Abstract
A glutathione transferase (PfGST) isolated from Plasmodium falciparum has been associated with chloroquine resistance. A range of natural products including malagashanine (MG) were screened for inhibition of PfGST by a GST assay with 1-chloro-2,4-dinitrobenzene as a substrate. Only the sesquiterpene (JBC 42C), the bicoumarin (Tral-1), ellagic acid and curcumin, were shown to be potent inhibitors of PfGST with IC50 values of 8.5, 12, 50 and 69 μM, respectively. Kinetic studies were performed on PfGST using ellagic acid as an inhibitor. Uncompetitive and mixed types of inhibition were obtained for glutathione (GSH) and 1-chloro-2, 4-dinitrobenzene (CDNB). The Ki for GSH and CDNB were −0.015 μM and 0.011 μM, respectively. Malagashanine (100 µM) only reduced the activity of PfGST to 80% but showed a time-dependent inactivation of PfGST with a t1/2 of 34 minutes compared to >120 minutes in the absence of MG or in the presence of 5 mM GSH. This work facilitates the understanding of the interaction of PfGST with some plant derived compounds.
Introduction
Malaria, caused by Plasmodium parasites, is one of the world’s deadliest diseases, killing over a million people each year, mainly women and young children in Africa and South East Asia [Citation1]. The most deadly species, Plasmodium falciparum, is responsible for 80% of malaria infections and 90% of deaths [Citation2]. One of the reasons for this devastating effect of malaria is the emergence and spread of drug resistance to classical and affordable antimalarial drugs such as chloroquine (CQ) [Citation3]. During World War 2 public health workers had ambitious plans to eradicate malaria by various means, including DDT against the mosquito, and chloroquine against the parasite. These efforts failed and among the reasons for failure was the appearance and spread of chloroquine resistant malaria [Citation4]. Glutathione transferases (GSTs) [E.C. 2.5.1.18] are a versatile enzyme family whose main role is to inactivate a wide range of exogenous/endogenous toxic molecules and to turn them into water-soluble compounds [Citation5]. They occur abundantly in most organisms and are essentially involved in the intracellular detoxification of numerous substances.
The malarial parasite P. falciparum possesses a GST isoenzyme, P. falciparum GST (PfGST), which is highly abundant in the parasite. PfGST activity was found to be increased in chloroquine-resistant cells, and it has been shown to act as a ligandin for parasitotoxic haemin [Citation6]. Thus, PfGST represents a promising target for antimalarial drug development. Crystal analysis of PfGST revealed that this enzyme is homodimeric. This homodimeric protein of 26 kDa per subunit represents a GST form that cannot be assigned to any of the known GST classes [Citation7]. In comparison to other GSTs, and in particular to the human isoforms, PfGST possesses a shorter C-terminal section resulting in a more solvent-accessible binding site for the hydrophobic and amphiphilic substrates [Citation8]. Furthermore, the structure reveals features in this region that could be exploited for the design of specific PfGST inhibitors. PfGST only shares the highest sequence similarities with the pi-class GSTs from Dirofilaria immitis and Onchocerca volvolus (∼ 35% identity) [Citation7].
The therapeutic effect of CQ is based on the fact that when it reaches the food vacuole of the parasite it binds haemin (a toxic compound) and inhibits conversion of this compound to haemozoin (a non toxic compound). Accumulation of haemin inside the food vacuole of the parasite results in the parasite’s cell death. However, in CQ resistant malaria, PfGST binds haemin more effectively than CQ and detoxifies it (Ki = 6.5 µM) [Citation9], thus, it prevents the activity of CQ in CQ resistant malaria. Chloroquine was found to be a very weak inhibitor of PfGST (IC50>200 µM) [Citation9]. Based on the activity of PfGST in extracts of P. falciparum, the enzyme represents between 1% and 10% of cellular protein and might, therefore serve as an efficient in vivo buffer for parasitotoxic haemin [Citation10].
An increase in the resistance of P. falciparum to conventional treatments is a worldwide problem and a few alternative drugs are under development, necessitating urgent efforts to identify new classes of antimalarial drugs [Citation11]. GSTs have been investigated in parasite protozoans with respect to their biochemistry and they have been identified as potential vaccine candidates in protozoan parasites and as targets in synthesis of new antiparasitic agents [Citation12].
Plant extracts have been found to act at different vulnerable metabolic sites of PfGST, disturbing glutathione (GSH) dependent detoxification processes, increasing cytotoxic peroxides levels and possibly increasing the concentrations of toxic haemin in the parasites [Citation14]. The crude alkaloids of Strychnos myrtoides Gilg and Busse, empirically used as an adjuvant to CQ in Malagasy herbal remedies, were found to be practically devoid of intrinsic in vitro and in vivo antimalarial activity. Malagashanine (MG, ) is the parent compound of a series of Nb,C[2I]-secocuran alkaloids isolated from two Malagasy Strychnos species [Citation15]. It was found to display a good pharmacological profile by enhancing chloroquine (CQ) action in vitro against the CQ-resistant strain Fc29 of P. falciparum in a follow-up study of the empirical uses of Strychnos species as CQ adjuvants in the treatment of chronic malaria [Citation16]. When combined with CQ at a dose level much lower than their IC50 value, they markedly enhanced the in vitro effectiveness of the synthetic drug against a CQ-resistant strain of P. falciparum.
Figure 1. Structures of natural plant products used in this study to investigate the inhibition of PfGST.
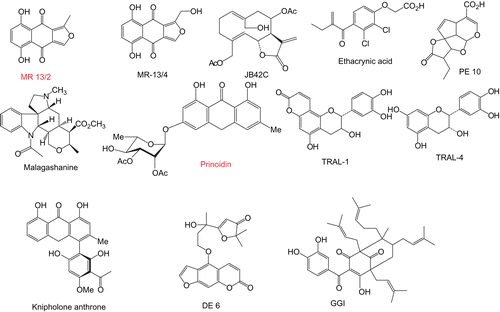
The aim of this paper is to study the effects of natural plant products on PfGST to identify any with inhibitory activity that could be employed as chloroquine-potentiating additives to enhance the effectiveness of this malaria control measure. The natural plant products () were selected on the basis of novelty, availability and also on having diverse classes of secondary metabolites, including phenolic polyketides, terpenoids, and coumarin derivatives, many of which contain α,β-unsaturated carbonyl groups, which may be responsible for the GST inhibiting properties of many compounds.
Materials and methods
Chemicals
All the reagents used were of analytical grade. The following chemicals and biochemicals were obtained from Sigma-Aldrich Chemical Companies, (St Louis MO, USA): 1-chloro-2, 4-dinitrobenzene (CDNB), glutathione (GSH), hexylglutathione, ethacrynic acid, EDTA, DDT, NaN3, BSA, tris-(hydroxymethyl) amino methane (Trizma base), yeast extract, glycerol, tryptone, ampicillin, isopropyl thiogalactoside (IPTG), chicken egg white lysozyme, molecular weight markers, genistein, etoposide, reserpine, quercetin, caffeic acid, flavones, ellagic acid, resveratol, (+)-catechin hydrate, (-)-epicatechin, curcumin, daidzein, kaempferol, ferulic acid, mitoxantrone, vinblastine, and vincristine.
Malagashanine was obtained from Professor Rasoanaivo (Institut Malgache de Recherches Appliquees, Madagascar). Escherichia coli cells with the gene for human PfGST were obtained from Professor Katja Becker (Justus-Liebig University, Germany). Diospyrin was isolated from the stem bark of Diospyros montana Roxb which was collected from Bolangir district, Orissa, India, and the voucher specimen was authenticated at the Botanical Survey of India, Calcutta. The compound was isolated and purified meticulously as previously described [Citation17]. Its structure has been established through routine spectroscopic methods, and it was reconfirmed to be 2,6′-bis(5-hydroxy-7-methyl-1,4-naphthoquinone) through total synthesis [Citation18]. Geshoidin was isolated as described by Abegaz and Kebede [Citation19]. The purity of the compound was checked by comparing physical and spectroscopic data. These were as reported in the literature [Citation19]. The isofuranonaphthoquinones, 5,8-dihydroxy-1-hydroxymethylnaphtho[2,3-c]furan-4,9-dione (Mr13/4) and 5,8-dihydroxy-1-methylnaphtho[2,3-c]furan-4,9-dione (Mr13/2) were extracted from Bulbine frutescens [Citation20], 14-hydroxy, 8, 15-diacetoxy-1(10)4,11(13) germacratrien-12,6-olide (Jb42c) was extracted from Dicoma anomala, knipholone anthrone, (12KA) was extracted from Knipholfia foliosa, prinoidin (2QG1) and geshoidin were extracted from Rhamnus prinoides [Citation21], the iridoid derivative (PE10) was extracted from Plumera rubra [Citation22], the monoterpene substituted furocoumarin (DE6) was from Dorstenia elliptica [Citation23], the polyprenylated benzophenone derivative (GGI) was isolated from Garcinia smeathmannii collected in Cameroon, and the 3-hydroxyflavans (Tral-1 and Tral-4) were extracted from Garcinia species. The natural products GG1, JB42c, Tral1 and Tral-4 were extracted from the above mentioned plants using the following general protocol. The sun-dried plant material (about 1 kg) was soaked in a mixture of dichloromethane-methanol (1:1) and pure methanol for 24 h and 2 h, respectively, at room temp. Concentration of the combined organic extract gave a residue (about 50–65 g). Part of this residue was chromatographed on a silica gel column eluting with hexane-ethyl acetate mixtures, to give fractions of 250 ml each. The fractions were concentrated and monitored by TLC and 1H NMR and similar fractions were combined. The first fractions examined by TLC (Hexane-ethyl acetate; 9:1) contained mainly mixtures of hydrocarbons and phytosterols, which were not investigated further. More polar fractions were passed through a Sephadex LH-20 column (CHCl3/methanol, 2:1). The post chlorophyll fractions were subjected to repeated silica gel column chromatography and PTLC to yield the various metabolites. The molecular structures of the pure metabolites were established by spectroscopic techniques such as NMR, MS, and IR.
Expression and purification of recombinant PfGST
A 2 ml aliquot of 2TYA medium (tryptone, yeast extract, NaCl and glycerol) containing 100 µg/ml ampicillin and 50 µg/ml kanamycin was inoculated with E. coli M15 (pQE30PfGST) cells. The culture was incubated in a shaking incubator (Labcon, Labotec, South Africa) operating at 170 rpm and 37°C overnight. Three 2000 ml conical flasks containing 600 ml 2TYA medium containing 100 µg/ml ampicillin and 50 µg/ml kanamycin were inoculated with 200 µl of the overnight culture and incubated in the shaking incubator at the same settings. When an A600 of 0.5 was obtained, isopropylthiogalactoside (IPTG) was then added to a final concentration of 1 mM in each flask to induce expression of the PfGST gene and the cells were grown for 4 hours. The bacteria were harvested by centrifugation and resuspended in an equal volume of buffer A (50 mM sodium phosphate pH 8, 300 mM NaCl, 10 mM imidazole), followed by sonication 3 times (Dawe Soniprobe, England, London) at setting 5 for 30 s, on ice. Polymethylsulphonylfluoride (PMSF) was added to a final concentration of 170 μM to inhibit the proteases and then any cellular debris was removed by centrifugation at 35 000 rpm for 1 h using a 70Ti rotor in an ultracentrifuge to obtain the cell supernatant fraction.
The protein was purified by an immobilised metal affinity chromatography (IMAC) method using a Ni-CAM affinity resin (Sigma). The cell supernatant was combined with 5 ml of the resin and the mixture was kept on ice with gentle periodic swirling for 1 hour to allow the GST to bind. The resin was packed into a column and the non bound fraction was collected. The column was then washed with buffer A. The PfGST was eluted with buffer B (50 mM sodium phosphate pH 8, 300 mM NaCl, 250 mM imidazole). The fractions collected from the column were tested for GST activity using 1-chloro-2,4-dinitrobenzene (CDNB) as a substrate. The fractions that exhibited activity were pooled (affinity pool fraction) and dialysed against 9 L of dialysis buffer (50 mM sodium phosphate pH 8, 1 mM EDTA, 0.2 mM DTT, 0.02 % NaN3) using 2 × 3 L buffer changes.
The various fractions collected during purification i.e. the cell supernatant, non bound, affinity pool, and dialysate fractions, were tested for protein content using the Lowry procedure [Citation24]. The protein content of the dialysate was also determined using the A280 value.
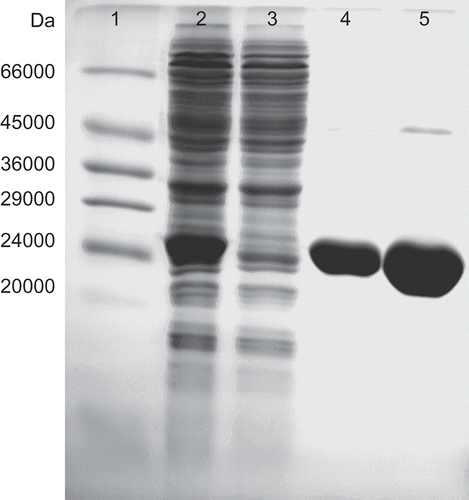
The molecular weight of the purified PfGST was determined by SDS-PAGE on 15% slab gels with the help of molecular weight markers (14 200 to 66 000 Da) using a BioRad Protean system (BioRad Laboratories, California). The protein bands were stained using Coomassie G stain (0.025% Coomassie G250, 40% methanol, 7% acetic acid) overnight, destained for 1 hour in 50% methanol, 10% acetic acid and then further destained in 5% methanol, 7% acetic acid solution ().
Figure 2. SDS-PAGE gel for determination of the molecular weight of PfGST. The proteins were analysed by SDS-PAGE and stained with Coomassie Blue to determine the molecular weight of PfGST. Lane 1, Molecular weight markers (Sigma), lane 2, E. coli lysate IPTG induced, lane 3, unbound fraction of Ni-NTA affinity chromatography, lane 4, bound affinity fraction of Ni-NTA affinity chromatography and lane 5, the concentrated affinity pool.
Inhibition of PfGST by natural products
A range of natural products were screened for inhibition of PfGST by GST assay with CDNB as a substrate, using a 96-well SpectraMax 340 microplate spectrophotometer equipped with a kinetics mode (Molecular Devices, California). The final concentration of CDNB was 0.5 mM and of GSH was 1mM. Ethacrynic acid (ETA) and haemin were used as positive controls. ETA is a known mammalian GST inhibitor whilst haemin is known to inhibit this enzyme [Citation9]. The incubation temperature was 30°C. The final concentration of organic solvent in the inhibition assays was 2.5%. Initially the natural plant compounds were tested at arbitrary concentrations of 33 μM and 100 µM. These concentrations were chosen so as to cover potent as well as moderate inhibitors. Stock solutions of the natural compounds were prepared in DMSO. Once a compound was found to be a potent inhibitor, various concentration ranges of natural product were tested to generate inhibition curves from which IC50 values could be determined, the IC50 value being the concentration required for 50% inhibition of enzyme activity. The IC50 value was determined by plotting sigmoidal dose response curves of enzyme activity versus the log of the natural product concentration using GraphPad Prism™ version 4.00 for Windows, (GraphPad™ Software Inc., San Diego, California).
Determination of kinetic properties
The effects of ellagic acid on the kinetics of PfGST were determined as described by Mukanganyama et al. [Citation25] using the SpectraMax 340 microplate spectrophotometer equipped with the kinetics mode (Molecular Devices, California). Since GSTs have two substrate binding sites, the kinetic parameters were determined for each site. In one case, the concentration of CDNB was varied from 0.05–1.5 mM at a fixed concentration of GSH of 5 mM and in the second case; the concentration of GSH was varied from 0.05–5 mM at a fixed concentration of CDNB of 1.5 mM. Michaelis-Menten plots were used to determine the kinetic parameters Km and Vmax using the GraphPad Prism 4 software. The Km(app) and Vmax(app) were determined using GraphPad Prism™ version 4.00 for Windows, (GraphPad™ Software, San Diego, California). The Ki values with respect to GSH and CDNB, as well as the type of inhibition were determined. The type of inhibition was deduced by determination of the trends of the Km and Vmax values with increase in natural product concentration. To determine the trend, the means of the Km (or Vmax) values with increase in inhibitor concentration were compared by performing a one-way ANOVA with Dunnett’s post test using GraphPad InStat™ version 3.00 for Windows 95, (GraphPad™ Software). The inhibition constant (Ki) was determined by means of re-plots [Citation25]. The type of re-plot depends on the type of inhibition, for example, plotting 1/Vmax versus inhibitor concentration for non-competitive inhibition will give Ki as the intercept on the baseline [Citation26].
Inactivation of PfGST and effect of GSH
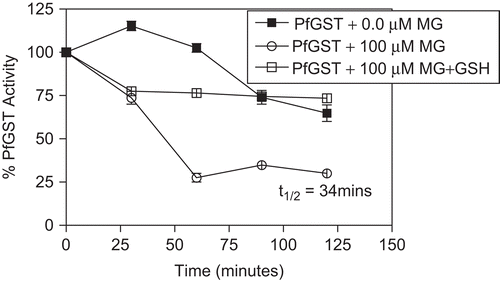
Incubation mixtures containing PfGST (0.48 μM), 100 mM HEPES buffer (pH 6.5), and varying concentrations of MG (between 0 and 100 μM) were prepared. The incubation temperature was 30°C. At 30 minute intervals, 25 µl of the incubation mixture was withdrawn and assayed for GST activity. These incubations were run in parallel with a negative control containing PfGST and buffer. The inactivation parameter t1/2 (the half life) was obtained by plotting graphs of the percentage remaining enzyme activity with time.
The possible role of the thiol groups of GSH in the protection of the enzyme PfGST from inactivation by MG was investigated by incubating the enzyme with MG (100 µM) including 5 mM GSH. An incubation mixture containing enzyme and MG was also run to determine the effect of MG alone. The enzyme alone was used as a control. The incubation temperature was 30°C. At 30 minute intervals, 25 µl were withdrawn and assayed for PfGST activity spectrophotometrically at 340 nm.
Results
Purification of PfGST
Plasmodium falciparum recombinant PfGST was over expressed and isolated from E. coli cells and purified by metal affinity chromatography on a nickel column. The specific activity was found to be 0.18 U/mg, this value was found to be comparable with that previously published of 0.2 U/mg [Citation9]. The molecular mass of the enzyme was found to be 26.424 kDa which was comparable with the calculated molecular mass of the His-tagged protein of 26,000Da [Citation9] ().
Effect of some natural products on PfGST
The effect of various natural products on the CDNB conjugating activity of PfGST was determined. For the novel chemical compounds extracted from plants, the activity was screened at 33 and 100 µM. These concentrations were chosen so as to cover potent as well as moderate inhibitors. Typical results are shown for JB42c, Tral1, ETA, GG1 and haemin (). The results for the determination of the IC50 values are summarised in and . Of the commercial natural plant products, the most potent inhibitor was found to be ellagic acid.
Table 1. The effect of commercially purified natural products on the PfGST activity. The extent of inhibition is shown at the two concentrations of 33 μM and 100 μM. Values are mean values for quadruplicate determinations. Data are the means of quadruplicate determination as shown in .
Table 2. The effect of novel natural products from medicinal plants on PfGST activity. The extent of inhibition is shown at the two concentrations of 33 μM and 100 μM. Values are mean values for quadruplicate determinations. Data are the means of quadruplicate determination as shown in .
Figure 3. The inhibition of PfGST by JB42c, Tral1, ETA, GG1 and haemin. The % of activity refers to controls shown by the basal line. The controls contained an equal amount of solvent in which the inhibitor was dissolved. Values + SD for n =6”. ** P<0.01 significantly different from the control.
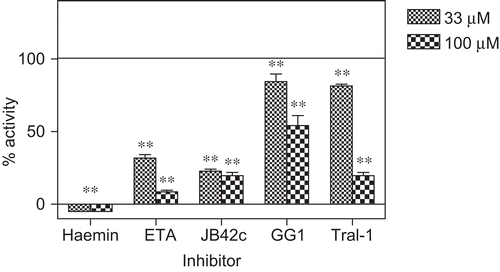
Malagashanine inhibited heterologously expressed human PfGST in vitro under different conditions. At a concentration of 100 µM, MG reduced the activity of PfGST by only 20% indicating that at 100 μM MG is not an effective inhibitor of PfGST, compared to haemin, a potent inhibitor of PfGST, which had an IC50 of 4 µM (). However, malagashanine showed a time-dependent inactivation of PfGST () which was prevented by GSH.
Figure 4. The time-dependent inactivation of PfGST by MG. The incubation mixtures contained PfGST (0.48 μM), 100 mM HEPES buffer (pH 6.5), and concentrations of MG (0 and100 μM). The incubation temperature was 30°C. At 30 minute intervals, 25 µl of the incubation mixture was withdrawn and assayed for GST activity.
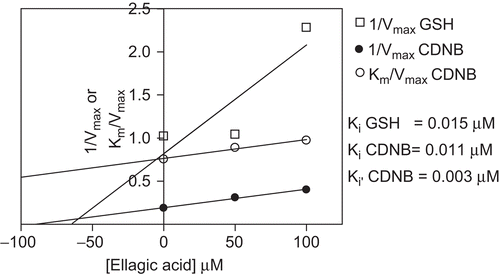
Uncompetitive and mixed types of inhibition were obtained for GSH and CDNB respectively (). The Ki values for GSH and CDNB were 15 nM and 11 nM, respectively (). Diospyrin, genistein, quercetin, catechin hydrate, curcumin, mitoxantrone, geshoidin and kaempferol were also inhibiting to a lesser extent. Of the novel natural plant compounds, the germacratriene sesquiterpene lactone JB42C was found to be the most potent.
Table 3. The kinetics of the inhibition of PfGST by ellagic acid. The Km values for GSH and CDNB were observed to increase with increase in the ellagic acid concentration. The Vmax decreased too as the ellagic concentration was increased for both GSH and CDNB. The Kcat and Kcat/Km both decreased with increased ellagic concentration for both CDNB and GSH indicating that ellagic acid decreased both catalysis and specificity of the PfGST. The type of inhibition was noted to be mixed type for both GSH and CDNB variations.
Figure 5. Determination of Ki for GSH and CDNB for cDNA expressed PfGST in the presence of ellagic acid. Replots of slope (Km/Vmax) and 1/Vmax versus were used to determine Ki GSH and Ki(□) CDNB values for ellagic acid which showed mixed type of inhibition for both substrates.
Discussion
Chloroquine has been the drug of choice for the treatment of malaria until recently as there has been a spread of the chloroquine resistant strains of Plasmodium. A glutathione transferase (PfGST) has been isolated from Plasmodium falciparum and it has been implicated in the development of drug resistance [Citation3]. The malarial parasite may depend on this functional enzyme, particularly because it is the only GST which is present in this parasite [Citation8]. PfGST activity is increased in chloroquine-resistant strains and it has been shown to act as a ligandin for the parasitotoxic haemin [Citation27]. In order to restore the effectiveness of chloroquine, it may be possible to co-administer chloroquine and a chemomodulator to inhibit the action of PfGST.
Although combinatorial chemistry and bioinformatics are revolutionising drug discovery, natural compounds also offer structural diversity that is not rivalled by the creativity or synthetic ingenuity of medicinal chemists. Historically, the two powerful antimalarial drugs of quinine and artemisinin come from medicinal plants traditionally used to treat malaria or fever and new synthetic or semisynthetic antimalarial drugs have been derived from these lead compounds. It has been reported that GSTs are able to interact covalently and non-covalently with various compounds that are not substrates for enzymatic activity [Citation28]. GST activity has been shown to be modulated by natural plant products [Citation29,Citation30]. Flavonoids have been shown to inhibit GSTs in human blood platelets as well as in cancer cell lines [Citation31]. In our study we have screened a range of naturally-occurring compounds encompassing a large structural diversity for their ability to inhibit expressed PfGST in vitro. Initial screening at 33 and 100 uM showed that Mr13/2, Tral 1, GG1, Jb42C, ellagic acid, KA, curcumin were potential inhibitors of this enzyme ( and ). The most potent inhibitors of PfGST were JB42C, Tral-1, ellagic acid and curcumin with IC50 values of 8.5 μM, 12 μM, 50 μM, 69 μM, respectively. Of the 11 plant derived medicinal compounds that were successful on the initial screening, JB42C proved to be the most potent inhibitor of PfGST as shown by its very low IC50 value relative to the IC50 values of the other four compounds. The α,β-unsaturated carbonyl structural moiety which is present in the structure of JB42C and Tral-1 resembles that which is found in the structure of ethacrynic acid, a well known inhibitor of GSTs [Citation32]. This could explain why these two compounds isolated from medicinal plants were potent inhibitor of PfGST. Diospyrin, genistein, quercetin, catechin hydrate, curcumin, mitoxantrone, geshoidin and kaempferol were also inhibiting to a lesser extent.
The effect of ellagic acid on the kinetic parameters of the enzyme was investigated. Generally the two concentrations of 50 and 100 µM reduced the Kcat/Km of the enzyme for GSH by 66% and 79%, respectively, and for CDNB by 15% and 22%, respectively. The Kcat/Km, described the catalytic efficiency of the enzyme [Citation33]. Just as expected, ellagic acid exhibited inhibitory effects on the enzyme and lowered this value with an increase in concentration. The Km for GSH was observed to increase with increase in ellagic acid concentration whilst for CDNB the Km decreased with an increase in ellagic acid (). The Vmax also decreased as the ellagic concentration was increased for both GSH and CDNB. The Kcat and Kcat/Km both decreased with increased ellagic concentration for both CDNB and GSH. This indicates that ellagic acid decreased both the catalysis and the specificity of the PfGST. The type of inhibition was noted to be of the mixed type for both GSH and CDNB.
Malagashanine (MG) is the parent compound of the Nb-C(21) seco-curan alkaloids isolated hitherto from Madagascan Strychnos. On account of its promising chemosensitising activity against chloroquine (CQ)-resistant Plasmodium falciparum strains, the interaction of malagashanine with heterologously expressed PfGST was investigated in vitro. Malagashanine (100 µM) only reduced the activity of PfGST to 80% indicating that at 100 µM, it is not a potent inhibitor. However, MG also showed a time-dependent inactivation of the PfGST (). The half life of PfGST in the presence of MG was found to be 34 minutes. This suggests that PfGST is able to bind irreversibly to MG resulting in loss of catalytic activity. Glutathione was shown to be capable of protecting PfGST from inactivation by MG. This suggests that the over-expression of GSH in the parasite might help overcome MG. However, the effects of MG has been shown to prevent CQ efflux from and stimulated CQ influx into drug resistant Plasmodium falciparum [Citation34] suggesting that its effects are more on the plasma membrane than inside the parasite. These results facilitate the understanding of the interaction of MG with PfGST with regards to its role as a chemomodulator in cases of PfGST over-expression in chloroquine resistant strains.
Since there is elevated expression of GSTs in chloroquine resistance [Citation27], it is possible that compounds that inhibit the functioning of GSTs might actually restore the utility of chloroquine. In order to restore the effectiveness of chloroquine, it may be possible to co-administer chloroquine and a chemomodulator to inhibit the action of PfGST. It would seem that the inactivation of PfGST by malagashanine would be dependent on the concentration of glutathione in vivo as data obtained in this in vitro study showed that excess GSH protected the enzyme from inactivation by malagashanine. Data from studies with P. berghei have also shown that in chloroquine resistant strains, GSH can detoxify haemin within the food vacuole, thus precluding its polymerisation and preventing the activity of chloroquine and other quinoline containing drugs [Citation10]. Our study shows that MG inhibits PfGST and it has been found to possess antimalarial activity by increasing the accumulation of CQ inside the parasite [Citation15]. These two properties of MG may then act as a double edged sword in malaria therapy. If MG was co-administered with chloroquine, it could prevent resistance to chloroquine by inhibiting PfGST-mediated detoxification of haemin, thus allowing CQ to bind haemin, leading to accumulation of haemin and hence toxicity to the parasite by allowing the accumulation of CQ inside the parasite.
Further work needs to be done to evaluate the use of natural compounds as reversal agents in resistance to antimalarial drugs. At this point, there are two approaches for drug discovery: the physiology-based approach which seeks to induce a therapeutic effect by reducing disease-specific symptoms or physiological changes, and the mechanism-based approach, which corresponds to the target-based approach, and which seeks to produce a therapeutic effect by targeting a specific mechanism. This latter approach has replaced the traditional physiology-based approach because it allows an increased screening capacity and can define rational drug discovery programmes. The best strategy would be to combine the advantages of the target-based approach with a strong physiology/disease-based approach in order to overcome some of the weaknesses of both methods. To this end, it is necessary to confirm the activity at the enzyme level observed with the test compounds, by the in vitro cellular models and then for the in vivo models using the drug combination. Secondly, there is a need to determine whether these compounds might interact with other macromolecules in the parasites. It has been suggested that PfGST, the only GST in Plasmodium falciparum, is essential for parasite survival by protecting the parasite against oxidative stress and/or acting as a buffer for detoxification of haem-binding compounds in vivo [Citation9]. Therefore, an inhibitor of the enzyme would be expected to act synergistically with the antimalarial drug chloroquine, not only to impair the general detoxification processes, but also to reduce the antioxidant capacity of the parasite. It has been suggested that PfGST evolved by optimising its binding property with the parasitoxic haemin rather than its catalytic efficiency towards the electrophilic compounds [Citation35]. Thus, studies of GSTs in the malarial parasites might help in understanding the mechanism of resistance to important antimalarial agents and guide the design of novel inhibitors to overcome antimalarial drug resistance. The results obtained in the present study showed that natural plant products interact with PfGST in vitro resulting in reversible and irreversible effects. The unique presence of a single GST in Plasmodium falciparum [Citation7] and its characterisation would provide for better understanding of its roles in the development of specific inhibitors as potential drugs. The plant natural compounds identified in this study could act as targets in the development of novel antimalarial drugs. There is a need for the development of toxicological assays in further studies in order to evaluate the safety of some of JBC 42c, Tral-1 in vitro and in vivo. It would be also interesting to measure the amount of haemin produced in the presence of the inhibitors as shown by Famin et al. [Citation36]. It was shown that antimalarial drugs inhibited glutathione-dependent degradation of ferritoprotoporphyrin (IX) in solution.
Acknowledgements
This study was supported by the International Program in the Chemical Sciences (IPICS), Uppsala University, Sweden, IPICS-ZIM01, and the International Foundation for Science (IFS), Stockholm, Sweden, F/3413-02.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Snow RW, Trape JF. Marsh K. The past, present and future of childhood malaria mortality in Africa. Trends in Parasitology 2007;17:593–597.
- Kathrin B, Boniface MM, Schirmer H, Katja B. Structure-Based Drug Development against Malaria. Frontiers in Drug design and Discovery, 2007; 3:225–255.
- Hiller N, Fritz-Wolf K, Deponte M, Wende W, Zimmermann H, Becker K. Plasmodium falciparum glutathione S-transferase-structural and mechanistic studies on ligand binding and enzyme inhibition. Protein Science 2006;15:281–289.
- Boger RA, Shapiro TA. Molecular mechanisms of resistance in antimalarial chemotherapy: The unmet challenge. Annual Rev Pharmacol 2005;45:565–85.
- Torres-Rivera A, Landa A. Glutathione transferase from parasites: A biochemical view. Acta Tropica 2008;105:99–112.
- Chikezie PC, Uwake AA, and Monago CC Glutathione S-transferase activity of erythrocytes HbAA, HbAS and HbSS in male volunteers administered with fansidar and quinine. African Journal of Biochemistry Research. 2009;3:210–214.
- Tripathi T, Rahlfs S, Berker K, Bhakuni V. Glutathione mediated regulation of oligomeric structure and function al activity of Plasmodium falciparum glutathione s-transferase. BMC Structural Biology 2007;7:67.
- Fritz-Wolf K, Becker A, Rahlfs S, Harwaldt P, Schirmer RH, Kabsch W, Becker K. X-ray structure of glutathione S-transferase from the malarial parasite Plasmodium falciparum. The National Academy of Science 2003;100:13821–13826.
- Harwaldt P, Rahlfs S, Becker K.Glutathione S-Transferase of the malarial parasite Plasmodium falciparum: characterization of a potential drug target. Biol. Chem 2002;383:821–830.
- Platel DFN, Mangou F. Tribouley-Duret J. Role of glutathione in the detoxification of ferriprotoporphyrin IX in chloroquine resistant Plasmodium berghei. Molec Biochem Parasitol 1999;98:215–223.
- Frederich M, Hayette M, Tits M, Mol PP, Angenot L. In vitro activities of Strychnos alkaloids and extracts against Plasmodium malaria. Antimicrob Agents Chemotherapy Alkaloids 1999;43:2328–2331.
- Ahmad R, Srivastava AK, Tripath RP, Batra S, Walter RD. Synthesis and biological evaluation of potential modulators of malaria Glutathione Transferases. J Enzyme Inhib Med Chem 2007;22:327–42.
- Tuteja R, Pradhan A. Unraveling the ‘DEAD-box’ helicases of Plasmodium falciparum. Gene 2006;376:1–12.
- Deponte M, Becker K. Glutathione S-transferase from malarial parasites: structural and functional aspects. Methods Enzymology 2005;401:241–253.
- Rafatro H, Ramanitrahasimbola D, Rasoanaivo P, Ratsimamanga S, Ratsimamanga AR, Frappier F. Reversal activity of the naturally occurring chemosensitizer malagashanine in Plasmodium malaria. Biochem Pharmacol 2000;59:1053–1061.
- Rasoanaivo P, Ratsimamanga-Urverg S, Milijaona R, Rafatro H, Rakoto-Ratsimamanga A, Galeffi C, Nicoletti M. In vitro and in vivo chloroquine-potentiating action of Strychnos myrtoides alkaloids against chloroquine-resistant strains of Plasmodium malaria. Planta Med 1994;60:13–6.
- Hazra B, Sur P, Roy DK, Sur B, Banerjee A. Biological activity of diospyrin towards Ehrlich ascites carcinoma in Swiss A mice. Planta Med 1984;51:295– 297.
- Yoshida M, Mori K. Synthesis of a diospryin, a potential agent against leishmaniasis, and related parasitic protozoan diseases. Eur J Org Chem 2000;1313–1317.
- Abegaz BM, Kebede T. Geshoidin. A bitter principle of Rhamnus prinoides and other constituents of the leaves. Bull Chem Soc Ethiop 1995;9:107–114.
- Bezabih M, Abegaz BM, Dufall K, Croft K, Skinner Adams T, Davis TME. Antiplasmodial and anti-oxidant isofuranonaphthoquinones from the roots of Bulbine capitata. Planta Medica 2001;67,297–390.
- Abegaz B, Dagne E. Anthracene derivatives of Rhamnus prinoides. Bull Chem Soc Ethiop 1988;2:15.
- Elsasser B, Krohn K, Nadeem Akhtar M, Ke UF, Kouam SF, Kuigoua MG, Ngadjui BT, Abegaz BM, Antus S, KurtΒn T. Revision of the Absolute Configuration of Plumericin and Isoplumericin from Plumeria rubra. Chem Biodiversity 2005;2:799–808.
- Abegaz BM, Ngadjui BT, Folefoc GN, Fotso S, Ambassa P, Bezabih M, Dongo E, Rise F, Petersen D. Prenylated Flavonoids, Monoterpenoid Furanocoumarins and other Constituents from the Twigs of Dorstenia elliptica (Moraceae). Phytochemistry 2004;65:221–226.
- Lowry OH, Rosebrough NR, Farr AL, Randall RJ. Protein Measurement with the Folin Phenol Reagent. J Biol Chem 1951:193:265–275.
- Mukanganyama S, Widersten M, Naik YS, Mannervik B, Hasler JA. Inhibition of glutathione S-transferases by antimalarial drugs possible implications for circumventing anticancer drug resistance. Int J Cancer 2002;97:700–705.
- Segel IH. Enzyme kinetics behavior and analysis of rapid equilibrium steady-state enzyme systems.. John Wiley and Sons. New York. USA. 100-107 pp.
- Na BK, Kang JM, Kim TS, Sohn WM. Plasmodium vivax: Molecular cloning, expression and characterization of GST. Experimental Parasitology 2007;116:414–418.
- Yu SJ, Abo-Elghar GE. Allelochemicals as inhibitors of glutathione S-transferases in the fall armyworm. Pesticide Biochem Physiol 2000;68:173–183.
- Muleya V, Hayeshi R, Ranson H, Abegaz B, Bezabih MT, Robert M, Ngadjui BT, Ngandeu F, Mukanganyama S. Modulation of Anopheles gambiae Epsilon glutathione transferase activity by plant natural products in vitro. J Enzyme Inhib Med Chem 2008;23:391–9.
- Hayeshi R, Mutingwende I, Mavengere W, Masiyanise V, Mukanganyama S. The inhibition of human glutathione S-transferases activity by plant polyphenolic compounds ellagic acid and curcumin. Food ChemToxicol 2007;45:286–95.
- Ghazali R, Waring RH. Effects of flavonoids on glutathione-S-transferase in human blood platelets, rat liver, rat kidney, and HT-29 colon adenocarcinoma cell-lines: potential in drug metabolism and chemoprevention. Med Sci Res 1999;27:449–451.
- Mannervik B, Wildersten M.Human Glutathione Transferase: Classification, Tissue Distribution, Structure and Functional Properties. In: Pacific GM, Fracchia GN. eds. Advances in Drug Metabolism in Man. Luxemburg: Commission of European Communities, 1995:14–16.
- Fersht A. Enzyme Structure and Mechanism. San Franscisco: WH Freeman, 1977:371.
- Ramanitrahasimbola D, Rasoanaivo P, Ratsimamanga S, Vial H. Malagashanine potentiates chloroquine antimalarial activity in drug resistant Plasmodium malaria by modifying both its efflux and influx. Mol Biochem Parasitol 2006;9775:1–10.
- Liebau E, Perbandt M, Turella P, Stella L, Federici G, Ricci G. Cooperativity and pseudo-cooperativity in the glutathione transferase from Plasmodium falciparum. J Biol Chem 2005;280:26121–26128.
- Famin O, Krugliak M, Ginsburg H. Kinetics of inhibition of glutathione-mediated degradation of ferriprotopophyrinIX by antimalarial drugs. Biochem Pharmacol 1999;58:59–68.