
Abstract
The main aim of this work was to develop rectal suppositories for better delivery of metoprolol tartrate (MT). The various bases used were fatty, water soluble and emulsion bases. The physical properties of the prepared suppositories were characterized such as weight variation, hardness, disintegration time, melting range and the drug content uniformity. The in vitro release of MT from the prepared suppositories was carried out. The evaluation of the pharmacological effects of MT on the blood pressure and heart rate of the healthy rabbits after the rectal administration compared to the oral tablets was studied. Moreover, the formulation with the highest in vitro release and the highest pharmacological effects would be selected for a further pharmacokinetics study compared to the oral tablets. The results revealed that the emulsion bases gave the highest rate of the drug release than the other bases used. The reduction effect of the emulsion MT suppository base on the blood pressure and heart rate was found to be faster and greater than that administered orally. The selected emulsion suppository base (F11) showed a significant increase in the AUC (1.88-fold) in rabbits as compared to the oral tablets. From the above results we can conclude that rectal route can serve as an efficient alternative route to the oral one for systemic delivery of MT which may be due to the avoidance of first-pass effect in the liver.
Introduction
Metoprolol tartrate, MT ((RS)-isopropylamino-3-p (2-methoxyethyl)) phenoxypropen-2-ol(2R,3R)-tartrate) a selective β-adrenoreceptor blocking agent () has been used widely for the management of hypertension and other cardiac disorders such as angina pectoris, arrhythmias and myocardial infarction (Moffat, Citation1986). Metoprolol tartrate (MT) is almost completely absorbed after oral administration but it is subjected to extensive first-pass metabolism with a reported systemic bioavailability of about 40% and a low half life of 3–4 h (Dollery, Citation1999).
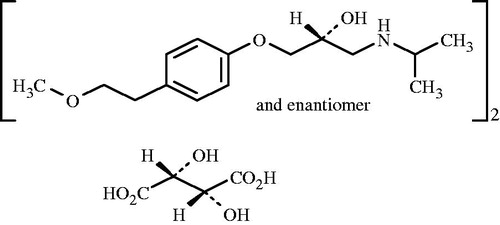
Figure 1. Chemical structure of metoprolol tartrate (Moffat, Citation1986).
Currently, there are no suppositories of metoprolol tartrate (MT) available in our market. In addition, no information was reported concerning rectal formulations of the drug. Only De Stoppelear et. al. (Citation1999) reported that metoprolol fatty suppositories appear to be a suitable alternative for the patients who are in need for beta-blocking medication and who are unable to take oral medication for a certain amount of time. Several attempts have been developed on the possibility of alternative routes of administration of MT to avoid the first-pass metabolism such as, intravenous (Dollery, Citation1999), transdermal (Aqil et al., Citation2004), intranasal (Coucke et al., Citation2009), buccoadhesive drug delivery systems (Abou el Ela et al., Citation2013).
Rectal route offers potential advantages for drug delivery. It is considered a convenient route of the drug administration especially in cardiovascular emergencies where the patient is unconscious and the parental routes are not possible. Also, it is convenient for patients with vomiting episodes and who may be unable or unwilling to swallow medication. In addition, it was proven to be advantageous over other routes because of the reduced side effects such as unwanted gastrointestinal tract side effects and the avoidance of the first-pass effect (Ansel, Citation1985).
Conventional suppositories are solid dosage forms at room temperature and intended to exert localized or systemic effect. They are commonly employed rectally or vaginally. Once inserted into the rectum, they melt, soften or even dissolve at rectal temperature (Zuheir et al., Citation2013). Cocoa butter and witepsols are commonly used as fatty bases that melt at 37 °C. However, glycerinated gelatin and polyethylene glycols (PEGs) are used as water soluble bases (Saleem et al., Citation2008). The release properties of many suppositories depend considerably on the physicochemical properties of the drug and the suppository bases used (Realdon & Ragazzi, Citation2001). The therapeutic use of rectal suppository dosage forms has been applied for some beta-adrenergic blocking agents (Ryu et al., Citation1999; Eman, et al., Citation2012).
Since the oral metoprolol tartrate (MT) has a low bioavailability, therefore, the objective of this study was to formulate MT in the rectal suppository delivery systems using different bases. The physical properties and the in vitro release of MT from the prepared suppositories were studied. In addition, the pharmacological effects of MT on the blood pressure and the heart rate of the healthy laboratory rabbits were carried out. Finally, the formulation with the optimized physical properties, the highest in vitro release and the highest pharmacological effects in the rabbits would be selected for a further pharmacokinetics study compared to the oral tablets.
Materials
Metoprolol tartrate (MT) was kindly supplied by Sid. Co. for Pharmaceutical and Chemical Industry, Cairo, Egypt. Polyethylene glycols (PEG) with molecular weights of 600, 1000, 1500, 4000 and 6000 were purchased from Sigma-Chem. Co., MO, USA. Witepsols, H15, H37, E75, W35 and Suppocire AM were obtained from Gattefosse Etablissements, Saint-Priest, France. Sodium alginate and sodium carboxymethyl cellulose were purchased from General Chemical & Pharmaceutical Co., Ltd., London, UK. Cocoa butter was bought from El Gomhouria Co., Cairo, Egypt. Urethrane was purchased from Sigma-Aldrich GmbH, Munich, Germany. All other reagents and ingredients used were of analytical grade.
Methods
Preparation of MT suppositories
MT suppositories, each containing 100 mg of the drug were formulated using different water soluble, emulsion and fatty bases as shown in and . Drug displacement values of the bases used were first determined and the amount of drug required was calculated (Ghorab et al., Citation2011). The fusion method was adopted to prepare the different suppositories. For fatty and water soluble suppositories, the base was melted first then MT powder was added to the melted base. Gentle stirring was continued to ensure complete mixing and to enhance cooling. The melted mass was then molded in a metal mould (1 g capacity) just before congealing. Moreover, the emulsified suppositories were prepared by dissolving the surfactant in either the hydrophilic or lipophilic phase, and the polymer was dissolved in the water phase using Witepsol E75 as a suppository base. All suppositories were kept in the refrigerator and were stored in a desiccator at room temperature for 24 h before use.
Table 1. Composition of different fatty and water soluble MT suppository basesa.
Table 2. Composition of different emulsion MT suppository basesa.
Physicochemical characterization of MT suppositories
Uniformity of the weight
Weight variation was determined as per the procedure of British Pharmacopoeia (British Pharmacopoeia, Citation2007).
Hardness
The Erweka method was used to measure the weight in kilograms a suppository can bear without breaking (Ghorab et al., Citation2011).
Melting range
The capillary method was utilized to measure the melting range of the prepared suppositories using the electrothermal melting point apparatus (1101D Mel-Tem® GallenKamp, England). A straight capillary tube which was 8–10 cm in length and 1–1.2 mm in internal diameter, opened at both ends was used. One end of the tube was dipped into the suppository bases and a gently sufficient amount was packed to fill about 1 cm column. The capillary tube was then placed in the apparatus attached to a thermometer. The melting range was recorded when the contents of the capillary tube started to melt (El-Majri & Sharma, Citation2010).
Disintegration time
Six suppositories were selected randomly from each formulation for the disintegration test. Disintegration test was performed in a phosphate buffer of pH 7.4 using USP disintegration apparatus (Erweka, GmbH, Heusenstamm, Germany). The disintegration time was recorded as soon as the suppositories placed in the basket either completely melted or dissolved (El-Shanawany & Aly, Citation1994).
Uniformity of drug content
Drug content uniformity was determined as per the procedure of British Pharmacopoeia (British Pharmacopoeia, Citation2007). Briefly, ten suppositories were randomly selected from each formula and assayed individually. The suppositories were melted and dissolved in 100 ml phosphate buffer of pH 7.4 and were allowed to rotate using a shaking water bath (Gesellschaft für, labortechnik GmbH, Germany) at 37 ± 0.5 °C for 2 h. Aliquots were withdrawn from the aqueous phase, filtered, suitably diluted and assayed spectrophotometrically (Shimadzu Seisakusho, Ltd., Kyoto, Japan) at 275 nm (Ramana et al., Citation2007) against blank.
In vitro MT release study
The in vitro release tests were carried out according to the dialysis method (Samy et al., Citation2000). Each suppository was placed in a glass tube opened in one side and the other side was sealed by a cellophane membrane (MW-cutoff 10000, Diachema, Germany) via rubber band. About 10 ml of a phosphate buffer pH 7.4 was added to the donor side and the glass tubes then lowered into glass beakers containing 50 ml of a phosphate buffer (pH 7.4) maintained at 37 ± 0.5 °C and rotated at 100 rpm using a shaking water bath (Gesellschaft für, labortechnik GmbH, Germany). Samples each of 2 ml were withdrawn at appropriate time intervals and replaced immediately by equal volumes of the fresh buffer. The amount of MT released by the time was assayed using a UV-visible spectrophotometer at 275 nm for MT. The experiment was repeated three times and the results were calculated as the mean ± SD.
Kinetic analysis of the release data
In order to describe the release model, the in vitro release data from MT suppositories were analyzed according to zero, first and Higuchi diffusion models. The model that consistently produced the highest correlation among the suppository preparations was used for the assessment of the drug release rates (Samy et al., Citation2000).
Pharmacological study in rabbits
The comparison of the pharmacological effects of MT on the blood pressure and heart rate of the healthy rabbits after the rectal administration of the selected suppository formulations of MT, F1, F10, F11 and the oral tablets, Betaloc® was carried out. The three selected formulations were administered at the dose level of 7 mg/kg of the drug corresponding to a 100 mg of the human dose. This equivalent dose for the rabbits was calculated by the aid of the surface area ratio, as the therapeutic dose of man was multiplied by a certain mathematical factor obtained from a special table for surface area ratios of some common laboratory species and man (Weibel et al., Citation2004; Geoffrey & James, Citation2005). The following equation was used: Dr = Dh (Wr/Wh)3/4, where Dr is the rabbit dose, Dh the human dose, Wr the rabbit weight and Wh the human weight (Abou el Ela et al., Citation2013).
Mean blood pressure
Twelve New Zealand rabbits (obtained from the Animal Care Center, College of Pharmacy, Assuit University) of either sex were used in this study. They had a body weight ranging from 1.8–2.0 kg and were kept on standard laboratory pellet-diet and tap water and were housed at a room temperature. All the experiments were performed in accordance to the ethical guidelines established and approved by the committee on the use and care of laboratory animals at our University (Issa et al., Citation2009). The rabbits were divided into four groups each of three animals. Each animal was anesthetized with intraperitoneal injection of urethane solution (25% v/v) at a dose of 6.4 mg/kg (El-Shanawany & Aly, Citation1994). After shaving the neck and canulating the trachea with a polyethylene tube, the animal was ventilated with a room air. Then it was prepared for i.v. injection of heparinized saline (1000 U/kg) through a cannula placed in the right jugular vein. The rabbits were divided as follows: the first group was given MT oral tablets, Betaloc® by gastric intubation. In the second, third, and fourth groups, the rabbits received the selected medicated rectal suppositories F1, F10 and F11, respectively. During the experiment, the rabbits breathe normally and the body temperature was maintained at 37 °C. The blood pressure was measured from the canulated left common carotid artery which was canulated by a special cannula attached to a blood pressure transductor and an amplifier of a four channel oscillograph connected to two channel recorders. The blood pressure was measured after 0 (control), 0.5, 1, 2, 3, 4, 5 and 6 h of the drug administration.
Mean heart rate
For the mean heart rate, ECG (electrocardiograph) pictures were obtained, that is the body surface manifestation of the depolarization and repolarization waves of the heart (Katzung, Citation1995). Cardisuny needles were putted subcutaneously and the pulse rate and the ECG pictures were measured at 0 (control), 0.5, 1, 2, 3, 4, 5 and 6 h of the drug administration.
Pharmacokinetics study in rabbits
Animal dosing and sampling scheme
The plasma concentrations of MT were evaluated from the healthy rabbits after the rectal administration of the suppository formula (F11) compared to the oral tablets, Betaloc® (MT, 100 mg). Nine healthy rabbits (1.5–2.25 kg) were used in the present study. The rabbits were fasted for 24 h before the administration of the drug and anesthetized as mentioned above. The dose level of 7 mg/kg of the drug corresponding to a 100 mg human dose was used. Rabbits were randomly divided into three groups each of three animals as follows: the first group, received MT oral tablets by a gastric intubation, the second group, was given the selected MT rectal suppository base (F11) and the third group, was given i.v. MT in an isotonic saline solutions. Control blood samples were taken from the rabbits immediately before administration of the drug. Multiple blood samples (1–2 ml) were collected in heparinized vacutainer tubes before (0 time) and at 0.5, 1, 1.5, 2, 3, 4, 6, 12 and 24 h following drug administration. The plasma was then separated after centrifugation and stored frozen at −20 °C until analysis (El-Shanawany & Aly, Citation1994).
Analysis of plasma samples
MT plasma concentration was measured using a sensitive high-performance liquid chromatography (HPLC) assay. The HPLC system (Knauer K-500 pump, Knauer, K-2500 UV detector, Shimadzu C-R6A, chromatopac integrator, Kyoto, Japan) was used with the reversed-phase mode. Analysis was performed on Aqua RP-C18 packed column (250 × 4.6 mm internal diameter, 5 μm particle diameters). The 50 μL aliquots were injected and eluted with a mobile phase containing 3.9 g ammonium acetate in 810 ml water, 2 ml triethylamine, 10 ml glacial acetic acid, 3 ml phosphoric acid and 146 ml acetonitrile. The flow rate was set at 1 ml/min and the eluent was monitored at 275 nm. A calibration curve of MT in the plasma was constructed using blank plasma spiked with standard MT solutions to obtain a concentration range of 0.3–30 μg/ml. The spiked plasma was then subjected to the same extraction procedure as that of the samples. Triplicate runs were made for each standard sample.
Pharmacokinetics analysis
After measuring MT concentrations in the plasma, MT pharmacokinetics in the plasma was assessed by fitting the plasma concentration-time data to the suitable model using Win Nonlin standard version 1.5 (Science Consulting, Apex, NC, USA) software. Absorption rate constant (Ka), absorption half-life (t½a), elimination rate constant (Kel), elimination half-life (t½) and the area under the plasma MT concentration versus time curve (AUC) were calculated. Also, the maximum concentration (Cmax) and the time to reach the maximum concentration (Tmax) were reported. The absolute bioavailability of a drug is generally measured by comparing the respective AUC after extravascular and i.v. administration.
Statistical analysis
Statistical evaluations of data were performed using Student’s t-test of unpaired data (MedCalc Statistical Software). Differences between means were considered statistically non-significant (NS) if the p value was > 0.05. When 0.05 > p ≥ 0.01 the parameters were taken as significantly (S) different and when 0.01 > p ≥ 0.001 they were regarded to be highly significantly (HS) different.
Results and discussion
Physicochemical characteristics of MT suppositories
Appearance
The prepared suppositories were well formed with a smooth shinning surface, white or creamy white in color. After slicing the suppositories longitudinally, they did not show any fissures, cracks or contraction holes.
Weight variation
The results in showed that all the prepared suppositories were within the pharmacopeial limits for the uniformity of the weight according to the procedure of British Pharmacopoeia (British Pharmacopoeia, Citation2007).
Table 3. Physicochemical parameters of different MT suppositoriesa.
Hardness
The prepared suppository formulations exhibited hardness ranging from 1.2 ± 0.12 to 4.6 ± 0.31 kg, as shown in . This is an important indication of their ability to withstand pressure during handling, shipping and insertion.
Melting range
There is a considerable variability of the melting point between the tested formulations. A narrow melting range is important for maintaining the shape of the suppository in an ambient temperature and also, for controlling the melting time of the suppository after insertion. The water soluble PEG bases showed higher melting ranges (50–55 °C) than the emulsion (35–39 °C) and the fatty bases (33–38 °C). Furthermore, Witepsol H15 fatty base had the lowest melting range (33–35 °C) as compared to the other fatty bases as shown in .
Disintegration time
The different MT formulations exhibited various disintegration times as shown in . They are either dissolved or softened and melted within the range of 5–15 min, 18–21 min and 25–30 min for fatty, polyethylene glycol and emulsion bases, respectively.
Drug content uniformity
The drug content was found to comply with the requirements of the British Pharmacopoeia and it ranged from 97.55% ± 0.27 to 101.9% ± 0.11 of the incorporated amount () (British Pharmacopoeia, Citation2007).
In vitro release studies
In our study, the dialysis technique was adapted. Three types of the suppository bases were used, fatty, water soluble and emulsion bases. The effect of the type of suppository base on the in vitro release of MT is presented in . The rank order of the drug release using various bases was as follows: emulsion bases > water soluble PEG bases > fatty bases. Generally, the emulsion bases (F11–F13) gave higher rate of the drug release than the water soluble PEG bases (F6-F10). This may be attributed to the presence of the hydrophilic (Tween 20) and lipophilic (Span 60) surfactants, which they have the ability to improve the wettability of the base matrix and enhancing the elution or dispersion of the embedded drug particles to the surrounding medium (Abd El-Gawad et al., Citation1988). On the other hand, the water soluble bases gave higher drug release than the fatty bases (F1–F5) which may be due to the rapid dissolution and the rapid solubility of the water soluble base, wherein the drug can be released by both diffusion and erosion mechanisms (Hussain et al., Citation1980). These results are in a good agreement with many other investigators (Asikoglu et al., Citation1995; El-Nabarawi et al., Citation2003).
Figure 2. In vitro release profiles of MT from different fatty suppository bases in phosphate buffer of pH 7.4. Results are the mean ± SD of 3 determinations (n = 3).
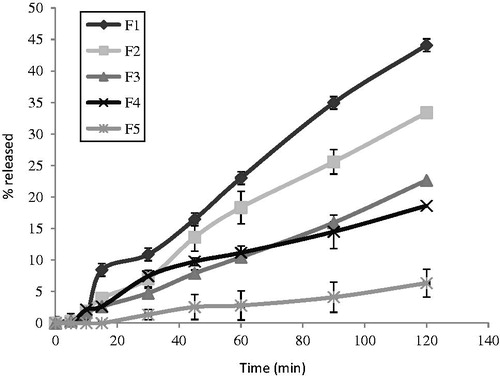
Figure 3. In vitro release profiles of MT from different PEG suppository bases in phosphate buffer of pH 7.4. Results are the mean ± SD of 3 determinations (n = 3).
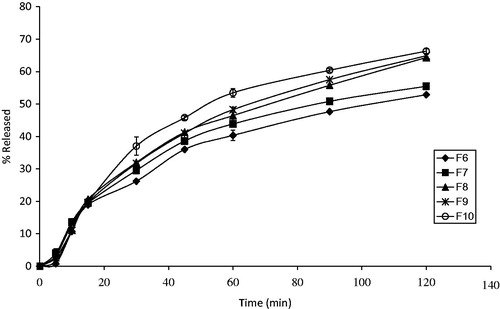
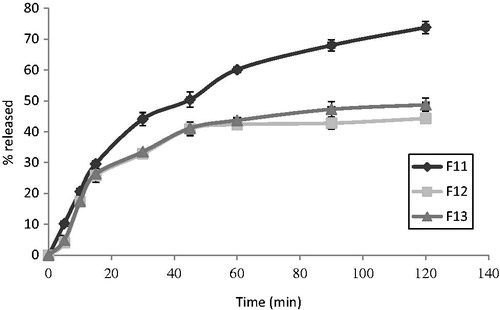
Figure 4. In vitro release profiles of MT from different emulsion suppository bases in phosphate buffer of pH 7.4. Results are the mean ± SD of 3 determinations (n = 3).
In vitro release from fatty suppository bases
The in vitro release of MT from the different prepared fatty suppository bases (F1–F5) is shown in . Ranking them in a descending order according to the percent drug release after 2 h was as follows: Suppocire AM (F1) > Witepsol H15 (F2) > Witepsol W35 (F3) > Witepsol H37 (F4) > Cocoa butter (F5). It is clear that the release pattern depends on the melting behavior and the chemical composition of the fatty bases used. Semi-synthetic fatty suppository bases are mixtures of the fatty acids and esters with certain amounts of glycerides. The hydroxyl values reported for the specific bases represent the proportions of the free mono-and diglycerides and the free hydroxyl groups that are available for the interaction. A high hydroxyl value is an indication of the potential for a base to absorb the water. The presence of high hydroxyl values in the fatty bases could thus favor the formation of a water-in-oil emulsion, which will generally result in a slow transfer of drug molecules from the inner aqueous phase and retarded the release of the drug (Hosny et al., Citation1996; Tukker, Citation2009). Suppocire AM (F1) gave a greater MT percent release, 44.09 ± 0.463 than Witepsol H15 (F2), 33.38 ± 1.935, which may be due to a low hydroxyl value of Suppocire AM compared to Witepsol H15 (Rowe et al., Citation2009). Generally, fatty bases with self-emulsifiers will give higher release of the water soluble drugs than Cocoa butter. It is clear that Cocoa butter (F5) gave a slower percent release (6.35 ± 3.376) than that from both Suppocire AM and Witepsol bases. This may be attributed to the presence of monoglyceride esters in the latter bases which act as self-emulsifiers resulting in a high emulsifying and water-absorbing capacities that responsible for enhancing the drug release (El-Shanawany & Aly, Citation1994). These results are in a good agreement with a higher release of ciprofloxacin hydrochloride and propranolol hydrochloride from Witepsol H15 than from Cocoa butter (El-Shanawany & Aly, Citation1994; Hassan & Mahfouz, Citation2001).
With respect to Witepsols, the percent of the drug release was found to be slower from Witepsol H37 (F4) 18.62 ± 2.651 than from Witepsol H15 (F2) 33.38 ± 1.935 and Witepsol W35 (F3) 22.65 ± 0.148. This may be due to a high melting range of Witepsol H37 (36–38 °C) compared to Witepsol H15 (33–35 °C) and Witepsol W35 (34–36 °C). Furthermore, Witepsol H15 (F2) has a lower hydroxyl value (≤15) compared to that of Witepsol W35 (F3) (40–50) which may be responsible for greater drug release from Witepsol H15 compared to Witepsol W35 in spite of having about the same melting range (Calis et al., Citation1994).
In vitro release from PEG bases
The in vitro release of MT from the various formulated PEG suppository bases (F6–F10) is shown in . Ranking them in a descending order according to the percent of the drug release was as follows: F10 > F9 > F8 > F7 > F6. This arrangement could be explained on the basis of the molecular weights of PEG bases. As the amount of high molecular weight polyethylene glycols (PEG 4000, 6000) increases and that of the low molecular weight polyethylene glycols (PEG 600, 1000, 1500) decreases, this resulted in raising the melting point, increasing the hardness of the bases and consequently retarding the in vitro release of the drug (Realdon & Ragazzi, Citation2001). It was reported that, as the molecular weights of PEG increase, their water solubility and hygroscopicity decrease (Rowe et al., Citation2009). Moreover, it was reported that PEG of different molecular weights can be combined to achieve a suppository base with a specific drug release rate profile (Coben & Lieberman, Citation1986).
It was observed that F10 is accompanied by the highest percent of the drug release (66.31 ± 0.692) while F6 has the slowest percent (50.88 ± 0.303). These results are in agreement with that reported by Ghorab et al. (Citation2011) who found that the release rate of fenoterol hydrobromide from different PEG bases was dependent on their hydrophilic and lipophilic character.
In vitro release from emulsion suppository bases
The in vitro release of MT from the different emulsion suppository bases (F11–F13) are presented in . The rank order of the percent of drug release was as follows: F11 > F13 > F12. The obtained results revealed an inverse relationship between the amount of the drug released and the melting point range and the dissolution time of suppositories (). It was observed that the components of the base were found to determine the melting range as well as the dissolution time of the suppository. When PEG 1500 and PEG 600 were used in F11, a significant (p < 0.05) higher drug release percent was observed (73.79 ± 0.445) compared to the other emulsion suppositories, F12 and F13, respectively (44.30 ± 0.606 and 48.74 ± 0.661). This may be attributed to the concomitant rapid dissolution of the suppository. Similar results were obtained from the release of propranolol hydrochloride using different emulsion bases (El-Shanawany & Aly, Citation1994). Moreover, suppositories containing sodium CMC (F12) showed a slight lower percent release as compared to the suppositories containing sodium alginate (F13), this may be due to the gelling behavior exhibited by sodium CMC (Rowe et al., Citation2009). In addition, the incorporation of non-ionic surfactants, namely, Tween 20 as an example of a hydrophilic surfactant and Span 60 as an example of a lipophilic surfactant into a Witepsol E75 suppository base (F11) affected the rate of the drug release depending on the nature and the concentration of a surfactant (Realdonet al., Citation2008). Percent drug release increased significantly (p < 0.05) upon incorporating both Tween 20 and Span 60 (F11). This is probably due to the fact that Tween 20 lowers the interfacial tension, and hence increases dispersibility of the suppository base in the dissolution fluid. Furthermore, one percent of Span 60 increased drug release significantly (p < 0.05), which may be attributed to the enhancement of the wetting of the matrix and subsequently increasing the dissolution of the drug in the suppository and in the dissolution medium (Hanaee et al., Citation2004). The enhancement of MT release rate produced by Span 60 and Tween 20 (F11) was significantly (p < 0.05) higher than that produced by Tween 20 alone (F12, F13) as shown in . This may be attributed to the much higher hydroxyl value of Span 60 compared to Tween 20 (Rowe et al., Citation2009) which may increase the ability of the surfactant in wetting the matrices and producing a greater number of channels for the dissolution fluid to leach out the drug (Bolourtchian et al., Citation2005).
Kinetic analysis of release data
The kinetic analysis of the in vitro release data according to zero-order, first-order and Higuchi models as shown in revealed that the best fit with the highest correlation coefficient (R2) was obtained with zero-order (0.9816–0.9987) for the fatty base suppositories. While, for PEG and emulsion suppositories, the best fit with the highest correlation coefficient was shown with Higuchi model (0.9822–0.9878 and 0.9342–0.9895, respectively).
Table 4. Kinetic parameters of MT release data according to different kinetic models.
Pharmacological effects of MT in rabbit
The pharmacological effects of MT on the blood pressure and heart rate of the healthy rabbits after the rectal administration compared to the oral tablets, Betaloc® (MT, 100 mg) were carried out. Three suppository formulations of MT were selected, the fatty (F1), the water soluble (F10) and the emulsion one (F11). These formulations were selected on the basis of their physical parameters (satisfactory level in terms of hardness, melting point range, dissolution time, weight variation and drug content). In addition, they gave the highest percent release of the drug in 2 h as shown in . The dose of the drug corresponding to a 100 mg human dose was used for the oral and rectal administrations.
The effect on blood pressure
The effect of both oral and rectal administration of MT on the mean blood pressure of the healthy rabbits is shown in . It is clear that a single dose of MT suppository formulations (F1, F10 and F11) resulted in a reduction in the blood pressure after 30 min (119 ± 5.78, 114 ± 8.67 and 92 ± 4.44 mm Hg, respectively). This reduction in the blood pressure was highly significant for the emulsion base (F11) and non-significant for both the fatty and the water soluble bases, F1 and F10, respectively. The maximum effect was observed at 2 h with a decrease in the blood pressure of ∼15, 19 and 54 mm Hg (90 ± 5.97, 101 ± 4.67 and 68 ± 6.22 mm Hg) for fatty, water soluble and emulsion suppository bases, respectively. This antihypertensive effect was highly significant, significant and non-significant for F11, F1 and F10, respectively. While, a single dose of oral MT tablets evoked a non-significant reduction in the blood pressure after 1 h (115 ± 4.44 mm Hg) and a maximum effect was seen at 4 h, with a decrease in the blood pressure of ∼14 mm Hg (105 ± 4.44 mm Hg). The reduction in the blood pressure remained till the end of the experiment (6 h) which is longer than expected from the plasma half life of the drug. It was reported that MT has a significant antihypertensive effect which persists for 24 h (Moffat, Citation1986). The obtained results indicate that the effect of MT by using suppository rectal delivery system is considerably faster and greater than that of the oral tablets. These results may be attributed to the avoidance of the degradation of the drug by-passing the liver. These results are in agreement with De Stoppelaar et al. who stated that the metoprolol concentration in plasma and urine gave an indication for a partial avoidance of the first pass effect after rectal administration (De Stoppelaar et al., Citation1999). The reducing effect of MT suppository formulations on the blood pressure was ranked as follows: emulsion base (F11) > fatty base (F1) > PEG base (F10).These results revealed that the pharmacological effect of the drug was greatly influenced by the type and nature of the suppository base used (Asikoglu et al., Citation1995; Hosny et al., Citation1996). Moreover, MT formulated in the emulsion suppository base (F11) gave the highest significant antihypertensive effect in the rabbits at the end of the experimental time (6 h) compared to the other two tested formulations (F1, F10). These results are in accordance with Abd El-Gawad et al who reported that the use of the surfactants in the suppositories improved the in vitro release as well as the in vivo bioavailability of sulphamethoxazole (Abd El-Gawad et al., Citation1988). The high availability of the drug from the fatty base (F1) compared to the water soluble base (F10) is related to its rapid melting at body temperature and spreading after insertion, leading to the rapid dissolution of the drug in the rectal fluid. On the other hand, water soluble base liquefies by the absorption of water resulting in swelling and dissolution of the suppository. This leads to a dispersion process that is slower than the melting of the fatty base (Asikoglu et al., Citation1995; Hosny et al., Citation1996).
Table 5. Effect of rectal administration of MT suppositories on blood pressure of healthy rabbits compared to the oral tablets.
The effect on heart rate
The heart rate was calculated from the corresponding ECG (electrocardiograph) pictures and reported in . A single dose of MT suppository formulations (F1, F10 and F11) showed a reduction in the heart rate after 30 min (258 ± 5.78, 318 ± 7.56 and 228 ± 3.78 beat/min, respectively). This decrease in the heart rate was highly significant for both F1 and F11 but it was non-significant for F10. A highly significant maximum effect was observed at 2 h with a decrease in the heart rate of ∼84, 72 and 135 beat/min for fatty, water soluble and emulsion suppository bases, respectively (234 ± 9.33, 252 ± 3.33 and 180 ± 3.56 beat/min). While a single dose of oral MT tablets caused a non-significant reduction in the heart rate after 30 min (315 ± 6.00 beat/min) and the maximum effect was seen but after 4 h with a decrease in the heart rate of ∼42 beat/min. The decrease in the heart rate following the rectal administration of MT was greater and faster as compared to the oral tablets along the time of the experiment (6 h). The reduction in the heart rate also remained till the end of the experiment (6 h) which is longer than expected from the plasma half life of the drug. The reduction effect of MT suppository formulations on the heart rate was ranked as follows: emulsion base (F11) > PEG base (F10) > fatty base (F1). These results are in a good agreement with the other reported data (Grant et al., Citation1983; Nair & Bhargava, Citation1999). The emulsion suppository base (F11) gave the highest reducing effect (225 ± 5.56 beat/min) on the heart rate after the end of the experiment (6 h) as compared to F1 and F10, respectively (258 ± 5.78 and 258 ± 5.56 beat/min).
Table 6. Effect of rectal administration of MT suppositories on the heart rate of healthy rabbits compared to the oral tablets.
Pharmacokinetics of F11 and Betaloc® in rabbits
The emulsion suppository base (F11) was selected to determine the plasma level of MT in the rabbits compared to the oral MT tablets, Betaloc®. This selection was based on its physical parameters, the highest in vitro drug release percent and also, it gave a significant reduction in the blood pressure and heart rate of the healthy rabbits. MT was completely separated from the endogenous constituents, as a sharp MT peak with no interfering peaks appeared and the retention time was found to be 9.3 min.
The rectal formula (F11) of MT showed a plasma detectable concentration of 0.539 ± 0.125 µg/ml after 15 min post-treatment and the concentration was detectable for 24 h (). The plasma drug concentration increased progressively to reach a maximum concentration Cmax of 3.3 ± 0.821 µg/ml at Tmax of 1.5 ± 0.44 h, then decreased to 0.34 ± 0.095 µg/ml at 24 h after rectal administration (). In comparison, an oral dose of MT gave a detectable concentration of 0.177 ± 0.113 µg/ml after 30 min post-treatment and the concentration remained detectable for 24 h (). The plasma concentration increased progressively to reach the maximum peak concentration Cmax of 1.26 ± 0.223 µg/ml at Tmax of 4.223 ± 0.234 h, then descending to 0.012 ± 0.073 µg/ml at 24 h after oral administration ().
Figure 5. MT plasma concentration time profiles in rabbits (mean ± SD, n = 3) after oral administration of Betaloc® and rectal suppository (F11).
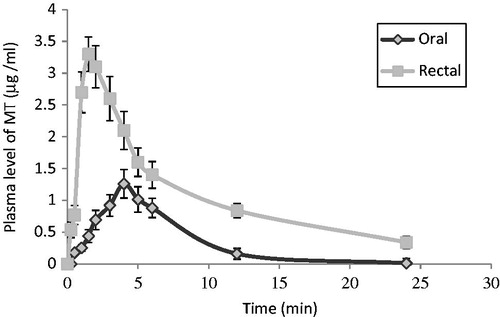
Table 7. Comparative pharmacokinetic parameters of MT after oral and rectal administration in three rabbits.
It could be noticed that F11 exhibited faster absorption rate that could be reflected by its shorter Tmax value (1.5 ± 0.44 h) in comparison with the oral one which has Tmax value of 4.223 ± 0.234 h. In addition, the peak serum concentration Cmax of the drug was markedly higher in the rectally treated animals (3.3 ± 0.821 µg/ml) than that of the oral one (1.26 ± 0.223 µg/ml). This reflects the higher rate of absorption after rectal delivery of the drug. Moreover, the mean AUC after F11 was higher (19.483 ± 2.27 µg ml−1 h) than that after the oral tablets (10.734 ± 3.48 µg ml−1 h). Also, the absolute bioavailability (FAbsolute) after F11 was found to be 71.452% while that following the oral tablets was 39.368% (). As the bioavailability was compared using AUC, Cmax and Tmax, so it is clear that the systemic bioavailability of MT after rectal administration was higher than that after oral one. This may be due to the enhancement of the rate and extent of absorption after rectal administration and this reflects the avoidance of first-pass metabolism. These results are in a good agreement with the earlier finding of De Stoppelaar et al. (Citation1999) who reported that metoprolol tartrate in a fatty suppository base has a considerable higher bioavailability after rectal administration as compared to oral metoprolol capsules using human volunteers (De Stoppelaar et al., Citation1999). The significance of difference between the pharmacokinetic parameters of F11and the oral tablets were also calculated (). The results showed that the differences between Cmax and AUC were significant while the differences between their Tmax, Ka and t½a were highly significant. However, the differences between their Kel and t½ were non-significant. A relationship between the plasma MT concentration and its pharmacological response after F11 was conducted, wherein the lowering in mean blood pressure and the reduction in the heart rate were plotted against the log plasma concentrations of MT for 6 h.
Good relationships with correlation coefficients of r = 0.9327 and 0.8864 were found between log plasma concentration of MT and both the antihypertensive effect and the reduction in heart rate, respectively. Finally, it could be concluded that the selected MT rectal suppository delivery system not only improved the pharmacological effects of MT but also its oral bioavailability in rabbits.
Conclusion
Metoprolol tartrate (MT) was successfully prepared in the different rectal suppositories using fatty, water soluble and emulsion bases. The physical characteristics of the prepared MT suppositories were found to be complying with the requirements of British Pharmacopoeia Citation2007. The selected emulsion suppository base (F11) gave the highest in-vitro drug release percent and also pronounced reduction in blood pressure and heart rate of the healthy rabbits. The absolute bioavailability after F11 was found to be higher than that following the oral tablets, Betaloc®. Finally, the rectal route can serve as an efficient alternative route to the oral one for systemic delivery of the beta-blocker drug, MT, which may be due to avoidance of first-pass effect in the liver.
Acknowledgements
The authors acknowledge the Research Center of the Center for Female Scientific and Medical Colleges, deanship of scientific research in King Saud University for supporting this research by a grant.
Declaration of interest
The authors have no conflict of interests to disclose.
References
- Abd El-Gawad AH, Zin El-Din E, Abd El-Alim H. (1988). Effect of surfactant incorporation techniques on sulphamethaxazole suppository formulations. Pharmazie 43:624–7
- Abou El Ela AESF, Ibrahim EH, Allam AA. (2013). Bucco-adhesive tablets containing metoprolol tartarate: formulation, in vitro and in vivo characterization. J Drug Del Sci Tech 23:171–9
- Ansel HC. (1985). Suppositories and other rectal, vaginal and urethral preparations. In: Williams and Wilkins, eds. Introduction to Pharmaceutical Dosage Forms, 4th ed. Philadelphia, USA: Lea & Febiger, 342–58
- Aqil M, Sultana Y, Ali A, et al. (2004). Transdermal drug delivery systems of a beta-blocker: design, in-vitro and in-vivo characterization. Drug Del 11:27–31
- Asikoglu M, Ertan G, Cosar G. (1995). The release of isoconazole nitrate from different suppository bases: In-vitro dissolution, physicochemical and microbiological studies. J Pharm Pharmacol 47:713–16
- British Pharmacopoeia. (2007). Vol. IV. Appendix XII G, A304. London, UK: The Stationary Office
- Bolourtchian N, Javid FS, Dadashzadeh S. (2005). The effect of various surfactants on release behavior of procainamide HCl from ethylcellulose based matrices. Iran J Pharm Res 1:13–9
- Calis S, Sumnu M, Hincal AA. (1994). Effect of suppository bases on the release properties of a potent antimicrobial agent (C31g). Pharmazie 49:336–9
- Coben LJ, Lieberman HA. (1986). Suppositories. In: Lachman L, Lieberman HA, Kanig JL, eds. The Theory and Practices of Industrial Pharmacy. 3rd ed. Philadelphia, USA: Lea and Febiger, 564–88
- Coucke D, Vervaet C, Foreman P, et al. (2009). Effect on the nasal bioavailability of co-processing drug and bioadhesive carrier via spray drying. Int J Pharm 379:67–71
- De Stoppelaar FM, Stolk LML, Beysens AJ, et al. (1999). The relative bioavailability of metoprolol following oral and rectal administration to volunteers and patients. Pharm World Sci 21:233–8
- Dollery C. (1999). Metoprolol Tartrate. In: Boobis A, Rawlins M, Thomas S, Wilkins M, eds. Therapeutic Drugs, Vol. 2, 2nd ed. New York, USA: Churchill Livingston, M139–46
- El-Majri MA, Sharma RK. (2010). Formulation and evaluation of piroxicam suppositories. Int J Drug Del 2:108–12
- El-Nabarawi MA, Nesseem DI, Sleem AA. (2003). Delivery and analgesic activity of tramadol from semisolid (topical) and solid (rectal) dosage forms. Bull Fac Pharm Cairo Univ 41:25–31
- El-Shanawany S, Aly SA. (1994). Formulation of propranolol hydrochloride suppositories and pharmacological evaluation in rabbits. Eur J Pharm Biopharm 40:327–32
- Eman G, Mokhtar M, El Ghamry H, Ghazy F. (2012). Sustained release rectal suppositories as drug delivery systems for atenolol. J Am Sci 8:323–32
- Geoffrey B, James H. (2005). The origin of scaling laws in biology from genomes to ecosystems: towards a quantitative unifying theory of biological structure and organization. J Exp Bio 208:1575–92
- Ghorab D, Refai H, Tag R. (2011). Preparation and evaluation of fenoterol hydrobromide suppositories. Drug Dis Therap 5:311–8
- Grant DJW, Liversage GG, Bell J. (1983). Influence of physicochemical interactions on the properties of suppositories. The in-vitro release of ketoprofen and metronidazole from various fatty suppository bases and the relations with in-vivo plasma levels. Int J Pharm 14:251–61
- Hanaee J, Javadzadeh Y, Taftachi S, et al. (2004). The role of various surfactants on the release of salbutamol from suppositories. Farmaco 59:903–6
- Hassan MA, Mahfouz NM. (2001). Formulation and evaluation of ciprofloxacin hydrochloride suppositories. Bull Pharm SciAssiut University 24:47–54
- Hosny EA, Abdel-Hady SS, El-Tahir KEH. (1996). Formulation, in-vitro release and ex-vivo spasmolytic effects of mebeverine hydrochloride suppositories containing polycarpophil or polysorbate 80. Int J Pharm 142:163–8
- Hussain A, Hirai S, Bawarshi R. (1980). Nasal absorption of propranolol from different dosage forms by rats and dogs. J Pharm Sci 69:1411–13
- Issa M, Mohamed A, Seham B. (2009). Synthesis and pharmacological evaluation of 2-substituted benzo[b]thiophenes as anti-inflammatory and analgesic agents. EurJ Med Chem 44:1718–25
- Katzung BG. (1995). Basic and Clinical Pharmacology, 6th ed. Appleton & Lange, Middle East Edition, 205–29
- Moffat AC. (1986). Metoprolol tartrate. In: Jackson JV, Moss MS, Widdop B, eds. Clarke's isolation and identification of drugs, 2nd ed. London, UK: The Pharmaceutical Press Inc., 779–80
- Nair L, Bhargava HN. (1999). Comparison of in-vitro dissolution and permeation of fluconazole from different suppository bases. Drug Dev Ind Pharm 25:691–94
- Ramana MV, Nagda C, Himaja M. (2007). Design and evaluation of mucoadhesivebuccal drug delivery systems containing metoprolol tartrate. Ind J Pharm Sci 69: 515–18
- Realdon N, Ragazzi E. (2001). Effect of drug solubility on in vitro availability rate from suppositories with polyethylene glycol excipients. Pharmazie 56:163–7
- Realdon N, Dal zotto M, Morpurgo M, Franceschinis E. (2008). Effects of surfactant characteristics on drug availability from suppositories. Pharmazie 63:459–63
- Rowe RC, Sheskey PJ, Weller PJ. (2009). Handbook of Pharmaceutical Excipients, 6th ed. London, UK: Pharmaceutical Press
- Ryu JM, Chung SJ, Lee MH, et al. (1999). Increased bioavailability of propranolol in rats by retaining thermally gelling liquid suppositories in the rectum. J Control Rel 59:163–72
- Saleem MA, Taher M, Sanaullah S, et al. (2008). Formulation and evaluation of tramadol hydrochloride rectal suppositories. Ind J Pharm Sci 70:640–4
- Samy EM, Hassan MA, Tous SS, Rhodes CT. (2000). Improvement of availability of allopurinol from pharmaceutical dosage forms I-Suppositories. Eur J Pharm Biopharm 49:119–27
- Tukker J. (2009). Rectal and vaginal drug delivery. In: Aulton ME, ed. Pharmaceutics, the science of dosage form design. Edinburgh, UK: Churchill Livingstone, 534–43
- Weibel E, Bacigalupe L, Schmitt B, Hoppeler H. (2004). Allometric scaling of maximal metabolic rate in mammals: muscle aerobic capacity as determined factor. J Physiol Neurobiol 140:115–32
- Zuheir A, Samein LH, Aiash N. (2013). Preparation and in vitro evaluation of metoclopramide HCL hollow-type suppository. Int J Pharm Pharm Sci 5:660–6