biological systems in the human.
Appendix A
Continuous Models
All the continuous models were run using a BMR of one standard deviation.
Hill model
Parameters:
μ(d) is the mean value of the response at dose d,
γ is the background response value,
k is the slope,
n is the power (restricted to >= 1), and
ν indicates the sign or direction of the change in response with increased dose.
Linear Model
Parameters:
μ(d) is the mean value of the response at dose d,
β0 is the background response value, and
β1 is the linear coefficient or slope.
Polynomial Model
Parameters:
μ(d) is the mean value of the response at dose d,
β0 is the background response value,
β1…βk are the polynomial coefficients, and
k is the polynomial degree.
Note that the coefficients are for the polynomial are restricted to all non-negative or all non-positive (depending on the direction of change of the response with increasing dose) to prevent wavy dose response curves.
Power Model
Parameters:
μ(d) is the mean value of the response at dose d,
γ is the background response value, and
β is the slope, and
α is the power term (restricted to > = 1 to prevent supralinear curves where the dose–response curve has an infinite slope as the dose approaches zero).
Dichotomous (Quantal) Models
All dichotomous models were run using a BMR of 0.1 (10%) extra risk.
Gamma Model
Parameters:
P(d) is the probability of the response occurring at dose d,
γ is the background probability value,
β is the slope, and
α is the power term (restricted to >= 1).
Logistic model
Parameters:
P(d) is the probability of the response occurring at dose d,
γ is the background probability value, and
β is the slope.
Logistic model
Parameters:
P(d) is the probability of the response occurring at dose d,
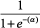 is the background probability value, and
is the background probability value, andβ is the slope.
Log-Logistic model
Parameters:
P(d) is the probability of the response occurring at dose d,
γ is the background probability value, and
β is the slope.
Multistage Model
Parameters:
P(d) is the probability of the response occurring at dose d,
γ is the background response value,
β1…βk are the polynomial coefficients (restricted to be non-negative), and
k is the polynomial degree.
Probit model
Parameters:
P(d) is the probability of the response occurring at dose d,
Φ is the normal distribution function,
Φ (α) is the background probability value, and
β is the slope.
Log-Probit model
Parameters:
P(d) is the probability of the response occurring at dose d,
Φ is the normal distribution function,
γ is the background probability value,
α is the intercept, and
β is the slope (restricted to >= 1).
Weibull Model
Parameters:
P(d) is the probability of the response occurring at dose d,
γ is the background response value,
β is the slope, and α is the power term (restricted to > = 1).
Appendix B: Model Validation and Parameterization of the Rat Gestation and Lactational Model
The final DEHP model contains four inter-connected sub-models, each with the necessary amount of detail to adequately describe the chemical species: diester, monoester, monoester-glucuronide, and the combined oxidative metabolites. The individual sub-models interact at sites of metabolism (hydrolysis of the diester, glucuronidation, hydrolysis of the glucuronide, and oxidation). The models for each chemical species in the adult rat are described below, followed by the modifications made to describe gestation and lactation.
Intact diester. Enzymes responsible for the hydrolysis of the diesters are present in the intestinal mucosa, blood and liver (Rowland et al., Citation1977; Tanaka et al., Citation1978). Hydrolysis in the blood and liver are described as a first order rates as none of the tested doses were sufficient to overwhelm hydrolysis. Hydrolysis in the upper GI (stomach + small intestine; GC1), on the other hand, is described as a saturable process based on the in vitro data of Rowland et al. (Citation1977) and the apparent saturation of oral uptake at the highest doses (>500 mg/kg) (Kessler et al., Citation2004). Some diester may also enter circulation intact via oral absorption or be passed into the lower intestine (GC2), where it is excreted in the feces. Oral absorption is described as a first order process. Movement through the intestine and fecal excretion are described as clearance rates (L/hr). Diester that is taken up into the gut wall is passed to the liver via the portal blood where it is hydrolyzed, released into systemic circulation or excreted into the bile. Biliary diester is excreted into the upper intestine.
Free monoester. Oral absorption of the monoesters is described as a first order process. Movement through the GI and fecal excretion are clearance rates. Unlike the diesters, their monoester metabolites MEHP and MBP are readily absorbed in the gut. Glucuronidation and oxidation of the monoester in the liver are described using saturable kinetics. The free monoesters may be excreted into the bile (recirculated to small intestine) or released into systemic circulation. Transport of monoester into the tissues is modeled using diffusion-limitation. Secretion into the urine is a saturable process, based on non-linear behavior of MBP excretion data at low doses (Payan et al., Citation2001).
Monoester-glucuronide conjugates. Because MEHP is not glucuronidated in vivo, this section of the model only applies to DBP.
Oxidative Metabolites. Metabolites of MEHP and MBP formed by P450 metabolism in the liver are released into the body via the venous blood. In the original DBP model, a one-compartment volume of distribution model was used to describe the combined oxidative metabolites. This description was sufficient for DBP, due the fact the majority of the dose exists as free MBP or MBP-G in the rat. However, in the case of DEHP, the metabolite profile is quite different. In fact, the majority of the dose (>90%) undergoes oxidation, free MEHP is only a minor metabolite, and the glucuronide conjugate does not exist at detectable levels. Thus the description of the oxidative metabolites was expanded to better describe DEHP kinetics.
Oxidative metabolism is described in the liver using a saturable Michealis-Menten description. The oxidized monoesters are then excreted into the bile or released into systemic circulation. Biliary metabolites are released into the upper intestine (GC1), where they may be reabsorbed (described as a first order rate) or passed in the feces (described as a first order clearance rate). A three compartment model is used to describe the oxidative metabolites in the blood, liver and other tissues. Distribution of into the tissues is modeled using flow-limitation, assuming distribution with body water. Urinary excretion is modeled using a first order clearance rate from the plasma compartment.
Gestation Kinetics
During gestation, all of the chemical species are allowed to move freely between the arterial and placental blood. However, only the free monoester was allowed to cross the placenta based on the previous DBP model and metabolite data in the fetal rat. While MBP-G was found in the fetal blood, the kinetic behavior suggests that it is formed in the fetus rather than maternally. Transfer of MEHP and MBP between the placental blood and the fetal blood are described as diffusion-limited processes (Gentry et al., Citation2003). Based on fetal MBP kinetic data, as well as published data on UDPGT and β-glucuronidase activities in fetal tissues (Lucier and McDaniel, Citation1977; Lucier et al., Citation1975; Wishart, Citation1978), glucuronidation and hydrolysis of the glucuronide conjugates were included in both dam and fetus. P450 activity, however, is negligible in the fetal liver (Neale and Parke, Citation1973) and was therefore excluded from the fetal model. Metabolite transfer between the fetus and amniotic fluid were described as first-order processes. Transfer between the fetal plasma and testes tissue is described using flow-limited transport. Postnatal Kinetics
The structure of neonatal model is identical to that of the adult non-pregnant rat. In vitro data suggests that the enzymes responsible for both glucuronidation and oxidative metabolism are present and increasing in concentration during the postnatal period. Thus, both metabolic processes were included in the neonatal model. Uptake of the diesters and monoesters are described as flow-limited. Transfer in the milk is described using first order clearance rates based on the milk concentrations and published suckling rates (Stolc et al., Citation1966).
Model Parameterization:
Physiological Parameters:
Physiological parameters were obtained from measured values in the literature as described in Clewell et al. (Citation2008). Adult male rat, body weight, cardiac output, and fractional tissue volumes and blood flows were available from Brown et al. (Citation1997). Fractional tissue volumes were scaled by BW and blood flows were scaled by BW0.75.
Gestation. During gestation, mammary gland (VM) and fat (VF) tissue growth were described as a linear processes based on the data of Hanwell and Linzell, (1973) and Andrade et al. (Citation2006b), Knight and Peaker (Citation1982), and Naismith et al., (Citation1982), as described in Clewell et al. (Citation2003). Placental volume (VPl) was described as the sum of the yolk sac and chorioallantoic placenta based on the model of O’Flaherty et al. (Citation1992). Growth equations were available in the cited papers. The total body weight of the dam was made equal to the initial body weight plus the change in volume of the uterus, fat, mammary gland, placenta, and fetus. Fetal volume (Vfet) was described using the equations of O’Flaherty et al. (Citation1992). Growth of fetal testes is proportional to the total body weight, accounting for approximately 0.1% of the total fetal volume from GD16 through the end of gestation (LaBorde et al., Citation1992; Naessany and Picon, Citation1982; Parks et al., Citation2000). Changes in amniotic fluid volume were described using a Table function in the simulation software, by linear interpolation between data points (Park and Shepard, Citation1994; Wykoff, Citation1971).
Maternal cardiac output was described as the sum of initial cardiac output (Brown et al., Citation1997) and the change in blood flow to the placenta, mammary and fat tissues, per the approach of O’Flaherty et al. (Citation1992). Changes in the fractional cardiac output to the mammary gland, fat and yolk sac were assumed to be proportional to changes in tissue volumes, with the exception of the chorioallantoic placenta which increased more rapidly than the tissue volume. Chemical transport within the fetus was modeled using diffusion, rather than blood-flow limitation. Thus, no assumptions were made as to proportional blood flows to fetal tissues.
Lactation. The physiological description of maternal and neonatal rats during lactation is based on the work of Clewell et al. (Citation2003). Maternal body weight increases by 12% between PND 1 and 10 (Clewell et al., Citation2003). The relative volume of the mammary tissue increased from 4.4% on PND 2 to 5.6, 6.3 and 6.6% of the maternal body weight on PND 7, 14 and 21, respectively (Knight et al., Citation1984). Maternal body fat increased from 12.4 to 15.2% of the body weight between parturition and PND 2, with a subsequent decrease to 6.9% of the body weight from PND 2 to 16 (Naismith et al., Citation1982). The rate of milk production was assumed to be equal to the suckling rate in Stolc et al. (Citation1966). Values for neonatal body fat increase from 2.7 to 11% BW between PND 2 and 16 and a subsequent decrease to the adult value of 4.61(Brown et al., Citation1997; Naismith et al., Citation1982). Changes in neonatal body weight and relative tissue volumes have been described previously (Clewell et al., Citation2003).
Changing maternal cardiac output and fractional blood flow to the mammary tissue throughout lactation are described according to the data of Hanwell and Linzell (Citation1973). Neonatal cardiac output, hematocrit and regional blood flows are based on the data of Rakusan and Marcinek (Citation1973).
Kinetic Parameters:
Kinetic parameters were scaled allometrically as is typical for intra- and inter-species extrapolation (Dedrick, Citation1973). PA, Vmax, and clearance constants were scaled by BW0.75. Whenever possible parameters were taken from published values or calculated from in vitro studies. However, the lack of specific tissue and metabolism data required that some model parameters be fitted to in vivo kinetic data. A detailed description of the process used to determine the original DBP model parameters is available elsewhere (Clewell et al., Citation2008).
While an attempt was made to keep the model parameters as similar as possible between the DEHP and DBP models, it was necessary to refine a number of the kinetic parameters in order to recapitulate the DEHP kinetic data. In fact, when the DBP model was run “as is” against the MEHP iv and DEHP po data, the model over-predicted serum MEHP levels by an order of magnitude (not shown). Several observations can be made about the kinetic differences in the two phthalates based on available in vitro and in vivo data. 1) DEHP is not hydrolyzed as efficiently as DBP in the gut (Rowland et al., Citation1977), which leads to a greater loss of unmetabolized diester in the feces as well as some circulation of DEHP in the blood. 2) Both DEHP and MEHP also are more poorly absorbed in the gut than DBP and MBP. 3) The high lipophilicity of DEHP and the reduced hydrolysis in the blood (when compared to DBP) also results in reduced clearance of the diester from the blood after iv dosing. Iv doses of DBP, on the other hand are metabolized within minutes of dosing. 4) Metabolism of the monoester is also quite different for the two phthalates. A large portion of MBP is removed via glucuronidation and oxidation is only important at low doses (Payan et al., Citation2001). In contrast, oxidation, not glucuronidation, is the dominant metabolic pathway for MEHP. Oxidative metabolites make up >90% of the total urinary metabolites, while glucuronide conjugates of MEHP have not been detected in the urine or plasma of rats after DEHP dosing (Albro and Moore, Citation1974).
Based on these observations, UGT activity (VmaxLc) was turned off in the DEHP model. The Vmax for oxidative metabolism (VmaxOc = 28 mg/hr-kg BW) was adjusted by visually fitting the model simulations to MEHP iv data in the adult male rat (Pollack et al., Citation1985). The rate of oral absorption of MEHP (kam = 0.4 hr−1) was adjusted based on the fit of the model to MEHP oral gavage studies (Teirlynck and Belpaire, Citation1985). The maximum capacity for hydrolysis in the gut (VmaxGc = 80 mg/hr-kg BW), oral absorption of DEHP (kad = 0.015 hr−1), and intestinal clearance of DEHP (kgic1 = 0.1L /hr) and the other metabolites (kgic2 = 0.05 L/hr), and the urinary excretion rate of MEHP (VmxUc = 1 mg/hr-kg BW, KmU = 450 mg/L) were fit to DEHP oral gavage data (blood, feces, urine) in the non-pregnant female rat (Kessler et al., Citation2004). With the exception of those parameters listed above, the kinetic parameters were identical to those used in the DBP model and are available elsewhere (Clewell et al., Citation2008). Final model simulations are shown versus various data sets after iv and po doses of MEHP or DEHP in the non-pregnant rat in Figures B1− B3.
Figure B1. Free MEHP in the blood of the adult male rat after an iv dose of 20 or 50 mg/kg MEHP. Lines indicate model simulations. Points represent measurements from individual animals administered (•) 20 mg/kg MEHP (Sjoberg et al., Citation1985), (▾) 50 mg/kg MEHP (Pollack et al., Citation1985), or (Δ) 50 mg/kg MEHP (Kessler et al., Citation2004).
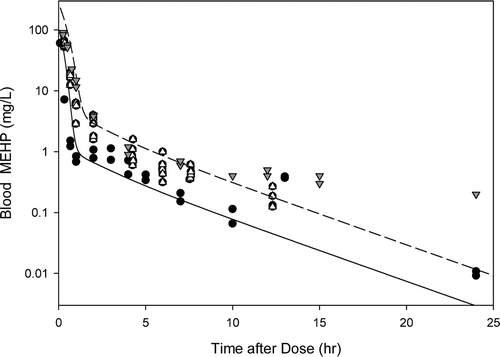
Figure B2. Free MEHP in the (A) blood of adult male rat after an oral dose of 70, 100, or 400 mg/kg MEHP or (B) excreta of adult male rat after an oral dose of 70 mg/kg MEHP. (A) Lines indicate model simulations. Points represent measurements from individual animals administered (o) 70 mg/kg MEHP (Chu et al., Citation1978), (▪) 100 mg/kg MEHP (Pollack et al., Citation1985), or (•) 400 mg/kg MEHP (Teirlynck and Belpaire, Citation1985). (B) Bars represent model simulations (gray) or mean + SD of measured (black) MEHP in the urine and feces rats administered 70 mg/kg MEHP (Chu et al., Citation1978).
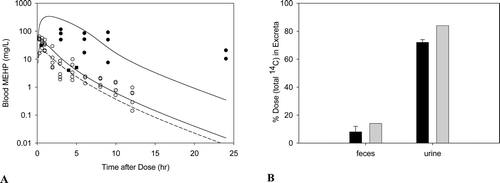
Figure B3. Free MEHP in the blood of adult rats after an (A)iv dose of 100 mg/kg DEHP, or (B) oral dose of 30, 300, or 500 mg/kg DEHP. Line indicates model simulation. Points represent measurements from individual animals administered (A) 100 mg/kg DEHP iv (Kessler et al., Citation2004) or (B) (o) 30 mg/kg, (Δ) 300 mg/kg, or (•) 500 mg/kg DEHP po (Kessler et al., Citation2004).
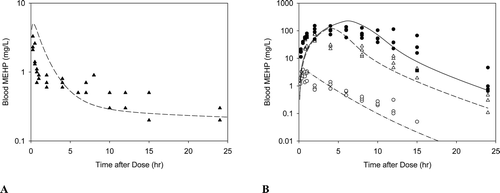
Gestation. Similar to DBP (Clewell et al., Citation2008), only one kinetic parameter was adjusted before using the model in the pregnant dam based on published in vitro studies: VmaxOc. Because glucuronidation is set to zero in the DEHP model, it was not necessary to adjust that parameter for the reduced capacity for glucuronide conjugation during gestation (Lucier et al., Citation1975; Luquita et al., Citation2001). However, similar studies with microsomes obtained from the liver of pregnant and non-pregnant rats showed approximately 40% reduction in the capacity of oxidative metabolism in the pregnant rat liver regardless of the substrate used (Neale and Parke, Citation1973). Thus, the maximum capacity for oxidative metabolism of MEHP (VmaxOc) was reduced by 40% when applied to the pregnant rat. All other maternal parameters were scaled allometrically from the adult male rat. Since MEHP is not significantly glucuronidated and oxidative metabolism is negligible in the rat fetus, both VmaxGcf and VmaxOcf were set to zero in the fetus. Transfer of MEHP between the maternal and fetal blood, and between the fetal blood and amniotic fluid were assumed to be the same as MBP. Model simulations are shown with data collected in the pregnant and fetal rat after oral administration of DEHP (Figure B4).
Figure B4. Free MEHP in the (A) maternal blood or (B) placenta and fetal tissues after the last oral dose of 30 or 500 mg/kg/day administered from GD 14 - 19. (A) Lines indicate model simulations. Points represent measurements from individual animals administered (•) 30 mg/kg/day or (o) 500 mg/kg/day DEHP po (Kessler et al., Citation2004). (B) Bars represent model simulations (black) or mean + SD of measured concentrations (gray) in the placenta and fetal tissues 2 hrs after the last daily dose of 500 mg/kg/day (Kessler et al., Citation2004).
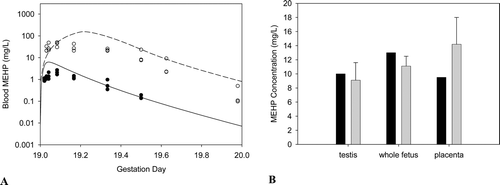
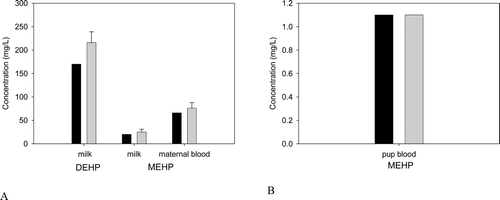
Figure B5. (A) DEHP and MEHP in the milk and MEHP in the blood of the lactating dam, and (B) MEHP in the suckling rat after the last oral dose of 2000 mg/kg/day administered on days 15− 17 of lactation. Bars represent model simulations (black) or mean + SD of measured concentrations (gray) in the milk, maternal blood and pup blood 3-6 hrs after the last dose of 2000 mg/kg/day DEHP (Dostal et al., Citation1987).
Lactation. During lactation, both DEHP and MEHP were assumed to move freely between the blood and the mammary gland. Transfer between the mammary gland and milk was described using diffusion-limited uptake. In the absence of rat data, partition coefficients for DEHP (pmk = 200) and MEHP (pmmk: 0.3) were obtained from human blood:milk measurements (Hogberg et al., Citation2008). Development of oxidative metabolism (VmaxOn) in the neonate was estimated using in vitro data for total P450 activity using the same method described for fetal UGT development in the DBP model (Equation B1; (Clewell et al., Citation2008). All other neonatal parameters were scaled allometrically from the adult male rat values.
VmaxOn = VmaxO × RAnL × MPC × BWn × numpups (Equation B1)
where VmaxO is the maximum capacity for oxidative metabolism in the adult rat (after scaling for BW), RAnL is the in vitro relative activity expressed as the ratio of neonatal to maternal activity per mg microsomal protein, MPC is the ratio of the microsomal protein content of the fetal liver to maternal liver, LW is the ratio of the fetal: maternal liver weight, and numpups is the number of pups per litter. Model simulations are shown with data collected in the lactating and suckling rat after oral administration of DEHP to the dam are shown in Figure B5.
Notes
1When considering cancer endpoints, the use of a species scaling factor based on comparative surface area still forms the basis for the extrapolation across species for the vast majority of chemicals listed in IRIS. This is the case because the original assessment of the carcinogenic potency of these chemicals was derived prior to the recommended and subsequently implemented change to a comparative body weight scaling USEPA. (1996). Proposed Guidelines for Carcinogen Risk Assessment. EPA/600/P-92/003C. Office of Research and Development, Washington, DC. April, 1996.. Further, not all regulatory bodies, such as the State of California’s Prop 65 program, have adopted the newest species scaling approach of body weight3/4.
2A “point of departure” (POD) marks the beginning of extrapolation to lower doses. The POD is an estimated dose (usually expressed in human-equivalent terms) near the lower end of the observed range, without significant extrapolation to lower doses.
3This classification scheme is still in use and reported in the USEPA’s IRIS for the vast majority of chemicals that have not been re-evaluated using the newest guidelines.