
Abstract
10-hydroxy camptothecin (10-HCPT) is an antitumor agent effective in the treatment of several solid tumors but its use is hampered by poor water solubility, low lactone stability, short plasma half-life and dose-limiting toxicity. In this study, 10-HCPT-hydroxyethyl starch (HES) conjugate was prepared to overcome these limits of 10-HCPT. The solubility of 10-HCPT conjugate was 0.72 mg/ml, about 100 times to free 10-HCPT. The 10-HCPT conjugate showed good sustained release effect in phosphate-buffered saline (PBS), rat plasma and liver homogenate. Meanwhile, 10-HCPT–HES conjugate achieved much lower IC50 and higher cytotoxicity effects than the free 10-HCPT on Hep-3B liver cancer cells. The pharmacokinetics results of 10-HCPT–HES conjugate demonstrated that the biological half-life of 10-HCPT was increased from 10 min to 4.38 h and the bioavailability was 40 times higher than the commercial 10-HCPT injection. The pharmacodynamics results indicated that 10-HCPT–HES conjugate had a better antitumor efficiency against nude mouse with Hep-3B tumor than the commercial 10-HCPT injection, and the inhibition ratio of tumor was 83.9 and 27.8%, respectively, at the same administration dosage. These findings suggest that 10-HCPT–HES conjugate is a promising drug delivery system providing improved long circulating effect, greater stability and better antitumor effect.
Introduction
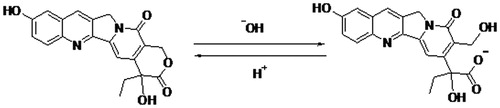
10-hydroxy camptothecin (10-HCPT) is one of the camptothecin (CPT) analogs with a powerful cytotoxic effect for cancer therapies and a broad range of anticancer activity in animal models (Mendichi et al., Citation2002; Deshmukh et al., Citation2010; Venditto & Simanek, Citation2010). The molecular target of the CPTs family is the enzyme DNA topoisomerase I (topo I), a ubiquitous enzyme which is over-expressed in a variety of tumor cell lines and involve in the regularization of DNA topology during replication, recombination and transcription period (Botella et al., Citation2011). However, several major drawbacks of 10-HCPT have made them less attractive for clinical use, such as, low aqueous solubility and poor lactone stability in vitro and in vivo, short plasma half-life and severe clinical toxicity and side effects (Subramanian et al., Citation2011). It is known that 10-HCPT has a pH-dependent equilibrium between its lactone form and carboxylate form under physiological conditions, and the structure of 10-HCPT is shown in (Lorence & Nessler, Citation2004). Generally speaking, the lactone form of 10-HCPT is essential for antitumor activity and the carboxylate form is inactive. In addition, the carboxylate form can induce severe cumulative hematological toxicity, diarrhea and chemical or hemorrhagic cystitis, which are often formidable and unpredictable. Meanwhile, serum albumin preferentially binds the carboxylate form of 10-HCPT and accelerates in vivo elimination of 10-HCPT (Burke & Mi et al., Citation1994). Therefore, several approaches have been used to stabilize the lactone ring prior to entry into the site of action and increase the solubility to maximize therapeutic activity and minimize toxicity (Cheng et al., Citation2003). For example, some 10-HCPT analogs possessing stabilized lactone structures and high water solubility have been developed to overcome this challenge, and topotecan and irinotecan are currently approved for the treatment of many types of cancer in humans.
Figure 1. 10-HCPT structure and pH-dependent equilibrium of the lactone form and carboxylate form. 46 × 10 mm (300 × 300 DPI).
In addition to the above analogs strategy, an alternative approach to overcome the pharmaceutical and pharmacokinetic shortcomings of 10-HCPT is to covalently attach the drug to water soluble polymers via the 10-OH or 20-OH position (Lerchen et al., Citation2001). It has been reported that the substitution on 20-OH group of 10-HCPT can substantially reduce the tendency for lactone ring opening and increase the solubility, and a variety of conjugates that link 10-HCPT via 20-OH group have been performed, including PEG-camptothecin conjugate, polyglutamate–camptothecin conjugate, PEG-SN38 conjugate and PAMAM–camptothecin conjugate, some of them have entered into the phase of clinical research (Bhatt et al., Citation2003; Cheng et al., Citation2004; Fleming et al., Citation2004; Samor et al., Citation2008; Joralemon et al., Citation2010; Tong & Cheng, Citation2010). Except improving the solubility and stability of 10-HCPT, polymer conjugate can also prolong circulation time through avoiding rapid clearance by the renal and reticuloendothelial systems (RES), decrease side effects, passive target tumor tissues via the enhanced permeability and retention (EPR) effect and improve bioavailability (Li & Wallace, Citation2008; Maeda et al., Citation2009; Torchilin, Citation2011).
Hydroxyethyl starch (HES) was selected to conjugate 10-HCPT in this study. As we all known, HES is a hydroxyethylated glucose polymer which used in medicine for the treatment of hypovolemic shock, artery occlusive disease, cerebral ischemia or apoplectic insult as HES molecules can improve the microcirculation within the organism (Baier et al., Citation2012; Noga et al., Citation2012, Citation2013). It is degraded in vivo by serum α-amylase, and the degradation behavior can be regulated by fine tuning the molar mass and degree of hydroxyethylation. In addition, it has a high water solubility, low hypersensitivity and protein repellent characteristics. These optimal properties make HES become an important biomedical material, which has been tested as a substitute for PEG in the circulation time extension of drugs with short half-life, and as cryoprotectant of peptides, proteins and other nanoparticulate systems.
In this study, 10-HCPT and HES conjugate (10-HCPT–HES conjugate) was prepared as a novel drug delivery system to transport and release 10-HCPT. The molecular weight (Mw) and the degree of substitution (Ds) of the selected HES were 20 kDa and 0.5 respectively. The aqueous solubility of 10-HCPT is increased by more than 100 times when it is conjugated with HES, and the conjugated 10-HCPT remains in the lactone form (confirmed by HPLC). Simultaneously, a series of studies were performed to evaluate the physical and chemical properties and antitumor effects of 10-HCPT–HES conjugate. In vitro release in PBS solution, stability in plasma and liver homogenate and cytotoxicity on Hep-3B liver cancer cells were investigated. In vivo pharmacokinetics in rats and pharmacodynamics in nude mouse with Hep-3B cancer cells were also studied. The results showed that 10-HCPT–HES conjugate significantly increased the solubility and lactone stability of 10-HCPT. Meanwhile, longer circulating effect in plasma, better cytotoxicity and higher anti-tumor effect were exhibited in comparison with free 10-HCPT.
Materials and methods
Materials and reagents
10-hydroxy camptothecin and 10-hydroxy camptothecin injection (1.0 mg/mL) were purchased from Wuhan Lishizhen Pharmaceutical Co., Ltd. (Wuhan, China). Hydroxyethyl starch (average Mw ∼200 000 Da, Ds = 0.5) were obtained from Chongqing Daxin Pharmaceutical Co., Ltd. (Chongqing, China). 1-Ethyl (3-dimethyllaminopropyl) carbodiimide hydrochloride (EDC HCl, 99%), 4-dimethylaminopyridine (DMAP, 99%) and N-hydroxysuccinimide (NHS, 99%) were purchased from Shanghai Medpep Co., Ltd. (Shanghai China). Acryloylchloride (98%), di-tert-butyl-dicarbonate (99%), pyridine (99%), succinic anhydride (99%) and ethanedithiol (97%) were obtained from Shanghai Darui Finechemical Co., Ltd. (Shanghai, China). 3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was furnished from Sigma. Dichloromethane (DCM) and dimethyl sulfoxide (DMSO) were dried with 4 Å molecular sieve before use. All chemicals and reagents used were of analytical or chromatographic quality. Deionized water was used throughout the study.
Measurements
1H- and 13C-NMR spectra were recorded on a Bruker AV 400 NMR spectrometer. The MALDI-TOF MS experiment was performed on an Auto ex III TOF/TOF Analyzer (Bruker Dal-tonics Inc., Germany). Acetonitrile of HPLC grade was purchased from Fisher Scientific (Fair Lawn, NJ). Distilled water was purified in a Barn-stead EASY pure II RF/UV ultrapure water system (Dubuque, Lowa) and filtered through a 0.22 μm filter prior to use in all the studies.
Animals and cell lines
Twelve male Wistar rats weighting 210 ± 20 g were obtained from the Laboratory Animal Center of Shenyang Pharmaceutical University. Rats were housed in groups of two or three with 12 h light/12 h dark cycle in an air-conditioned room (25 ± 2 °C) at a relative humidity of 45–60% for 1 week. The rats were fasted for 12 h but had free access to water prior to the administration.
Twenty four nude mouse weighting 20–25 g were obtained from the Laboratory Animal Center of Shenyang Pharmaceutical University. Mouse was housed in an air-conditioned room (25 ± 2 °C), at a relative humidity of 45–60%. All animal-use procedures were in accordance with the regulation for animal experimentation issued by the State Committee of Science and Technology of the People’s Republic of China.
Human liver cancer cell lines Hep-3B was obtained from the American Type Culture Collection (Manassas, VA). They were maintained as monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Invitrogen) supplemented with 10% fetal bovine serum (FBS) and maintained at 37 °C in a humidified incubator with 5% CO2. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Sigma(St. Louis, MO).
Synthesis and characteristics of 10-HCPT–HES conjugate (Zhao et al., 2008; Zhang et al., 2013)
Synthesis of 10-HCPT–HES conjugate
Reaction 1: To a suspension of 10-HCPT (1.002 g, 2.74 mmol) in 100 mL of anhydrous DCM were added di-tert-butyl dicarbonate (0.704 g, 3.21 mmol) and anhydrous pyridine (6.2 mL, 77.0 mmol). The suspension was stirred overnight at room temperature. The solution was washed with 0.5 N HCl (3 × 50 mL) and saturated NaHCO3 (1 × 50 mL). The organic phase was dried over MgSO4, filtered and evaporated under vacuum at 30 °C. The resulting solids were dried under vacuum at 40 °C to give 10-HCPT-(Boc)2. Reaction 2: To a solution of 10-HCPT-(Boc)2 (1.013 g, 2.03 mmol) of anhydrous DCM at 0 °C, acryloylchloride (AC, 0.308 g, 3.31 mmol) solution was added dropwise. The mixture was stirred at 0 °C for 30 min and then warmed up to room temperature, and maintained at this temperature for 2 h. The solution was washed with 0.5 N HCl (3 × 50 mL) and saturated NaHCO3 (1 × 50 mL). The organic phase was dried over MgSO4, filtered and evaporated under vacuum at 30 °C. The resulting solids were dried under vacuum at 40 °C to give 10-HCPT-BOC-AC. Reaction 3: To a solution of ethanedithiol (ESH, 0.271 g, 2.87 mmol) of anhydrous DCM, 10-HCPT-BOC-AC (0.807 g, 1.46 mmol) solution was added dropwise. The solution was stirred overnight at room temperature. After the reaction was completed, the reacted solution was added to the hexane and the 10-HCPT-SH precipitate was collected. The 10-HCPT-BOC-AC-ESH precipitates were dried under vacuum at 40 °C. Reaction 4: To a solution of anhydrous DCM, 10-HCPT-BOC-AC-ESH (0.709 g, 1.20 mmol), succinic anhydride (0.365 g, 3.60 mmol) and DMAP were added, and the solution was stirred at 30 °C for 24 h. The solution was washed with 50 °C distilled water (3 × 50 mL). The organic phase was dried over MgSO4, filtered and evaporated under vacuum at 30 °C. The resulting solids were dried under vacuum at 40 °C to give 10-HCPT-BOC-AC-ESH-COOH. Reaction 5: 10-HCPT-BOC-AC-ESH-COOH (0.654 g, 0.958 mmol) was dissolved in 50 mL of 30% TFA in anhydrous DMSO. The mixture was stirred for 3 h, and then the solution was added to the hexane and the precipitate was collected. The solids were dried under vacuum at 40 °C to give 10-HCPT-AC-ESH-COOH. Reaction 6: Briefly, EDC.HCl (0.602 g, 3.14 mmol) and DMAP (0.107 g, 0.882 mmol) were added to the solutions of 10-HCPT-AC-ESH-COOH (0.5012 g, 0.860 mmol) to activate the carboxyl groups, and the solutions were stirred continuously for 3 h at 40 °C. Subsequently, HES (1.498 g, 8.32 mmol) was added to the above solutions, and allowed to react for 48 h at 40 °C. After the reaction was completed, the reacted solution was added to the anhydrous ethanol and the precipitate was collected. The solids were dried under vacuum at 40 °C to give 10-HCPT–HES conjugate.
Characterization of 10-HCPT–HES conjugate
Following the synthesis of 10-HCPT–HES conjugate, TOF-MS and NMR techniques were performed to confirm the structure of products. In addition, there are three different forms of the 10-HCPT in pH 7.4 PBS solution, such as lactone ring form, carboxylate form and the conjugate form. Three forms of the 10-HCPT were monitored simultaneously by HPLC due to their polarity differences.
In vitro release of 10-HCPT–HES conjugates
In vitro 10-HCPT release
To determine the drug release of 10-HCPT from the 10-HCPT–HES conjugate, the conjugate was suspended in phosphate buffer saline (PBS) at a final concentration of 0.5 mg/mL in the dialysis bag (MWCO 3500 Da), and immersed in 50.0 mL PBS solution at 37 °C with constant shaking. At selected time intervals, 1.0 mL of solution outside the dialysis bag was removed for HPLC analysis and replaced with the same volume of fresh PBS. The concentrations of 10-HCPT present in the dialysate were determined using HPLC method. The HPLC system contained a pump (Hitachi 5110), an UV detector and a C18 column (Agilent, 250 mm × 4.6 mm). The mobile phase consisted of a mixture of water and methanol (v/v, 55/45) pass through a 0.45 μm membrane filter before use. The mobile phase was eluted at a flow rate of 1.0 mL/min and monitored by the UV detector at a wavelength of 384 nm.
Release of 10-HCPT from conjugate in plasma and liver homogenate
Fresh rat plasma and liver homogenate were diluted with physiological saline to 10, 30 and 60%, respectively. Then, 5.0 mg samples of 10-HCPT–HES conjugate were dissolved in 10 mL diluted rat plasma and liver homogenate and kept at 37 °C with constant shaking. Then, 0.2 mL samples were transferred to tubes at pre-determined intervals to determine the percentage release. The concentrations of 10-HCPT present in the dialysate were determined using HPLC method. The HPLC system contained a pump (Hitachi 5110), an UV detector, and a C18 column (Agilent, 250 mm × 4.6 mm). The mobile phase consisted of a mixture of water and methanol (v/v, 55/45) pass through a 0.45 μm membrane filter before use. The mobile phase was eluted at a flow rate of 1.0 mL/min and monitored by the UV detector at a wavelength of 384 nm. The experiments concerning rat plasma and liver homogenate were approved by the Committee on Ethical Use of Animals of Shenyang Pharmaceutical University Animal Center.
Cytotoxicity assay
The cytotoxicities of 10-HCPT–HES conjugate against Human liver cancer cell lines Hep-3B was evaluated by a MTT assay. Hep-3B cells were seeded into a 96-well plate at 6 × 103/well. Cells were incubated in an incubator in 5% CO2 at 37 °C environment for 24 h, subsequently the medium was removed, and the conjugate with varying concentration was added to cell wells for 48 and 72 h. Positive control treatments included varying concentrations of free 10-HCPT, and negative control with medium alone. About 50 μL of MTT (0.25 mg/mL) was added to each well at pre-determined times. After 4 h of incubation at 37 °C, the supernatant was removed, and 100 μL of DMSO was added to dissolve precipitated formazan crystals. The plate was shaken for 15 min. OD values was measured with ELISA reader at a wavelength of 492 nm. The apparent cytotoxicity was calculated according to the following equation, and cytotoxicity data were expressed as IC50, and the concentrations of the test agent producing a 50% reduction in cell numbers were compared with control cultures.
Pharmacokinetics of 10-HCPT–HES conjugate after intravenous administration
SD rats weighting 200–220 g were randomly divided into two groups and six rats for each. 10-HCPT–HES conjugate solution was administered via the femoral vein (1.5 mg/kg). As a reference group, commercial 10-HCPT injection was given at the same dose. The rats were anesthetized at appropriate intervals (5, 10, 15, 30, 60, 90 min, 2 h, 4 h, 6 h, 8 h, 12 h, 24 h, 36 h and 48 h) and 0.5 mL blood samples were collected from the retro-orbital sinus and transferred into heparin treated tubes, and then immediately centrifuged at 4000 rpm for 10 min to obtain plasma sample. The samples were stored at −20 °C until required for analysis. The experiments concerning procedures and the animal use and care protocols were approved by the Committee on Ethical Use of Animals of Shenyang Pharmaceutical University Animal Center.
The plasma samples were determined by LC/MS/MS (Waters) method, and the pharmacokinetics data were analyzed by drug and statistics (DAS) version software 2.0 (Rozet et al., Citation2011; Li et al., Citation2014).
The pharmacodynamic test of 10-HCPT–HES conjugate after intravenous administration
To assess anti-tumor activity in vivo of the conjugate, the pharmacodynamic test of 10-HCPT–HES conjugate was performed in tumor bearing nude mouse. Nude mouse weighting 20–25 g were randomly divided into four groups, consisting of six rats in each group, and subcutaneously transplanted with 1 × 105 human Hep-3B cells on the right flank on day 0. Mice which developed palpable tumors reaching 10–12 mm in diameter within 5 days after the implantation began to receive injections via the femoral vein. 10-HCPT–HES conjugate solutions at the dosage of 0.75 and 1.5 mg/kg, normal saline as a blank control, and commercial 10-HCPT injection at the dosage of 1.5 mg/kg as a reference were administered to each group separately. The injection was given twice a week for consecutive 3 weeks, on Monday and Thursday, respectively. The weight and tumor volume of the mice were measured every 2 days. After the tests were completed, the mice were sacrificed and the tumors were totally excised. The pharmacodynamics data were analyzed by SPSS software (SPSS Inc., Chicago, IL). Based on the weight of tumors, the inhibition ratios of tumor were calculated according to the following equation:
Results and discussion
Synthesis and characterization of 10-HCPT–HES conjugate
The synthesis process of 10-HCPT–HES conjugate is shown schematically in Supplementary Figure S1. The reaction between –OH groups from HES and –COOH groups from 10-HCPT occurs. The products are validated by NMR and TOF MS, and the results are as follows:
10-HCPT-(Boc)2: 1H-NMR (400 MHz, CDCl3): δ1.00 (3H, t), 1.65 (9H, s), 1.80–1.95 (2H, m), 5.23 (2H, d), 5.75 (1H, d), 7.63 (1H, dd), 7.69 (1H, s), 8.17 (1H, dd), 8.23 (1H, d). TOFMS (ESI) calculated for C34H40N3O10(M + H)+: 493.1975, found: 493.1971.
10-HCPT-BOC-AC: 1H-NMR (400 MHz, CDCl3): δ: 1.15 (3H, t), 1.75 (9H, s), 1.95–2.15 (2H, m), 2.452 (2H, m), 3.842 (2H, q), 5.38 (2H, d), 5.91 (1H, d), 6.22 (1H, d), 6.48 (1H, m), 7.83 (1H, dd), 7.91 (1H, s), 8.35 (1H, dd), 8.50 (1H, d). TOFMS (ESI) calculated for C37H42N3O11(M + H)+: 547.1875, found: 547.1871.
10-HCPT-BOC-AC-ESH: 1H-NMR (400 MHz, CDCl3): δ: 1.08 (3H, t), 1.60 (9H, s), 1.68 (2H, m), 1.85–1.95 (2H, m), 2.362 (2H, t), 2.842 (2H, t), 3.72 (2H, q), 5.28 (2H, d), 5.75 (2H, d), 7.68 (1H, dd), 7.77 (1H, s), 8.25 (1H, dd), 8.36 (1H, d). TOFMS(ESI) calculated for C39H48N3S2O10(M + H)+: 583.7058, found: 583.7053.
10-HCPT-BOC-AC-ESH-COOH: 1H NMR (400 MHz, CDCl3): δ: 1.11 (3H, t), 1.78 (9H, s), 2.18 (2H, m), 2.43 (2H, m), 2.75 (2H, t), 3.18 (2H, t), 3.72 (2H, q), 5.24 (2H, d), 5.49 (2H, d), 7.38 (1H, dd), 7.55 (1H, s), 8.36 (1H, dd), 8.45 (1H, d). TOFMS(ESI) calculated for C43H52N3 S2O13(M + H)+: 683.7758, found: 683.7752.
10-HCPT-AC-ESH-COOH: 1H-NMR (400 MHz, DMSO): δ: 1.08 (3H, t), 2.15 (2H, m), 2.41 (2H, m), 2.73 (2H, t), 3.12 (2H, t), 3.68 (2H, q), 5.23 (2H, d), 5.45 (2H, d), 7.39 (1H, dd), 7.57 (1H, s), 8.38 (1H, dd), 8.44 (1H, d). TOFMS (ESI) calculated for C38H43N3 S2O11(M + H)+: 582.6307, found: 582.6305.
The final 10-HCPT–HES conjugate was characterized by 13C NMR and the spectrum is shown in Supplementary Figure S2.
There were several types of C atoms with different chemical environments, such as carboxyl C atoms, alkyl C atoms and aromatic C atoms in the 10-HCPT–HES conjugate. For HES, there were lots of alkyl C atoms, and the absorption peaks of its C atoms appeared at a chemical shift of about 30 ppm (alkyl C atoms) and 60–80 ppm (alkyl C atoms connected to O atom). For 10-HCPT, the chemical shift of carboxyl C atoms of lactone (C19) and C17 was 167.50 and 156.72, and the chemical shift of aromatic C atoms was 149.50, 146.74, 145.52, 142.98, 129.37, 122.62, 117.96 and 108.70. For 10-HCPT–HES conjugate, it can be seen all the absorption peaks of 10-HCPT and HES from Supplementary Figure S2. Meanwhile, the absorption peaks of linker’s carboxyl C atoms (C77, C81 and C28) were observed, and its chemical shift was 171.40 and 169.76. In addition, there were three different forms of the 10-HCPT in pH 7.4 PBS solution, such as lactone ring form, carboxylate form and the conjugate form. Three forms of the 10-HCPT were monitored simultaneously by HPLC due to their polarity differences, and the retention time were 3.8, 6.0 and 14.8 min, respectively. In this study, the absorption peak of 10-HCPT–HES conjugate appeared at 6.0 min, and there were no other peaks appeared at HPLC spectra. Thus, we inferred that the conjugation reaction between 10-HCPT and HES occurred successfully.
Drug loading and water solubility
It was found that the water solubility and drug loading of the conjugate had important influence on its in vivo pharmacokinetics and antitumor activity. In this study, we prepared series of 10-HCPT–HES conjugates with different drug loadings (1.8, 3.5, 5.7, 8.1 and 9.5%) and investigated the relationship between drug loading and water solubility of the conjugate. The concrete results are shown in .
Table 1. The relationship between drug loading and water solubility of 10-HCPT–HES conjugate.
It can be seen that the solubility of 10-HCPT–HES conjugate decreased with the increase of the drug loading. The reason may be that the –COOH group of 10-HCPT would react to the hydroxyethyl group of HES preferentially due to low steric hindrance and high reactivity of hydroxyethyl group. The number of hydroxyethyl group would decrease with the increase of the drug loading, which may lead to the solubility of 10-HCPT–HES conjugate decreased because the amount of hydroxyethyl group had a significant impact on the solubility and in vivo behavior of HES. In addition, it was found that the conjugate showed minor value when the drug loading was 1.8% or even 3.5% due to the increase in dosing volume, and excessive HES exhibited a higher viscosity. Meanwhile, the high drug loading (9.5%) leaded to a significant decrease in the solubility of the conjugate, thus it also was not a good choice. Finally, we obtained a suitable solubility when the drug loading was 8.1%.
In vitro release of 10-HCPT–HES conjugate
The 10-HCPT release behavior from the 10-HCPT–HES conjugate was investigated in PBS solutions, plasma and liver homogenate using the dialysis method. The cumulative release percentages of 10-HCPT versus time are plotted in and .
Figure 2. Cumulative release (%) of 10-HCPT from 10-HCPT–HES conjugate in phosphate buffer saline (PBS) solution at 37 °C, -: pH 8.4; ▴: pH 7.4; ♦: pH 4.0; ▪: pH 6.0.
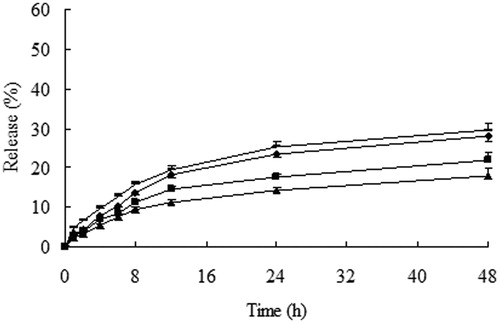
Figure 3. Cumulative release (%) of 10-HCPT from 10-HCPT–HES conjugate in plasma and liver homogenate at 37 °C, ♦: 10% plasma; -: 10% liver homogenate; ▪: 30% plasma; ▴: 30% liver homogenate; -: 60% plasma; ▪: 60% liver homogenate.
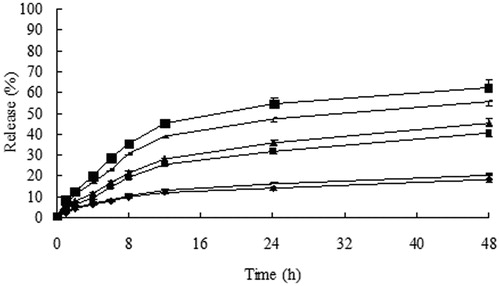
It can be seen from that minimal (less than 20%) loaded 10-HCPT was released from the conjugate in 48 h at pH 6.0 and 7.4. while the release degree of 10-HCPT increased when the conjugate was dissolved in pH 4.0 and pH 8.4 PBS solutions. This may be due to the hydrolysis of ester bonds more easily under strong acid and alkaline conditions. However 10-HCPT cannot release completely in PBS solutions, these results may be explained that HES can provide stereo-protective effect to 10-HCPT because HES had large molecular weight and lots of branching chains, meanwhile HES cannot be degraded in PBS solutions, which may protect the linkers from hydrolysis further. Whereas showed that the release rate and quantity of 10-HCPT from the conjugate increased significantly in rat plasma and liver homogenate with the increase of plasma and liver homogenate concentration. In addition, the release rate and quantity of 10-HCPT in liver homogenate were higher than in rat plasma at the same concentration. The reason may be that there were a variety of enzymes in plasma and liver homogenate, such as starch hydrolysis enzymes, esterase, which accelerated the breaking of ester linkers and promoted the degradation of HES polymer. Moreover, the stereo-protective effect of HES to 10-HCPT decreased in pace with the decrease of HES molecule mass. Separately, the release of 10-HCPT in liver homogenate was higher than in plasma, the results could be attributed to the types and quantity of enzymes in liver homogenate were higher than in plasma, which may promote the degradation of HES and the release of 10-HCPT.
Sum up, 10-HCPT–HES conjugate exhibited apparent sustained release effect in PBS solutions, plasma and liver homogenate. Meanwhile, it was found that the pH value of release medium and enzyme can influence the release of 10-HCPT from 10-HCPT–HES conjugate.
In vitro cytotoxicity test
The biocompatibility of HES was estimated using a MTT assay toward Hep-3B cells. The effect of 10-HCPT–HES conjugate concentrations on the proliferation of the cells and IC50 are shown in and , respectively. The results displayed that the HES molecule did not exhibit apparent inhibition effects on cell proliferation at all concentrations, which indicated good cytocompatibility of the HES. It can be seen that the cytotoxicity effect increased with the dose increase. A similar trend was observed for the 10-HCPT–HES conjugate, which exhibited lower IC50 than free 10-HCPT. The possible reason may be that the 10-HCPT–HES conjugate had an excellent cell penetrating property and lactone stability, while the free 10-HCPT was rapidly hydrolyzed to the carboxylate form under physiological conditions and the carboxylate form was difficult to enter the cell due to higher polarity. As we all know that polymer–drug conjugate usually shows higher IC50 than the free drug due to changes in ways of entering cells, such as slower endocytic capture compared with rapid transmembrane passage of the free drug. However, in this study, 10-HCPT–HES conjugate exhibited lower IC50 in comparison with free 10-HCPT, which may result from the structure characteristics of 10-HCPT. On the one hand, 10-HCPT held a pH-dependent equilibrium in aqueous medium between the lactone and the carboxylate forms. In general, the lactone form is essential for anti-tumor activity and the carboxylate form is almost inactive. While the majority of free 10-HCPT would exist in carboxylate form because the pH value of the cell culture solution is about 7.4, which may result in the decrease of cytotoxicity. On the other hand, the polarity of 10-HCPT would increase when 10-HCPT existed in the form of carboxylate form, which may hinder the access of 10-HCPT into cell.
Figure 4. Results of MTT assay on Hep-3B cells after incubation of 48 and 72 h with 10-HCPT and 10-HCPT–HES conjugate at various 10-HCPT concentrations.
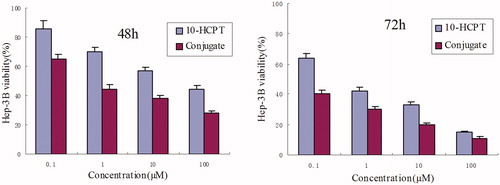
Table 2. IC50 values determined in Hep-3B cell line cultured with 10-HCPT–HES conjugate and free 10-HCPT for 48 and 72 h.
Pharmacokinetics of 10-HCPT–HES conjugate
The mean plasma concentration–time curves of 10-HCPT injection and 10-HCPT–HES conjugate are shown in , and the pharmacokinetic parameters are presented in . It can be seen that 10-HCPT injection was rapidly eliminated from rat plasma with t1/2 about 10 min, and this was consistent with the result reported by literature. In contrast, 10-HCPT–HES conjugate significantly extended the half-life of 10-HCPT to 4.38 h. Furthermore, it was noteworthy that the bioavailability of conjugate groups was 40-fold higher than that of 10-HCPT injection group. It was well known that the extension of the half life of 10-HCPT was a great advantage since it can increase the opportunity of drug arriving at tumor site.
Figure 5. The rat plasma concentration versus time curves of 10-HCPT–HES conjugate and 10-HCPT injection after intravenous administration at a dose of 1.5 mg/kg, ♦: 10-HCPT injection; ▪: 10-HCPT–HES conjugate.
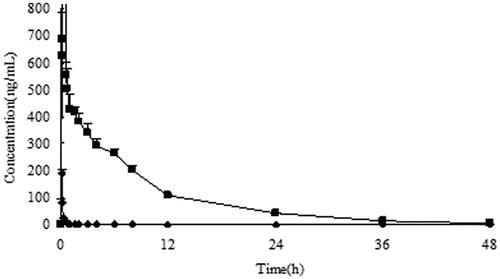
Table 3. Pharmacokinetic parameters of 10-HCPT after intravenous administration of different formulations (mean ± SD).
The long circulating effect of the conjugate can be explained by the characteristics of 10-HCPT–HES conjugate. First, the ester linkers exhibited good stability under neutral plasma environment, and the cracking of linkers would require long circulating time. Second, HES was a macromolecule with large molar mass and lots of branched chain structure. After the 10-HCPT–HES conjugate were prepared, 10-HCPT can be shielded by HES, thus the stability of 10-HCPT was increased significantly. In addition, there were a large number of hydroxyl groups in HES molecule, so the hydrogen bonds may be formed between HES and the hydroxyl group of 10-HCPT, which can increase the protective effects to 10-HCPT further. Third, HES showed a slow degradation rate in vivo after the introduction of hydroxyethyl groups in amylopectin structure, thus the retention time of HES in vivo was extended significantly in comparison with natural amylopectin. The in vivo behavior of HES molecule can be controlled by α-amylase, which was a dynamic balance, concluding hydrolysis, supplement and elimination process. Therefore, the release of 10-HCPT from the conjugate had a close relationship with the degradation process of HES in vivo. Meanwhile, the hydratability of HES would decrease in pace with the introduction of hydrophobic 10-HCPT molecule because hydrophobic materials can produce significant stacking phenomenon, which may hinder the access of other plasma materials. In addition, 10-HCPT–HES conjugate solution had higher viscosity than water solution due to polymer solution property of HES, so the colloid osmotic pressure of the blood will change after administration of 10-HCPT–HES conjugate, which may slow down the contact rate between conjugate and other plasma components. All of these properties made 10-HCPT–HES conjugate showed obvious long circulating effect.
The pharmacodynamic test of 10-HCPT–HES conjugate
In vivo antitumor activity of 10-HCPT–HES conjugate was performed in nude mouse bearing Hep-3B liver cancer cells, and the results are shown in and . It can be seen that the growth of tumor volume was rapid in negative control groups (treated with saline solution). In contrast, growth of tumor in mice treated with any 10-HCPT formulations significantly slowed down. 10-HCPT–HES conjugate found to be more effective in inhibiting the growth of tumor in comparison with 10-HCPT injection group. For example, the average tumor weight of the negative control group was 4.385 g. For mice treated with 0.75 and 1.5 mg/kg 10-HCPT–HES conjugate, the mean tumor weight was 2.033 ± 0.236 and 0.706 ± 0.085 g, respectively, which were significantly smaller than that of HCPT injection group (3.024 g). The inhibition rate of 10-HCPT–HES conjugate was 83.9%, which was 3.0-fold of 10-HCPT injection. Weight loss was observed after mice injected with 10-HCPT injection in the first 2 days, while 10-HCPT–HES conjugate groups did not show significant weight loss. In addition, it was found that 10-HCPT injection showed severe irritation to mice, such as temperature drops, loss of locomotor activity, etc. In contrast, 10-HCPT–HES conjugate group showed no important irritation. Thus, we concluded that 10-HCPT–HES conjugate can decrease the irrigation of the drug and increase the tolerance of the mice significantly. The reason may be as follows: 10-HCPT–HES conjugate increased the solubility and lactone stability of 10-HCPT, and showed better long circulating effect in vivo, which can promote the accumulation of 10-HCPT in tumor. The phagocytosis effect of macrophage may be decreased as the soluble branching chains of HES can protect the conjugate from recognizing by macrophage to some extent. In addition, 10-HCPT–HES conjugate exhibited better sustained effects during the transportation, which can alter organ distribution and subsequent clearance of 10-HCPT and promote the tumor targeting through the EPR effect. The characteristics of polymer may play a key role in the effect of conjugate. HES can increase the lactone ring stability of 10-HCPT, which made 10-HCPT can exhibit antitumor activity in the lactone form. Thus the antitumor effect of 10-HCPT–HES conjugate was better than 10-HCPT injection.
Figure 6. The tumor growth curve of nude mouse after intravenous administration of 10-HCPT injection and 10-HCPT–HES conjugate, ♦: negative group; ▪: 10-HCPT injection (1.5 mg/kg); ▴: 10-HCPT–HES conjugate solution (0.75 mg/kg); -: 10-HCPT–HES conjugate solution (1.5 mg/kg).
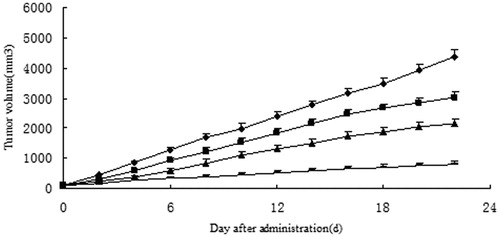
Table 4. Anti-tumor effects of 10-HCPT injection and 10-HCPT–HES conjugate against Hep-3B cell in nude mouse.
Conclusions
In this study, the 10-HCPT–HES conjugate with controlled drug loading and release profile has been prepared and characterized. HES can significantly improve the solubility and increase lactone stability of 10-HCPT. The prepared conjugate exhibited apparent sustained release effect in PBS solutions, plasma and liver homogenate. The 10-HCPT–HES conjugate showed much lower IC50 and higher cytotoxicity against Hep-3B cancer cells in comparison with the parent drug. The in vivo pharmacokinetic results showed that the conjugate produced much higher bioavailability and longer half-life than the pristine 10-HCPT. The pharmacodynamic study showed that the conjugate increased the anti-tumor effect of 10-HCPT and reduced the side effects. Overall, 10-HCPT–HES conjugate can significantly overcome the disadvantages of 10-HCPT and exhibit a better pharmacological activity. In addition, this is a preliminary research, and further studies on applications of the conjugate are underway in our team.
Supplemental Material.pdf
Download PDF (45.1 KB)Acknowledgements
Dr. David B. Jack is gratefully acknowledged for correcting the manuscript.
Declaration of interest
The authors report no conflicts of interest. The authors are responsible for the content and writing of this article. This work is supported by the National High-tech R&D Program of China 863 Program (2012AA020305).
References
- Baier G, Baumann D, Siebert JM, et al. (2012). Suppressing unspecific cell uptake for targeted delivery using hydroxyethyl starch nanocapsules. Biomacromolecules 13:2704–15
- Bhatt R, Vries P, Tulinsky J, et al. (2003). Synthesis and in vivo antitumor activity of poly(L-glutamic acid) conjugates of 20(S)-camptothecin. J Med Chem 46:190–3
- Botella P, Abasolo I, Fernandez Y, et al. (2011). Surface-modified silica nanoparticles for tumor-targeted delivery of camptothecin and its biological evaluation. J Controlled Release 156:246–57
- Burke GT, Mi Z. (1994). The structural basis of camptothecin interactions with human serum albumin: impact on drug stability. J Med Chem 37:40–6
- Cheng JJ, Khin KT, Davis ME. (2004). Antitumor activity of β-cyclodextrin polymer-camptothecin conjugates. Mol Pharm 3:183–93
- Cheng JJ, Khin KT, Jensen GS, et al. (2003). Synthesis of linear, β-cyclodextrin-based polymers and their camptothecin conjugates. Bioconjug Chem 14:1007–17
- Deshmukh M, Chao P, Kutscher HL, et al. (2010). A series of α-amino acid ester prodrugs of camptothecin: in vitro hydrolysis and A549 human lung carcinoma cell cytotoxicity. J Med Chem 53:1038–47
- Fleming AB, Haverstick K, Saltzman WM. (2004). In vitro cytotoxicity and in vivo distribution after direct delivery of PEG-camptothecin conjugates to the rat brain. Bioconjug Chem 15:1364–75
- Joralemon MJ, McRae S, Emrick T. (2010). PEGylated polymers for medicine: from conjugation to self-assembled systems. Chem Commun 46:1377–93
- Lerchen HG, Baumgarten J, Bruch K, et al. (2001). Design and optimization of 20-O-linked camptothecin glycoconjugates as anticancer agents. J Med Chem 44:4186–95
- Li C, Wallace S. (2008). Polymer-drug conjugates: recent development in clinical oncology. Adv Drug Deliv Rev 60:886–98
- Li G, Cai C, Ren T, Tang X. (2014). Development and application of a UPLC-MS/MS method for the pharmacokinetic study of 10-hydroxy camptothecin and hydroxyethyl starch conjugate in rats. J Pharm Biomed Anal 88:345–53
- Lorence A, Nessler CL. (2004). Camptothecin, over four decades of surprising findings. Phytochemistry 65:2735–49
- Maeda H, Bharate GY, Daruwalla J. (2009). Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm 71:409–19
- Mendichi R, Rizzo V, Gigli M, Schieroni AG. (2002). Fractionation and characterization of a conjugate between a polymeric drug-carrier and the antitumor drug camptothecin. Bioconjug Chem 13:1253–8
- Noga M, Edinger D, Klager R, et al. (2013). The effect of molar mass and degree of hydroxyethylation on the controlled shielding and deshielding of hydroxyethyl starch-coated polyplexes. Biomaterials 34:2530–8
- Noga M, Edinger D, Rodl W, et al. (2012). Controlled shielding and deshielding of gene delivery polyplexes using hydroxyethyl starch (HES) and alpha-amylase. J Controlled Release 159:92–103
- Rozet E, Marini RD, Ziemons E, et al. (2011). Advances in validation, risk and uncertainty assessment of bioanalytical methods. J Pharm Biomed Anal 55:848–58
- Samor C, Guerrini A, Varchi G, et al. (2008). The role of polyamine architecture on the pharmacological activity of open lactone camptothecin-polyamine conjugates. Bioconjug Chem 19:2270–9
- Subramanian N, Sundaraganesan N, Sudha S, et al. (2011). Experimental and theoretical investigation of the molecular and electronic structure of anticancer drug camptothecin. Spectrochim Acta Part A 78:1058–67
- Tong R, Cheng JJ. (2010). Controlled synthesis of camptothecin-polylactide conjugates and nanoconjugates. Bioconjug Chem 21:111–21
- Torchilin V. (2011). Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev 63:131–5
- Venditto VJ, Simanek EE. (2010). Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol Pharm 7:307–49
- Zhang H, Wang J, Mao W, et al. (2013). Novel SN38 conjugate-forming nanoparticles as anticancer prodrug: in vitro and in vivo studies. J Controlled Release 166:147–58
- Zhao H, Rubio B, Sapra P, et al. (2008). Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug Chem 19:849–59