Abstract
The biological evaluation of carbocyclic minor groove binders 1–6 is described. The cytotoxicity of the obtained compounds was tested on MDA-MB-231 breast cancer cells. The mechanism of action of compounds 1–6 was studied employing the topoisomerase I/II inhibition assay and ethidium displacement assay using pBR322. Determination of association constants was done using calf thymus DNA, T4 coliphage DNA, poly(dA-dT)2, and poly(dG-dC)2. The effect of compounds 1–6 on the amidolytic activity of plasmin, trypsin, thrombin, and urokinase was also examined.
Introduction
The clinical significance of DNA-binding compounds can hardly be overstated, as many anticancer regimens include the compound that binds to and/or modifies DNA.
Netropsin, bis-netropsin, or bis-amidines (e.g. pentamidine) () have been extensively studied due to their ability to bind to the minor groove of the DNA double helix in a sequence-specific manner, and have served as models for biochemical and physical studies of drugs that bind to the DNA minor grooveCitation1. In particular, it has been shown that they bind DNA reversibly through hydrogen bonds, van der Waals contacts, and electrostatic interactions at sequences of four or more consecutive AT pairs, and strongly discriminate against GC pairsCitation2. The rapidly increasing knowledge in molecular biology affords possibilities for the observation that this large family of sequence-specific ligands nonintercalatively binding within the minor groove of B-DNA is very important in the antitumor drug searchCitation3. The inhibition of many cellular processes and the cytotoxic effects of these antineoplastic agents are determined mainly by interference with the catalytic activity of important enzymes, such as DNA topoisomerases or a number of proteasesCitation4.
Figure 1. Structures of netropsin, pentamidine, and some carbocyclic minor groove binders.
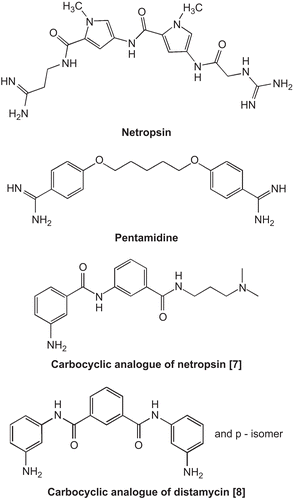
A model of binding of netropsin and distamycin with B-DNA became the inspiration for seeking new compounds with a similar interaction with DNA. The concept of information-reading molecules was introduced. This class of synthetic heteroaromatic oligopeptides, projected after modeling netropsin and the other minor groove binders, received the name lexitropsinsCitation5. Although there has been huge progress in designing distamycin and netropsin analogues, so far it has not resulted in compounds that can be applied in therapyCitation6.
The present work is in conjunction with our ongoing program on the synthesis and biological studies of carbocyclic potential minor groove binders. Such lexitropsins, which are readily available, can be modified easily, and are stable under most experimental conditions. We have found that carbocyclic analogues of netropsin and distamycin () can be used as carriers for the groove-specific delivery of functionalized groups to DNACitation7,Citation8. Molecular modeling of their interaction with d(CGCGAATTCGCG)2 showed that their structure is effectively isohelical with the DNA minor groove, however with decreased affinity for the minor groove of AT-rich regions in comparison to netropsin and distamycinCitation9. From energy analysis it appears that van der Waals and electrostatic interactions are more imortant than specific hydrogen bonds in stabilizing the ligand–duplex complexes. This has been confirmed by investigation of such distamycin analogues with a free aromatic amine group, which showed antiproliferative and cytotoxic effects against both MDA-MB-231 and MCF-7 cell linesCitation10.
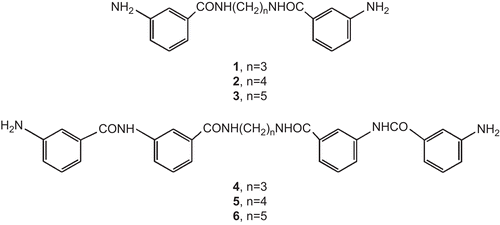
We present the biological evaluation of carbocyclic potential minor groove binders 1–6 with free aromatic amine groups (). Their synthesis and activity in the standard mammalian tumor cell line MCF-7 were described earlierCitation11. Here we present their antiproliferative and cytotoxic effects against MDA-MB-231 breast cancer cells. The ethidium displacement assay was used to show whether these compounds bind to plasmid pBR322. Determination of the association constants of drug–DNA complexes was done using calf thymus DNA, T4 coliphage DNA, poly(dA-dT)2, and poly(dG-dC)2 Citation12,Citation13.
Figure 2. Structures of compounds 1–6.
Because the antitumor activity of DNA-binding drugs is not due to the interaction with DNA per se but is, at least in part, the result of the inhibition of different enzymes, the mechanism of action of compounds 1–6 was also studied employing the topoisomerase I/II inhibition assay.
Plasmin and urokinase are involved in numerous biological processes, and also in tumor invasion, metastasis, and angiogenesisCitation14,Citation15. We reported earlier that some netropsin and pentamidine amino analogues inhibited the amidolytic activity of plasmin or trypsinCitation16. Here we present the effect of compounds 1–6 on the amidolytic activity of serine proteases: plasmin, trypsin, thrombin, and urokinase.
Materials and methods
Pharmacology
Ethidium bromide was purchased from Carl Roth GmbH, and topoisomerase I (calf thymus) and II (Escherichia coli containing a clone of human topoisomerase II gene) from Amersham Pharmacia Biotech, and were used without further purification. Stock cultures of MDA-MB-231 were purchased from the American Type Culture Collection, Rockville, MD, USA. Dulbecco’s modified Eagle’s medium, fetal bovine serum (FBS), distamycin, netropsin, pentamidine, streptomycin, and penicillin were products of Sigma. Plasmid pBR322 was purchased from Fermentas Life Sciences.
Urokinase, trypsin, and Bzl-l-Arg-pNA·HCl (Bzl = benzyl) were purchased from Sigma. Plasmin S-2444 (pyro-Glu-Gly-Arg-pNA·HCl), S-2238 (H-d-Phe-Pip-Arg-pNA), and S-2251 (H-d-Val-Leu-Lys-pNA) were obtained from Chromogenix. Thrombin was purchased from Lubelska Wytwórnia Szczepionek.
Cell culture
Human breast cancer MDA-MB-231 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, 50 μg/mL streptomycin, and 100 U/mL penicillin at 37°C, in an atmosphere containing 5% CO2. Cells were cultivated in Costar flasks, and subconfluent cells were detached with 0.05% trypsin and 0.02% ethylenediaminetetraacetic acid (EDTA) in calcium-free phosphate-buffered saline. The study was carried out using cells growing as a monolayer in six-well plates (Nunc) (5 × 10Citation5 cells per well) and preincubated for 24 h without phenol red.
Determination of IC50
The compounds were dissolved in dimethylsulfoxide (DMSO)/H2O (10:90) and used at concentrations of 5, 10, 15, 30, and 50 μM. Microscopic observations of cell monolayers were performed with a Nikon Optiphot microscope. Wright–Giemsa staining was performed using the Fischer Leuko Stat Kit. After 24 h of drug treatment, MCF-7 cells were mixed with a dye mixture (10 μM acridine orange and 10 μM ethidium bromide, prepared in phosphate-buffered saline). At the end of each experimental time point, all the medium was removed, and cells were harvested by incubation with 0.05% trypsin and 0.02% EDTA for 1 min, and then washed with the medium. Then, 250 μL of cell suspension was mixed with 10 μL of dye mix and 200 cells per sample were examined by fluorescence microscopy. The percentage of nonviable (apoptotic and necrotic) cells was calculated, and concentrations that inhibited 50% of colony formation (IC50 values) were determined. The results were submitted to statistical analysis using the method of least squares.
Relaxation assay of topoisomerase I and II
Native pBR322 plasmid DNA (0.20 μg) was incubated with 4 units topoisomerase I (reaction buffer: 50 mM Tris-HCl (pH 7.9), 1 mM EDTA, 0.5M NaCl, 1 mM dithiothreitol) or topoisomerase II (reaction buffer: 10 mM Tris-HCl (pH 7.9), 1 mM adenosine triphosphate (ATP), 50 mM KCl, 5 mM MgCl2, 50 mM NaCl, 0.1 mM EDTA, 15 μg/mL bovine serum albumin) in the absence or presence of varying concentrations of the test compounds (10, 50, and 100 μM) in a final volume of 10 μL. The mixture was incubated at 37°C for 30 min and the reaction was terminated by the addition of 2 μL of 10% sodium dodecyl sulfate (SDS). The reaction mixture was subjected to electrophoresis (3 h, 90 V) through a 1.0% agarose gel in TBE buffer (90 mM Tris-borate and 2 mM EDTA). The gels were stained for 30 min with ethidium bromide solution (0.5 μg/mL). The DNA was visualized using a 312 nm wavelength transilluminator and photographed under ultraviolet (UV) light. For the quantitative determination of topoisomerase activity, an area representing supercoiled DNA, migrating as a single band at the bottom of the gel, was measured using the AlphaEaseFC gel documentation and analysis system (Alpha Innotech, USA). The concentration of compound that converted 50% of the supercoiled DNA (IC50 values) was determined by averaging the data from at least three experiments.
Ethidium bromide assay
Each well of a 96-well plate was loaded with Tris buffer containing ethidium bromide (0.1 M Tris, 1 M NaCl, pH 8.0, 0.5 mM EtBr, final concentration, 100 μL). To each well was added 15 μg plasmid pBR322 as an aqueous solution (0.05 μg/μL). Then, to each well was added pentamidine, netropsin, or compound 1–6 (1 μL of 1 mM solution in water, 10 μM final concentration). After incubation at 25°C for 30 min, the fluorescence of each well was read on a Tecan Infinite M200 fluorescence spectrophotometer (excitation wavelength 546 nm, emission wavelength 595 nm) in duplicate experiments with two control wells (no drug = 100% fluorescence, no DNA = 0% fluorescence). Readings are reported as percentage fluorescence relative to control.
Ethidium displacement assay: determination of association constants
The fluorescence of the DNA solutions (calf thymus DNA, poly(dA-dT)2, T4 DNA, and poly(dG-dC)2) with the investigated compounds (at final concentrations of 10, 50, 75, 100, 150, and 200 μM) was measured on a Tecan Infinite M200 fluorescence spectrophotometer at room temperature, according to the procedure described above. Then, the concentration that reduced the fluorescence to 50% was determined. The fluorescence intensity data points were fit to theoretical curves with one or two different iterative nonlinear least-squares computer routines. The apparent binding constant was calculated from:
where [drug] is the concentration of test compound at a 50% reduction of fluorescence and KEtBr and [EtBr] are knownCitation11,Citation12. Compounds 1–6 and their DNA-bound complexes showed neither optical absorption nor fluorescence at 595 nm, and did not interfere with the fluorescence of unbound ethidium.
Antiamidolytic assay
Determination of the amidolytic activity was performed as previously describedCitation17. A detailed description of the method is given below. Quantities of 0.2 mL of examined preparation (as control, 0.15 M NaCl), buffer, and 0.1 mL of enzyme solution were mixed together. The mixture was incubated at 37°C for 3 min then the synthetic substrate solution in the same buffer was added. After 20 min of incubation, the reaction was stopped by adding 0.1 mL of 50% acetic acid, and the absorbance of the released p-nitroaniline was measured at 405 nm. Every value represents the average of triplicate determination. The IC50 value is considered as the concentration of inhibitor that decreases the absorbance by 50%, compared with the absorbance measured under the same conditions without an inhibitor.
enzyme: urokinase (50 units/mL), synthetic substrate: S-2444 (0.1 mL, 3 mM/L), Tris buffer: 0.6 mL (pH 8.8);
enzyme: thrombin (1 unit/mL), synthetic substrate: S-2238 (0.2 mL, 0.75 mM/L), Tris buffer: 0.5 mL (pH 8.4);
enzyme: plasmin (0.4 unit/mL); synthetic substrate: S-2251 (0.2 mL, 3 mM/L), Tris buffer: 0.5 mL (pH 7.4);
enzyme: trypsin (0.4 unit/mL), synthetic substrate: Bzl-l-Arg-pNA·HCl (0.2 mL, 8 mM/L), borane buffer: 0.5 mL (pH 7.5).
Our results were compared with the data obtained for 2-phenethyl-SO2-d-Ser-Ala-Arg-al, the irreversible urokinase plasminogen activator (uPA) inhibitor with the same tripeptide sequenceCitation18. The determination methods were identical.
Statistical analysis
In all experiments, the mean values for three assays ± standard deviations (SD) were calculated.
The results were submitted to statistical analysis using Student’s t test. Differences were considered significant when p < 0.05. Mean values, standard deviations, and the number of measurements in a group are presented in the tables.
Results and discussion
Antiproliferative and cytotoxic effects of compounds 1–6 in the standard human breast cancer cell line MCF-7 were investigated earlierCitation11. Their MCF-7 IC50 values together with in vitro antitumor activities in estrogen-independent human breast cancer cells MDA-MB-231 are presented in . All of the tested compounds showed concentration-dependent activity. Against MDA-MB-231 cells, the compounds were more cytotoxic than pentamidine with IC50 = 17.74 ± 2 μM and netropsin with IC50 = 228.80 ± 2 μM. The compound concentration that inhibited 50% of colony formation was in the range 8.10 ± 2 to 17.52 ± 2 μM. IC50 values against the MCF-7 cell line were in the range 209.8 ± 2 to 406.12 ± 2 μM, while IC50 of pentamidine was 14.31 ± 2 μM and of netropsin was 5.40 ± 2 μM. From these data we can see that compounds 1–6 were nearly 20 times more active against MDA-MB-231 than against MCF-7 cells.
Table 1. Antiproliferative activity of netropsin (NT), pentamidine (PN), and compounds 1–6 against breast cancer cells.
To test whether the cytotoxic properties were related to DNA-binding and topoisomerase I/II inhibition, the new minor groove binders were evaluated in a cell-free system. The ethidium bromide assay showed that the investigated compounds could bind to plasmid DNA, although relatively weaker than netropsin and pentamidine (). The topoisomerase DNA-inhibitory effects and binding affinities of compounds 1–6, netropsin, and pentamidine to calf thymus DNA, T4 coliphage DNA, and synthetic polymers poly(dA-dT)2 and poly(dG-dC)2 are presented in . These data demonstrate that all compounds could bind to the DNAs studied. The high-binding constant values for T4 coliphage DNA for 1–6 gave evidence of their minor-groove selectivity, because the major groove of T4 coliphage DNA is blocked by α-glycosylation of the 5-(hydroxymethyl)cytidine residuesCitation19. The DNA-binding data reported in characterize the affinity of compounds 1–6 for a more limited set of DNA-binding sites, and can give an indication of base-sequence specificity for DNA-binding molecules. These data indicate that compounds 1–6 interacted with a GC base pair, though the binding affinity was weak compared with that for an AT base pair. Since calf thymus DNA contains random sequences and therefore fewer AT sites than poly(dA-dT)2, the selectivity of 1–6 was further demonstrated by their much weaker binding to calf thymus DNA compared to poly(dA-dT)2. All of the compounds bound to AT-rich sequences similar to pentamidine, but more weak than netropsinCitation20.
Table 2. DNA-binding effect of netropsin (NT), pentamidine (PN). and compounds 1–6.
Table 3. Association constants (Kapp) and topoisomerase DNA inhibitory effects of netropsin (NT), pentamidine (PN), and compounds 1–6.
A number of minor-groove binding drugs inhibit the catalytic activity of isolated topoisomerases (both I and II)Citation21,Citation22. These data suggest that these topological enzymes read the DNA structure at least in part through the minor grooveCitation23. The ability of compounds 1–6 to inhibit topoisomerase I and II activity was quantified by measuring the action on supercoiled pBR322 DNA substrate as a function of increasing concentration of the ligands by the use of agarose gel electrophoresis. The concentrations of inhibitors that prevented 50% of supercoiled DNA from being converted into relaxed DNA (IC50 values) were determined (). These results demonstrated that 1–6 had topoisomerase I (topo I) inhibitory activity in the range from 10 to 40 μM and topoisomerase II (topo II) inhibitory activity in the range from 30 to 100 μM. Compounds 1–3 were more active, as both topo I and topo II inhibitors, compared to 4–6. Pentamidine had weak activity in this experiment; netropsin was more active than all other test compounds. The compounds that were less effective against topoisomerase I/II activity were also weaker DNA-binding ligands ().
The influence of compounds 1–6 on the amidolytic activity of urokinase, thrombin, plasmin, and trypsin is shown as IC50 values in . Compounds 1, 2, and 3 were ineffective as amidolytic activity inhibitors. None of the investigated compounds inhibited the activity of thrombin. Compounds 4–6 were inhibitors of plasmin; meanwhile, amidolytic urokinase activity was inhibited by 5 and 6. Trypsin activity was inhibited only by compound 6.
Table 4. Inhibition of amidolytic activity of proteolytic enzymes.
We were unable to establish a quantitative relationship between potency of enzyme (both topoisomerases and proteases) inhibition and cytotoxicity. The investigated compounds showed an interesting spectrum of activity. We can see that they bind to minor groove B-DNA and inhibit topo I and topo II activity. Some of them are also inhibitors of plasmin and urokinase. The differences in antiproliferative and cytotoxic effects against MCF-7 and MBA-MD-231 breast cancer cell lines demonstrate that the mechanism of action of our compounds is dependent not only on DNA-binding mode but can be partially connected with the fact that in the case of MDA-MB-231 cells, higher uPA/uPAR (urokinase plasminogen activator system) expression and higher plasminogen-binding was observed than in the MCF-7 cell lineCitation24.
The exact mechanism of action of the tested compounds and their structural investigation should be investigated in further studies.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Nelson SM, Ferguson LR, Denny WA. Non-covalent ligand/DNA interactions: minor groove binding agents. Mutat Res 2007;623:24–40.
- Neidle S. DNA minor-groove recognition by small molecules. Nat Prod Rep 2001;18:291–309.
- Baraldi PG, Bovero A, Fruttarolo F, Preti D, Tabrizi MA, Pavani MG, et al. DNA minor groove binders as potential antitumor and antimicrobial agents. Med Res Rev 2004;24:475–528.
- Bailly C. Sequence-specific recognition and modification of double-helical DNA by minor-groove binding conjugates structurally related to netropsin and distamycin. In: Palumbo M, ed. Advances in DNA Sequence-Specific Agents. London: JAI Press, 1998;3:97-156.
- Kopka ML, Yoon C, Goodsell D, Pjura P, Dickerson RE. The molecular origin of DNA-drug specificity of netropsin and distamycin. Proc Natl Acad Sci USA 1985;82:1376–80.
- Pindur U, Jansen M, Lemster T. Advances in DNA-ligands with groove binding, intercalating and/or alkylating activity: chemistry, DNA-binding and biology. Curr Med Chem 2005;12:2805–47.
- Bartulewicz D, Markowska A, Wołczyński S, Dąbrowska M, Różański A. Molecular modelling, synthesis and antitumour activity of carbocyclic analogues of netropsin and distamycin – new carriers of alkylating elements. Acta Biochim Polon 2000;47:23–35.
- Bartulewicz D, Bielawski K, Bielawska A, Różański A. Synthesis, molecular modelling, and antiproliferative and cytotoxic effects of carbocyclic derivatives of distamycin with chlorambucil moiety. Eur J Med Chem 2001;36:461–7.
- Bielawski K, Bielawska A, Bartulewicz D, Różański A. Molecular modelling of the interaction of carbocyclic analogues of netropsin and distamycin with d(CGCGAATTCGCG)2. Acta Biochim Polon 2000;47:855–66.
- Drozdowska D, Rusak M, Miltyk W, Midura-Nowaczek K. Synthesis and biological evaluation of distamycin analogues - new potential anticancer agents. Arch Pharm Chem Life Sci 2009;342:87-93.
- Bartulewicz D, Anchim T, Midura-Nowaczek K. Synthesis and cytotoxic effect of carbocyclic potential minor groove binders. Farmaco 2004;59:211–14.
- Morgan AR, Lee JS, Pulleyblank DE, Murray NL, Evans DH. Ethidium fluorescence assays 1. Physiochemical studies. Nucleic Acids Res 1979;7:547–69.
- Debart F, Periguad C, Gosselin D, Mrani D, Rayner B, Le Ber P, et al. Synthesis, DNA binding, and biological evaluation of synthetic precursors and novel analogues of netropsin. J Med Chem 1989;32:1074–83.
- Schmitt M, Wilhelm OG, Reuning U, Krüger A, Harbeck N, Lengyel E, et al. The urokinase plasminogen activator system as a novel target for tumour therapy. Fibrinolysis Proteolysis 2000;14:114–32.
- Andreasen PA, Kjoller L, Christensen J, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer 1997;72:1–22.
- Pućkowska A, Midura-Nowaczek K, Bruzgo I. Effects of netropsin and pentamidine amino analogues on the amidolytic activity of plasmin, trypsin and urokinase. Acta Polon Pharm 2008;65:213-215.
- Okada Y, Tsuda Y, Teno N, Wanaka K, Bohgaki M, Hijikata-Okunomiya A, et al. Synthesis of active center-directed peptide inhibitors of plasmin. Chem Pharm Bull 1988;36:1289–97.
- Tamura S, Weinhouse MI, Roberts CA, Goldman EA, Masukawa K, Anderson SM, et al. Synthesis and biological activity of peptidyl aldehyde urokinase inhibitors. Biorg Med Chem Lett 2000;10:983–7.
- Lown JW. Newer approaches to the study of the mechanisms of action of antitumor antibiotics. Acc Chem Res 1982;15:381–7.
- Bartulewicz D, Bielawski K, Bielawska A. Carbocyclic analogues of netropsin and distamycin: DNA-binding properties and inhibition of DNA topoisomerases. Arch Pharm Pharm Med Chem 2002;9:422–6.
- Capranico G, Borginetto ME, Cornarotti M, Zagni E, Palumbo M, Zunino F Sequence-specific poisons of type II DNA topoisomerases. In: Palumbo M, ed. Advances in DNA Sequence-Specific Agents. London: JAI Press, 1998;3:7–38.
- Gatto B, Fong Liu L. Topoisomerase I-targeting drugs: new developments in cancer pharmacology. In: Palumbo M, ed. Advances in DNA Sequence-Specific Agents. London: JAI Press, 1998;3:39–66.
- Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 2001;70:369–413.
- Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev 2008;34:122–36.