Abstract
An efficient and economical synthesis of some new fluorine substituted phthalides was accomplished from two γ-keto acids, 2-(4-fluorobenzoyl)benzoic acid and 2-(3,5-dinitro-4-flurobenzoyl)benzoic acid. Each acid was reacted with various phenolic compounds in presence of catalytic quantity of concentrated sulphuric acid to get the phthalides. The structures of the synthesized compounds were established on the basis of their elemental analysis, spectral data and chemical reactions. Some of the synthesized phthalides exhibited antibacterial and antifungal activity on antimicrobial screening against human pathogenic bacteria and fungi.
Introduction
Fluorinated organic compounds are the subject of topical interest due to their unique physical and biological propertiesCitation1,Citation2. It has been observed that presence of fluorine substituents in a lead molecule is capable of changing all the physical properties and influencing various drug-action related phenomenon such as adsorption, distribution, metabolism, and excretionCitation3. Consequently, the use of fluorine substituent in drug synthesis has increased considerably during the recent years. Aryl fluoride (Ar-F) moieties are synthetically useful functional groups broadly applicable in medicinal chemistry. They are isosteric to parent hydrocarbons and show improved lipophilicity as well as inertness to metabolic transformationsCitation4–6. Large number of fluorinated organic compounds exhibit antimicrobialCitation7,Citation8, antitumour and anti lung cancerCitation9 activities. Some fluorinated chalcone derivatives are reported to possess anti-inflammatory activity due to their influence on nitric oxide productionCitation10. Fluorinated 3-aminoindazoles have been identified as lead compounds for the treatment of psychiatric disorders such as obsessive compulsive disorder and attention deficit disorderCitation11. In addition, they are reported to be useful in the treatment of diseases associated with protein kinase, e.g., diabetes, cancer and Alzheimer’s diseasesCitation12. The paramount importance of fluorine substituted drugs in human health is evident by the fact that antidepressant fluoxetine (Prozac), the cholesterol–lowering drug atorvastatin (Lipitor), and the antibacterial ciprofloxacin (Ciprobay) are among the most widely used and top-selling fluorine containing drugsCitation3.
In the present synthetic programme in addition to incorporation of fluorophenyl group, attachment of hydroxyphenyl moiety with phthalide structure has been envisaged because phenols are also endowed with excellent biological properties. It is well recognized that antioxidant activity of several plants is due to their phenolic contentsCitation13. The use of phenol and m-cresol as antiseptics and disinfectants is quite old, and mechanism of their antimicrobial action is knownCitation14. Curcuphenol is a sesquiterpene phenol recently isolated from sponges and plant sources showed anticancer propertyCitation15. 1-(2-Ethyl-6-heptyl)phenol (EHP), a biologically active compound isolated from cumin (Cuminum cyminum) seeds has been found to exhibit anticancer and antibacterial activitiesCitation16. o-Dihydroxyphenols are considered the most active antioxidants and antimicrobial agentsCitation17. Catechol and pyrogallol are allelo chemicals which belong to phenolic compounds synthesized in plantsCitation18. Both of these two compounds are reported to be associated with antibacterialCitation19,Citation20 and anticancerCitation21,Citation22 properties. Resorcinol structure is present in large number of compounds of therapeutic value showing anticancerCitation23,Citation24 and antifungal activityCitation25,Citation26. A novel series of quinols with heteroaromatic or (arylsulphonyl) indole substitution at 4 position has been reported to show in vitro antitumour activityCitation27. Both synthetic and natural phloroglucinols exhibit a vast array of biological activities, viz., anti-inflammatory, anticancer, antimicrobial, antiallergic, enzyme inhibitory, neuroregenerative and antioxidant, etc.Citation28
Although organic compounds with phthalide moieties in their structures are well known as analytical reagents and for their photochromatic properties, during recent years, interest in this area has been regenerated and gained a new momentum due to diverse pharmacological properties associated with these compounds. Phthalide [1-(3H)-isobenzofuranone] structures are present in large member of natural products and biologically active compoundsCitation29,Citation30. A number of medicinal plants are found to contain various phthalides as their active principlesCitation31. Chiral 3-substituted phthalides are found to possess therapeutic properties and also used as versatile building blocks for large number of medicinally important compoundsCitation32,Citation33. There is a continuous interest in developing efficient and convenient methods for the synthesis of phthalidesCitation34–38.
Despite the above mentioned facts, to date, almost negligibleCitation38 attention has been paid towards the synthesis of fluorinated phthalides. To address this deficiency, we now document the synthesis of new fluorinated phthalides. In the present study, we have condensed two fluorine containing γ-keto acids, 2-(4-fluorobenzoyl)benzoic acid and 2-(3,5-dinitro-4-fluorobenzoyl)benzoic acid with various mono, di- and trihydroxyphenols in presence of catalytic quantity of concentrated sulphuric acid to afford the fluorine substituted phthalides of biological interest. The used synthetic procedure is simple, efficient, economical and environmentally benign as it does not involve the use of any solvent. To the best of our knowledge, this is the first report on the synthesis of fluorinated phthalides from fluorinated γ-keto acids.
Materials and methods
Melting points were determined in open capillary tubes and are uncorrected. UV spectra were recorded in methanol on a Perkin-Elmer Lambda 15 UV/Vis spectrophotometer (USA). IR spectra were taken in KBr on a Perkin-Elmer FT-IR Spectrum RX-I spectrometer (USA). The 1H NMR spectra were recorded in DMSO-d6 solutions on a JEOL ECX- 400 spectrometer (Japan) at 400 MHz. Chemical shift (δ) are reported as downfield displacement from TMS that is used as internal standard. Mass spectra were recorded on a SURVEYOR-MSQ (LC-MS) mass spectrometer. The chemicals and solvents were procured from E. Merck (Germany) and Qualigens (India) and used as received without further purification. TLC was used to access the progress of the reactions and to ascertain the purity of the synthesized products. TLC plates coated with alumina-silica gel G (1:1) layers were run in petroleum ether (bp 40–70°C) − acetone (60:40; v: v).
Preparation of 2-(4-fluorobenzoyl)benzoic acid (1a)
A vigorously stirred mixture of fluorobenzene (100 mL) and finely powdered phthalic anhydride (25.2 g, 0.17 mol) was heated to 40–50°C. To this, anhydrous aluminium chloride (52.3 g, 0.34 mol) was added in small parts with continued stirring. The dark brown reaction mixture was heated on a steam bath for 5 h and allowed to stand at room temperature for overnight. Thereafter, it was hydrolyzed with ice and concentrated hydrochloric acid, and steam distilled to remove excess of fluorobenzene that was used as solvent. The crude product after filtration was dissolved in sodium carbonate solution, charcolised and finally acidified with concentrated hydrochloric acid. The precipitated acid 1a was further purified by crystallization from a mixture of benzene and petroleum ether as colourless needles. Yield, 33.7 g (97%); mp, 138–139°C (lit,Citation39 137–137.5°C).
Preparation of 2-(3,5-dinitro-4-fluorobenzoyl)benzoic acid (1b)
2-(4-Fluorobenzoyl)benzoic acid (1a) (15.87 g, 0.065 mol) was dissolved in concentrated sulphuric acid (d, 1.83, 36 mL). To this, a mixture of concentrated nitric acid (d, 1.41, 9 mL) and sulphuric acid (d, 1.83, 11 mL) was added gradually with stirring and maintaining the temperature at 40–50°C. The reaction mixture was allowed to stand at 40–50°C for 1 h. After cooling at room temperature, it was poured into ice-cold water with vigorous stirring. The deposited crude acid 1b was filtered, washed with water and purified by crystallization from ethanol as a light yellow crystalline solid. Yield 12.5 g (57.5%); mp = 159–160°C; IR (KBr) νmax/ cm−1: 3400, 2850, 2750, 1780, 1711, 1680, 1615, 1590, 1530, 1348, 842; 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.69–8.6 (m, 6H), 8.69(s, 1H); Anal. Calcd. for C14H7FN2O7: C, 50.31; H, 2.11; N, 8.38; Found: C, 50.55; H, 2.20; N, 8.56%.
Synthesis of the fluorinated phthalides (4a-9a and 4b-9b)
Typical synthesis of 3-(2,4-dihydroxyphenyl)-3-(4-fluorophenyl)phthalide (5a)
An intimate and well dried mixture of 2-(4-fluorobenzoyl)benzoic acid 1a (4.8 g, 0.02 mol) and resorcinol (2.75 g, 0.025 mol) was heated to 110°C in an oil bath to obtain a homogeneous solution. Thereafter, concentrated sulphuric acid (5 drops) was added cautiously and the heating was continued between 110°C and 120°C for 1 h to get a hard and brittle mass on cooling. It was washed thoroughly with water to remove the excess of resorcinol, extracted with 2% aq. sodium hydroxide and the extract was filtered. The filtrate was cooled and acidified with dilute hydrochloric acid to precipitate the phthalide, 5a. It was filtered, washed well with water and finally crystallized with aq. ethanol as a pale yellow microcrystalline solid. Yield 5.3 g (78.9%); mp = 145–146°C; UV λmax (MeOH)/ nm: 206, 281; IR (KBr) νmax/ cm−1: 3367, 1740, 983, 756, 694; 1H NMR (400 MHz, DMSO-d6): δ 6.13–7.83(m, 11H), 9.47 (s, 2H); MS: ESI-MS, m/z (%):337 [M+H]+ (100), 359 [M+Na]+ (24), 391 [M+Na+MeOH]+ (30); Anal. Calcd. for C20H13FO4: C, 71.43; H, 3.90; Found, C, 71.58; H, 3.82 %.
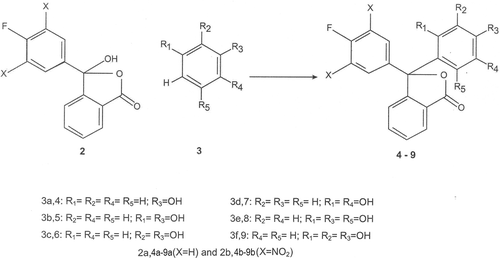
The other phthalides were synthesized by condensing appropriate γ-keto acid (1a and 1b) with phenols (phenol, resorcinol, catechol, quinol, phloroglucinol and pyragallol) following the procedure described above. In case of the condensation of γ-keto acids with phenol, the unreacted phenol after condensation was removed by steam distillation. The relevant data of the synthesized phthalides are given below.
3-(2,4-Dihydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (5b)
A mixture of the acid 2-(3,5-dinitro-4-fluorobenzoyl)benzoic acid (1b) (3.34 g, 0.01 mol) and resorcinol (1.65 g, 0.015 mol) was heated at 120–130°C for 0.5 h in presence of concentrated sulphuric acid (5 drops) to get phthalide 5b as green solid. Yield 2.75 g (64.6%); mp = 210–212°C; UV: λmax (MeOH)/ nm 207, 252, 382; IR (KBr): νmax/ cm−1 3368, 1763, 1256, 983, 750,685; 1H NMR (400 MHz, DMSO-d6): δ 6.97–8.60 (m, 9H), 9.80 (s, 2H). MS: ESI-MS, m/z (%): 427 [M+H]+ (100), 465 [M+K]+ (5); Anal. Calcd. for C20H11FN2O8: C, 56.35; H, 2.60; N, 6.57; Found, C, 56.50; H, 2.71; N, 6.63%.
3-(4-Hydroxyphenyl)-3-(4-fluorophenyl)phthalide (4a)
The acid 1a (2.44 g, 0.01 mol) and phenol (1.41 g, 0.015 mol) were mixed with each other and heated at 80–90°C for 5 h using concentrated sulphuric acid (4 drops) as condensing agent to get 4a as pinkish white solid. Yield 2.21 g (69.2%); mp = 110–112°C; UV: λmax (MeOH)/ nm 210, 250, 300; IR (KBr): νmax/ cm−1 3351, 1754, 1300, 980, 755, 693; 1H NMR (400 MHz, DMSO-d6): δ 6.77–7.89 (m, 12H), 9.75 (s, 1H); Anal. Calcd. for C20H13FO3: C, 74.99; H, 4.09; Found, C, 74.77; H, 4.19%.
3-(4-Hydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (4b)
The acid 1b (2.17 g, 0.0065 mol) and phenol (0.66 g, 0.007 mol) were mixed with each other and subjected to condensation at 70–80°C for 3 h in presence of concentrated sulphuric acid (3 drops) to afford a yellow solid. Yield 1.60 g (60.0%); mp = 122–124°C; UV: λmax (MeOH)/ nm 210, 281, 305; IR (KBr): νmax/ cm−1 3400, 1710, 1320, 981, 760, 691; 1H NMR (400 MHz, DMSO-d6): δ 6.75–8.60 (m, 10H), 8.66 (s, 1H); Anal. Calcd. for C20H11FN2O7: C, 58.54; H, 2.70; N, 6.83; Found, C, 58.64; H, 2.78; N, 6.69%.
3-(3,4-Dihydroxyphenyl)-3-(4-fluorophenyl)phthalide (6a)
A mixture of the acid 1a (2.44 g, 0.01 mol) and catechol (1.65 g, 0.015 mol) was condensed in presence of concentrated sulphuric acid (3 drops) at 85–95°C for 2 h to afford a blackish grey solid. Yield 2.96 g (88.1%); mp = 110–112°C; UV: λmax (MeOH)/ nm 208, 260, 300; IR (KBr): νmax/ cm−1 3368, 1744, 1335, 994, 753, 694; 1H NMR (400 MHz, DMSO-d6): δ 6.42–7.89 (m, 11H), 9.13 (s, 2H). Anal. Calcd. for C20H13FO4: C, 71.43; H, 3.90; Found, C, 71.60; H, 3.82%.
3-(3,4-Dihydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (6b)
The acid 1b (2.17 g, 0.0065 mol) was mixed with catechol (0.77 g, 0.007 mol) and the resulting mixture was heated at 90–100°C for 40 minutes using concentrated sulphuric acid (3 drops) as condensing agent to obtain phthalide 6b as pinkish brown solid. Yield 1.44 g (52.0%); mp = 205–207°C; UV: λmax (MeOH)/ nm 210, 280, 350; IR (KBr): νmax/ cm−1 3403, 1773, 1250, 1000, 750, 690; 1H NMR (400 MHz, DMSO-d6): δ 6.46–8.63 (m, 9H), 9.19 (s, 2H); Anal. Calcd. for C20H11FN2O8: C, 56.35; H, 2.60; N, 6.57; Found: C, 56.19; H, 2.57; N, 6.61%.
3-(2,5-Dihydroxyphenyl)-3-(4-fluorophenyl)phthalide (7a)
The acid 1a (2.44 g, 0.01 mol) was condensed with quinol (1.65 g, 0.015 mol) in presence of concentrated sulphuric acid (5 drops) at 135–145°C for 2 h. The usual workup of the reaction mixture gave 7a as a dark brown solid. Yield 2.46 g (73.3%); mp = 140–142°C; UV: λmax (MeOH)/ nm 205, 260, 300; IR (KBr): νmax/ cm−1 3368, 1744, 1291, 995, 753, 693; 1H NMR (400 MHz, DMSO-d6): δ 6.58–7.98 (m, 11H), 8.82 (s, 1H), 8.97 (s, 1H); Anal. Calcd. for C20H13FO4: C, 71.43; H, 3.90; Found: C, 71.26; H, 3.83%.
3-(2,5-Dihydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (7b)
The acid 1b (2.17 g, 0.0065 mol) was condensed with quinol (0.77 g, 0.007 mol) at 120–130°C in presence of concentrated sulphuric acid (4 drops) for 1 h. The usual workup of the condensed mass gave 7b as a green solid. Yield, 1.41 g (50.9%); mp = 225–226°C; UV: λmax (MeOH)/ nm 208, 275, 325; IR (KBr): νmax/ cm−1 3385, 1774, 1350, 982, 750, 689; 1H NMR (400 MHz, DMSO-d6): δ 6.48–8.55 (m, 9H), 8.55 (s, 1H), 8.66 (s, 1H); Anal. Calcd. for C20H11FN2O8: C, 56.35; H, 2.60; N, 6.57; Found, C, 56.51; H, 2.67; N, 6.78%.
3-(2,4,6-Trihydroxyphenyl)-3-(4-fluorophenyl)phthalide (8a)
An intimate mixture of the acid 1a (2.44 g, 0.01 mol) and phloroglucinol (1.89 g, 0.015 mol) was heated at 170–180°C for 1 h in presence of concentrated sulphuric acid (3 drops) and the condensed mass was worked up to obtain 8a as a brick red solid. Yield 2.09 g (59.3%), mp = 245–246°C; UV: λmax (MeOH)/ nm 210, 280, 375; IR (KBr): νmax/ cm−1 3368, 1734, 1289, 1015, 755, 696; 1H NMR (400 MHz, DMSO-d6): δ 6.59–7.97 (m, 10H), 9.00 (broad and unresolved signal, 3H). Anal. Calcd. for C20H13FO5: C, 68.18; H, 3.72; Found, C, 68.18; H, 3.79%.
3-(2,4,6-Trihydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (8b)
An intimate mixture of the acid 1b (2.17 g, 0.0065 mol) and phloroglucinol (0.88 g, 0.007 mol) was heated at 155–165°C for 0.5 h using concentrated sulphuric acid (3 drops) to get 8b as a brown solid. Yield 2.07 g (72.3%); mp = >300°C; UV: λmax (MeOH)/ nm 210, 281, 380; IR (KBr): νmax/ cm−1 3377, 1717, 1350, 1000, 747, 687; 1H NMR (400 MHz, DMSO-d6): δ 6.10 (br s, 3H), 6.93–8.64 (m, 8H); Anal. Calcd. for C20H11FN2O9: C, 54.31; H, 2.51; N, 6.33; Found: C, 54.60; H, 2.58; N, 6.40 %.
3-(2,3,4-Trihydroxyphenyl)-3-(4-fluorophenyl)phthalide (9a)
The acid 1a (2.44 g, 0.01 mol) was intimately mixed with pyrogallol (1.89 g, 0.015 mol). Heating of this mixture at 115–125°C for 0.5 h in presence of concentrated sulphuric acid (3 drops) and work up in usual manner afforded the phthalide 9a as a yellow solid. Yield 2.06 g (58.5%); mp = 170–172°C; UV: λmax (MeOH)/ nm 203, 281, 382; IR (KBr): νmax/ cm−1 3422, 1730, 1300, 1000, 752, 695; 1H NMR (400 MHz, DMSO-d6): δ 6.51–7.82 (m, 10H), 8.41 (s, 3H). Anal. Calcd. for C20H13FO5: C, 68.18; H, 3.72; Found, C, 68.45; H, 3.83%.
3-(2,3,4-Trihydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (9b)
The acid 1b (2.17 g, 0.0065 mol) was intimately mixed with pyrogallol (0.88 g, 0.007 mol). On heating the mixture at 110–120°C in presence of concentrated sulphuric acid (3 drops) the phthalide 9b was instantly obtained within 10 minutes as a brown solid. Yield 1.77 g (64.2%). MP: >300°C; UV: λmax (MeOH)/ nm 203, 280, 382; IR (KBr): νmax/ cm−1 3365, 1718, 1261, 990, 759, 690; 1H NMR (400 MHz, DMSO-d6): δ 6.92 (br s, 3H), 7.00–8.10 (unresolved m, 8H); Anal. Calcd. for C20H11FN2O9: C, 54.31; H, 2.51; N, 6.33 Found: C, 54.12; H, 2.48; N, 6.45%.
Acetylation of phthalide 5a and 5b
Synthesis of 3-(2,4-diacetoxyphenyl)-3-(4-fluorophenyl)phthalide (10a)
The phthalide, 5a (1 g, 0.003 mol) was mixed with acetic anhydride (20 mL) and freshly fused sodium acetate (3.00 g). The resulting mixture was refluxed at 130–140°C for 3.5 h, poured into ice-cold water with stirring and allowed to stand for 2 h. The deposited crude diacetyl compound 10a was filtered and purified by crystallization from aq. acetone as an off white crystalline solid. Yield 0.82 g (65.5%); mp = 120–122°C; UV: λmax (MeOH)/ nm 204, 266, 287; IR (KBr): νmax/ cm−1 1777, 1288, 1189, 978, 759, 693; 1H NMR (400 MHz, DMSO-d6): δ 2.04 (s, 6H), 6.88–7.92 (m, 11H); Anal. Calcd. for C24H17FO6: C, 68.57; H, 4.08; Found, C, 68.70; H, 3.98%.
Synthesis of 3-(2,4-diacetoxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (10b)
Following the procedure given above, the phthalide 5b (1.02 g, 0.0024 mol) was acetylated with acetic anhydride (25 mL) in presence of fused sodium acetate (3 g). The resulting mixture was refluxed at 130–140°C to afford the diacetyl compound 10b. It was recrystallised from aq. acetone as a light brown solid. Yield 0.91 g (74.1%); mp = 165–167°C; UV: λmax (MeOH)/ nm 203, 260, 285; IR (KBr): νmax/ cm−1 1780, 1290, 1190, 978, 760, 693; 1H NMR (400 MHz, DMSO-d6): δ 2.05 (s, 6H), 7.08–8.64 (m, 9H); Anal. Calcd. for C24H15FN2O10: C, 56.48; H, 2.96; N, 5.49; Found, C, 56.05; H, 2.72; N, 5.92%.
Bromination of phthalide 5a and 5b
Synthesis of 3-(3,5-dibromo-2,4-dihydroxyphenyl)-3-(4-fluorophenyl)phthalide (11a)
The phthalide 5a (1 g, 0.003 mol) was finely powdered and dissolved in ethanol (10 mL), and bromine (2 mL) was added drop by drop with shaking. The resultant mixture was allowed to stand overnight at room temperature. The dibromo compound 11a separated as viscous oil. It was repeatedly washed with cold water and dissolved in aq. sodium hydroxide and filtered. The filtrate was acidified with dilute hydrochloric acid to get the dibromo compound as a brown powder which was finally crystallized from ethanol. Yield 0.91 g (61.4%); mp = 135–137°C; UV: λmax (MeOH)/ nm 204, 246, 285; IR (KBr): νmax/ cm−1 3400, 1725, 1282, 1020, 758, 713; 1H NMR (400 MHz, DMSO-d6): δ 7.28–7.92 (m, 9H), 9.94 (s, 2H); Anal. Calcd. for C20H11FO4Br2: C, 48.62; H, 2.24; Br, 32.34; Found: C, 48.83; H, 2.28; Br, 32.50%.
Synthesis of 3-(3,5-dibromo-2,4-dihydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (11b)
The phthalide 5b (1.02 g, 0.0024 mol) was dissolved in ethanol (20 mL) and treated with bromine (2 mL) to get the dibromo compound 11b following the procedure used for the synthesis of 11a as described above. It was a brown solid. Yield 0.99 g (71.2%); mp = 155–157°C; UV: λmax (MeOH)/ nm 211, 255, 300; IR (KBr): νmax/ cm−1 3448, 1774, 1350, 980, 750, 690; 1H NMR (400 MHz, DMSO-d6): δ 6.70–8.65 (m, 7H), 9.68 (s, 2H). Anal. Calcd. for C20H9FN2O8Br2: C, 41.13; H, 1.55; N, 4.80; Br, 27.36; Found, C, 41.30; H, 1.60; N, 4.75; Br, 27.40%.
Alkaline degradation of 5a and 5b
A mixture of phthalide 5a (1 g), KOH pellets (10 g) and water (10 mL) was strongly heated at 250°C for 3 h. The contents were cooled, dissolved in water and filtered. The filtrate was acidified with dilute hydrochloric acid when a solid compound (I) was obtained. It was filtered and the filtrate so obtained was extracted with ether (3 × 25 mL). The combined organic layer was dried over anhydrous sodium sulphate and distilled off to afford another solid compound (II). The compounds I and II were identified as 2-(4-hydroxybenzoyl)benzoic acid (12a) and resorcinol, respectively by direct comparison (mp, mixed mp, co-TLC and co-IR spectra) with their authentic samples. In a similar manner the phthalide 5b (1 g) was mixed with KOH pellets (10 g) and water (10 mL) to make a thick paste, and strongly heated at 250°C for 3 h. In this case also, the workup of the residue gave two compounds which were identified as 2-(3,5-dinitro-4-hydroxybenzoyl)benzoic acid (12b) and resorcinol. The authentic samples of 12a and 12b were prepared by the literature procedureCitation39 from 1a and 1b, respectively.
Procedure for biological activity
The fluorinated phthalides 6a, 6b, 9a and 9b were evaluated for their in vitro antibacterial activity against Staphylococcus aureus ATCC 29213 (Sa), methicillin-resistant S. aureus ATCC 33591 (MRS), Escherichia coli ATCC 35218 (Ec), Pseudomonas aeruginosa ATCC 27853 (Pa), and Mycobacterium intracellulare. The antifungal activity was tested against Candida albicans ATCC 90028 (Ca), C. glabrata ATCC 90030 (Cg), C. krusei ATCC 6258 (Ck), Cryptococcus neoformans ATCC 90113 (Cn), and Aspergillus fumigatus ATCC 204305 (Af).
All organisms were tested using modified versions of the CLSI (formerly NCCLS) methods. For all organisms excluding M. intracellulare and A. fumigatus, optical density was used to monitor the growthCitation40,Citation41. Media supplemented with 5% Alamar Blue™ (BioSource International, Camarillo, CA) was utilized for growth detection of M. intracellulareCitation42,Citation43 and A. fumigatusCitation44. Samples (dissolved in DMSO) were serially-diluted in 20% DMSO/saline and transferred (10 µL) in duplicate to 96-well flat bottom microplates. Inocula were prepared by correcting the OD630 of microbe suspensions in incubation broth [RPMI 1640/0.2% dextrose/0.03% glutamine/MOPS at pH 6.0 (Cellgro) for Candida spp., Sabouraud Dextrose for C. neoformans, cation-adjusted Mueller-Hinton (Difco) at pH 7.3 for Staphylococcus spp., E. coli, and P. aeruginosa, 5% Alamar Blue™(BioSource International, Camarillo, CA) in Middlebrook 7H9 broth with OADC enrichment, pH 7.0 for M. intracellulare, and 5% Alamar Blue™/RPMI 1640 broth (0.2% dextrose, 0.03% glutamine, buffered with 0.165 M MOPS at pH 7.0) for A. fumigatus to afford an assay volume of 200 µL and final target inocula of: Candida spp. and C. neoformans: 1.5 × 103, M. intracellulare: 2.0 × 106, Staphylococcus spp., E. coli, P. aeruginosa: 5.0 × 105 CFU/mL, and A. fumigatus: 2.7 × 104 CFU/mL. Final sample test concentrations are 1/100th the DMSO stock concentration.
Drug controls (Ciprofloxacin (ICN Biomedicals, OH) for bacteria and Amphotericin B (ICN Biomedicals, OH) for fungi) are included in each assay. All organisms are read at either 530 nm using the Biotek Powerwave XS plate reader (Bio-Tek Instruments, Vermont) or 544 ex/590 em, (M. intracellulare, A. fumigatus) using the Polarstar Galaxy Plate Reader (BMG LabTechnologies, Germany) prior to and after incubation: Candida spp. at 35°C for 46–50 h, Staphylococcus spp., E. coli, and P. aeruginosa at 35°C for 16–20 h, C. neoformans at 35°C for 70–74 h, A. fumigatus at 35°C for 46–50 h, and M. intracellulare at 37°C and 10% CO2 for 70–74 h. IC50 (concentrations that afford 50% inhibition relative to controls) were calculated using XLfit 4.2 software (IDBS, Alameda, CA) using fit model 201. The MIC was defined as the lowest test concentration that allows no detectable growth (for M. intracellulare and A. fumigatus, no colour change from blue to pink).
Results and discussion
During the course of our present work, it has been found that 2-(4-fluorobenzoyl)benzoic acid (1a) and 2-(3,5-dinitro-4-fluorobenzoyl)benzoic acid (1b) reacted with phenol, resorcinol, catechol, quinol, phloroglucinol and pyrogallol in presence of a few drops of concentrated sulphuric acid to produce new fluorine substituted phthalides (4a-9a and 4b-9b). In the synthesized phthalides, carbon- 3 of the phthalide structure is attached to two different phenyl rings, one being phenolic and the other non-phenolic having a fluorine atom with or without nitro groups. Thus, the synthesized compounds may be regarded as unsymmetrically substituted phthalides. The phenols (3) were taken in slight excess of molecular proportion than the γ-keto acids 1a and 1b. The occurrence of keto-lactol tautomerism in γ-keto and γ-formyl acids, and their participation in some chemical reactions through their cyclic lactol form is well documentedCitation45,Citation46. The open keto-acid (1) and the cyclic lactol (2) tautomeric forms of the 2-(4-fluorobenzoyl)benzoic acid and 2-(3,5-dinitro-4-benzoyl)benzoic acid maybe depicted as given in . These two acids (1) reacted with phenols (3) through their cyclic lactol tautomeric form (2) to give the fluorinated phthalides (4a-9a and 4b-9b) as shown in . In the light of the reported pharmacological activities associated with constituent moieties present in 4a-9a and 4b-9b, these compounds as well as their diacetyl (10a and 10b) and dibromo derivatives (11a and 11b) () are expected to find use as therapeutic agents.
Scheme 1. Keto-acid and lactol tautomeric forms of 2-(4-fluorobenzoyl)benzoic acid and 2-(3,5-dinitro-4-fluorobenzoyl)benzoic acids.
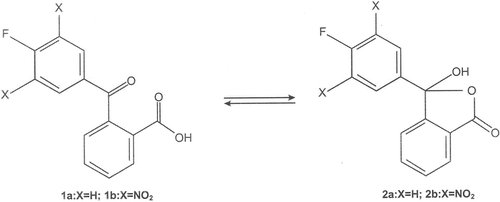
Scheme 2. Synthesis of fluorinated phthalides 4-9.
Scheme 3. Acetylation, bromination and KOH fusion of fluorinated phthalides 5a, b.
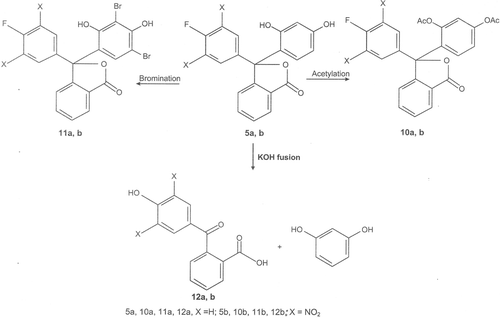
It has been found that efficient synthesis of the phthalides depends mainly on three factors such as proper condensation temperature, proper condensation time and proper quantity of the catalyst. Therefore, during the present study, these three factors have been worked out for each of the synthesized phthalides. It was also observed that during the reaction of the γ-keto acid 1b with phenols (3) the presence of electron withdrawing nitro groups in 1b considerably reduced the condensation time during the synthesis of the phthalides (4b-9b) in comparison to reaction of 1a with phenols giving phthalides (4a-9a). This can be attributed to the fact that presence of electron withdrawing groups in γ-keto acids augments the formation of cyclic lactol tautomer and thereby increase the rate of those reactions wherein the cyclic lactol form is involved. The structures proposed to the synthesized fluorine substituted phthalides were established on the basis of elemental analysis, UV, IR, 1H NMR and Mass spectral data and chemical reactions, viz., acetylation, bromination and fusion with potassium hydroxide. The representative phthalides 5a and 5b on acetylation and bromination gave their corresponding diacetyl derivatives (10a and 10b) and dibromo derivatives (11a and 11b), respectively. The potassium hydroxide fusion of 5a and 5b degraded them to γ-keto acids, 2-(4-hydroxybenzoyl)benzoic acid (12a) and 2-(3,5-dintro-4-hydroxybenzoyl)benzoic acid (12b), respectively and resorcinol. The acetylation, bromination, and potassium hydroxide degradation reactions of 5a and 5b are given in .
The starting 2-(4-fluorobenzoyl)benzoic acid (1a) was prepared by Friedel-Crafts condensation of fluorobenzene with phthalic anhydride in presence of anhydrous aluminium chloride following a literature methodCitation39 with some modifications in workup procedure in order to improve the yield and purity of the product. The acid 1a was nitrated by the method of De Tar and RelyeCitation47 to afford 2-(3,5-dinitro-4-fluorobenzoyl)benzoic acid (1b). These workers used this method for the synthesis of 2-(3-nitrobenzoyl)benzoic acid from 2-benzoylbenzoic acid. It is interesting to note that during the present study we have obtained a dinitro product (1b) instead of the mononitro acidCitation47.
The IR spectra (in KBr) of the synthesized fluorinated phthalides 4a-9a and 4b-9b and 11a and 11b showed a broad and strong absorption band in the region 3351–3448 cm−1 due to bonded OH stretching vibrations. The diacetyl compounds 10a and 10b did not exhibit any IR absorption in hydroxyl region. All the compounds (4a-11a and 4b-11b) displayed a sharp and strong band near 1730–1780 cm−1 which is characteristic of lactonic carbonyl group present in phthalide structures. Besides the presence of this band, two bands were noticed at 693–713 cm−1 and 750–760 cm−1 which can be attributed to o-disubstituted phthalide ring. The stretching vibrations of C-O-C of the lactone structure and C-O bond of phenolic group of the phthalides gave absorption peaks near 980–1015 cm−1 and 1288–1300 cm−1, respectively. The UV spectra (in methanol) of all the compounds revealed same pattern of absorption at 203–211, 250–281 and 285–382 nm. In the 1H NMR spectra (400 MHz, DMSO-d6) of the synthesized phthalides (4a-11a and 4b-11b) the aromatic protons formed a complex multiplet in the region δ 6.42–8.65. Their hydroxyl protons generally appeared as a singlet near δ 8.41–9.80. In 10a and 10b, protons of acetoxyl groups formed a singlet at about δ 2.04–2.05. The representative phthalides 5a and 5b were subjected to mass spectral analysis to confirm the proposed structures on the basis of fragmentation pattern and molecular weight obtained from molecular ion. The compound 5a gave a molecular ion at m/z 337, while in case of 5b molecular ion appeared at m/z 427. In both the compounds (5a and 5b) molecular ion peak appeared as base peak exhibiting 100% abundance.
Biological activity
The synthesized fluorinated phthalides (4a-11a and 4b-11b) are expected to possess diverse biological activities due to combination of fluorophenyl, hydroxyphenyl and phthalide moieties within a single entity. During the present study, the phthalides, 3-(3,4-dihydroxyphenyl)-3-(4-fluorophenyl)phthalide (6a); 3-(3,4-dihydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (6b); 3-(2,3,4-trihydroxyphenyl)-3-(4-fluorophenyl)phthalide (9a) and 3-(2,3,4-trihydroxyphenyl)-3-(3,5-dinitro-4-fluorophenyl)phthalide (9b) were tested for their in vitro antibacterial activity against S. aureus ATCC 29213 (Sa), methicillin-resistant S. aureus ATCC 33591 (MRS), E. coli ATCC 35218 (Ec), P. aeruginosa ATCC 27853 (Pa), and M. intracellulare ATCC 23068 (Mi) using DMSO as solvent. The in vitro antifungal activity of the phthalides 6a, 6b, 9a and 9b was determined against C. albicans ATCC 90028 (Ca), C. glabrata ATCC 90030 (Cg), C. krusei ATCC 6258 (Ck), Cryptococcus neoformans ATCC 90113 (Cn), and Aspergillus fumigatus ATCC 204305 (Af). The results of this antimicrobial assay are presented in and . The compounds selected for the antimicrobial screening contain catechol and pyrogallol structural units in their structures. The rationale for this choice is the fact that catechol and pyrogallol are allelo chemicals belonging to phenolic compounds synthesized in plantsCitation18, and significant antibacterial activity is associated with themCitation19,Citation20.
Table 1. Antifungal and antibacterial activity of some of the synthesized phthalides (IC50 µg/mL).
Table 2. Antifungal and antibacterial activity of some of the synthesized novel phthalides (MIC µg/mL).
In the present study, it has been noticed that presence of nitro groups in phthalides considerably decrease the antifungal and antibacterial activity. The antifungal activity of the dinitro phthalides is much lower than their antibacterial activity. Dihydroxy substituted phthalides were found to possess higher antibacterial activity in comparison of trihydroxy substituted phthalides.
Mode of action of phthalides on biological systems
Although there are numerous reports on the bioactivities of phthalides, but investigations concerning their mode of action are very scanty. However, it has been suggested that presence of five membered lactone ring in phthalides is responsible for their biological activityCitation48. Antibacterial activity of phthalides has been explained by proposing a reaction between phthalides and amino acid cysteine, and certain enzymes (containing mercapto group) which are present in bacterial proteins and are necessary for their growth and normal activity. As a consequence of this reaction, the growth process of bacteria is inhibited. In an experiment involving the action of butylidenephthalide on hairless mouse, formation of a cysteine adduct as a urinary metabolite has been detected in the urine of the mouseCitation49. Examination of the structures of the new phthalides (4a-11a and 4b-11b) synthesized during the present study reveals that owing to their suitable structural feature, they are also capable of reacting with cysteine to form their corresponding adducts, and thereby exhibiting antimicrobial activity.
The precise biological activity of 3-substituted phthalides is often crucially related with chiralityCitation30,Citation50. Chiral 3-substituted phthalides such as isochracinic acidCitation51, herbaranic acidCitation51, cytosporone ECitation52 have been found to exhibit antibacterial activity, and fuscinarinCitation53 is a potent human CCR5 antagonist, effectively blocking HIV entry into host cell. Since the phthalides reported in this paper possess a chiral centre (C-3) in their structure, and therefore, it may be assumed that the antimicrobial activity of these racemic compounds is potentially influenced by it.
Conclusion
The present report constitute the first successful attempt to synthesize new fluorine substituted phthalides from fluorine containing 2-aroylbenzoic acids by a simple and straightforward protocol without producing any byproducts. Besides having the advantages of simplicity, good yields, use of cheap and easily accessible starting chemicals, this method is environmentally benign as it does not involve the use of any solvent. The synthesized fluorinated compounds are anticipated to have diverse applications in medicinal fields. A few of them have been found to exhibit some antibacterial and antifungal activity on in vitro antimicrobial screening. Specially, 6a has shown promising activity against S. aureus and MRS. In view of the growing incidences of drug resistance in microorganisms, and remarkable bioenhancing property shown by some less active or even inactive compounds, further appropriate investigations on anticipated biological properties including in vivo antimicrobial screening of the synthesized fluorinated phthalides are warranted to explore their therapeutic potential.
Acknowledgements
We are thankful to Dr. D. S. Rawat, Department of Chemistry, Delhi University, Delhi, India, for his generous help in recording the NMR spectra, and Dr. G. P. Pandey, Head and Scientist-G, Division of Organic Chemistry, National Chemical Laboratory, Pune, India, for providing mass spectral data. Support for antimicrobial evaluation by the NIH, NIAID, Division of AIDS, Grant No. AI 27094 and the USDA Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009 is gratefully acknowledged. We also sincerely thank Prof. Alice M. Clark and Marsha A. Wright (antimicrobial evaluation), University of Mississippi, Mississippi (USA), for providing generous help for the antimicrobial studies.
Declaration of interest
The authors report no conflicts of interest.
References
- Shelkea SN, Dalvi NR, Kale SB, More MS, Gill CH, Karale BK. Environmentally benign synthesis of fluorinated pyrazolone derivatives and their antimicrobial activity. Indian J Chem 2007;46B:1174–1178.
- Mogilaiah K, Dhanaja K, Srivani N, Chandra AV. Efficient synthesis of 9-aryl-6-(2- fluorophenyl)-1,2,4-triazolo[4,3-α] [1,8]naphthyridines using chloramine-T under microwave irradiation. Indian J Chem 2010;49B:500–504.
- Müller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007;317:1881–1886.
- Wang X, Mei TS, Yu JQ. Versatile Pd(OTf)2 × 2 H2O-catalyzed ortho-fluorination using NMP as a promoter. J Am Chem Soc 2009;131:7520–7521.
- Shimizu M, Hiyama T. Modern synthetic methods for fluorine-substituted target molecules. Angew Chem Int Ed Engl 2004;44:214–231.
- Tredwell M, Gouverneur V. Electrophilic fluorination of organosilanes. Org Biomol Chem 2006;4:26–32.
- Joshi NS, Shaikh AA, Deshpande AP, Karale BK, Bhirud SB, Gill CH. Synthesis, characterization and antimicrobial activities of some fluorine containing 2-(1-phenyl-3-aryl-1Hpyrazol-4-yl)-3-chlorochromones, 2-(1-phenyl-3-aryl-1H-pyrazol-4-yl)chromones and 5-(1-phenyl-3-aryl-1H-pyrazol-4-yl)-3-(2-hydroxyphenyl)-4,5-dihydropyrazolines. Indian J Chem 2005;44B:422–425.
- Patel VM, Desai KR. Microwave-induced synthesis of fluorine containing 1, 5-disubstituted hydantoins and thiohydantoins and their antibacterial activity. Indian J Chem 2005;44B:1084–1087.
- Hammam AEG, El-Salam OAI, Mohamed AM, Hafez NA. Novel fluoro substituted benzo[b]pyran with anti-lung cancer activity. Indian J Chem 2005;44B:1887–1893.
- Rojas J, Payá M, Dominguez JN, Luisa Ferrándiz M. The synthesis and effect of fluorinated chalcone derivatives on nitric oxide production. Bioorg Med Chem Lett 2002;12:1951–1954.
- Watson TJ, Ayers TA, Shah N, Wenstrup D, Webster M, Freund D, Horgan S, Carey JP. Process improvements for the preparation of kilo quantities of a series of isoindoline compounds. Org Process Res Dev 2003;7:521–532.
- Piccionello AP, Pace A, Pierro P, Pilibri P, Buscemi S, Vivona N. On the reaction of some 5-polyfluoroaryl-1,2,4-oxadiazoles with methylhydrazine: synthesis of fluorinated indazoles. Tetrahedron 2009;65:119–127.
- Pormord F, Hosseinimehr SJ, Shahabimajd N. Antioxidant activity, phenol and flavonoid contents of some selected Iranian medicinal plants. Afr J Biotechnol 2006;5:1142–1145.
- Russel AD. Mechanisms of antimicrobial action of antiseptics and disinfectants: an increasingly important area of investigation. J Antimicrob Chemother 2002;49:597–599.
- Rodirgo G, Almanja GR, Cheng Y, Peng J, Hamann M, Duan RD, Akesson B. Antiproliferative effects of curcuphenol, a sesquiterpene phenol. Fitoterapia 2010;81:762–766.
- Mekawey AAI, Mokhtar MM, Farrag RM. Antitumor and antibacterial activities of [1-(2-ethyl, 6-heptyl)phenol] from Cuminum cyminum seeds. J Appl Sci Res 2009;5:1881–1888.
- Bendini A, Cerretani L, Pizzolante L, Tosch TG, Guzzo F, Ceoldo S, Marconi AM, Andretta F, Levi M. Phenol content related to antioxidant and antimicrobial activities of Passiflora spp. extracts. Eur Food Res Techol 2006;223:102–109.
- Kocaçalischan I, Talah I, Terzi I. Antimicrobial Activity of Catechol and Pyrogallol as Allelochemicals. Z Naturforsch 2006;61c:639–642.
- Tomc JF, Busch K, Minnasian B, Kolek B, Flamm R, Gradelski A, Bonner D. Antibacterial activity of BMS-180680, a new catechol-containing monobactam. Artimicrob Agents Chemother 1997;41:1010–1016.
- Taguri T, Tanaka T, Kouno I. Antibacterial spectrum of plant polyphenols and extracts depending upon hydroxyphenyl structure. Biol Pharm Bull 2006;29:2226–2235.
- McDonald RW, Bunjobpon W, Liu T, Fessler S, Pardo OE, Freer IK et al. Synthesis and anticancer activity of nordihydroguaiaretic acid (NDGA) and analogues. Anticancer Drug Des 2001;16:261–270.
- Mitsuhashi S, Saito A, Nakajima N, Shima H, Ubukata M. Pyrogallol structure in polyphenols is involved in apoptosis-induction on HEK293T and K562 cells. Molecules 2008;13:2998–3006.
- Barbini L, Lopez P, Ruffa J, Martino V, Ferraro G, Campos R et al. Induction of apoptosis on human hepatocarcinoma cell lines by an alkyl resorcinol isolated from Lithraea molleoides. World J Gastroenterol 2006;12:5959–5963.
- Sharp SY, Boxall K, Rowlands M, Prodromou C, Roe SM, Maloney A et al. In vitro biological characterization of a novel, synthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors. Cancer Res 2007;67:2206–2216.
- Cojocaru M, Droby S, Glotter E, Goldman A, Gottlieb HE, Jacoby B, Prusky D. 5-(12-heptadecenyl)-resorcinol, the major component of the antifungal activity in the peel of mango fruit. Phytochemistry 1986;25:1093–1095.
- Suzuki Y, Esumi Y, Hyakutake H, Kono Y, Sakurai A. Isolation of 5-(8′Z-heptadecenyl)-resorcinol from etiolated rice seedlings as an antifungal agent. Phytochemistry 1996;41:1485–1489.
- Chew EH, Lu J, Bradshaw TD, Holmgren A. Thioredoxin reductase inhibition by antitumor quinols: a quinol pharmacophore effect correlating to antiproliferative activity. Faseb J 2008;22:2072–2083.
- Singh IP, Sidana J, Bansal P, Foley WJ. Phloroglucinol compounds of therapeutic interest: global patent and technology status. Expert Opin Ther Pat 2009;19:847–866.
- Seki T, Tachikawa H, Yamada T, Hattori HH. Synthesis of phthalide-skeleton using selective intramolecular tishchenko reaction over solid base catalysts. J Catal 2003;217:117–126.
- Karnik AV, Kamath SS. Cascade Enantioselective Synthesis of 3-arylphthalides using chiral auxiliary route. Synthesis 2008;12:1832–1834.
- Lin G, Chem SS-K, Chung H-S, Li S-L. Chemistry and biological activities of naturally occurring phthalides. In: Rehman AU. (ed). Studies in Natural Product Chemistry. Bioactive Natutral Products (Part L). Amsterdam: Elsevier North Holland; 2005; 32. pp. 611–669.
- Knepper K, Zigert RE, Brase ST. Solid-phase synthesis of isoindolinones and naturallyoccurring benzobutyrolactones (phthalides) using a cyclative-cleavage approach. Tetrahedron 2004;60:8591–8603.
- Witulski B, Zimmermann A, Gowans ND. First total synthesis of the marine illudalane sesquiterpenoid alcyopterosin E. Chem Commun (Camb) 2002;34:2984–2985.
- Zhang B, Xu MH, Lin GQ. Catalytic enantioselective synthesis of chiral phthalides by efficient reductive cyclization of 2-acylarylcarboxylates under aqueous transfer hydrogenation conditions. Org Lett 2009;11:4712–4715.
- Lin H, Sun X-W. Highly efficient synthesis of 3-indolyl-substituted phthalides via friedel–crafts reactions in water. Tetrahedron Lett 2008;49:5343–5346.
- Zhou L, Jiang H-F. Synthesis of phthalides via Pd/CNTs-catalyzed reaction of terminal alkynes and O-iodobenzoic acid under copper- and ligand-free conditions. Tetrahedron Lett 2007;48:8449–8452.
- Chang HT, Jeganmohan M, Cheng CH. Highly efficient cyclization of o-iodobenzoates with aldehydes catalyzed by cobalt bidentate phosphine complexes: a novel entry to chiral phthalides. Chemistry 2007;13:4356–4363.
- Faigl F, Thurner A, Molnar B, Simig G, Volk B. Manufacturing synthesis of 5-substituted phthalides. Org Process Res Dev 2010;14:617–622.
- Hahn FC, Reid EE. Ortho-benzoyl-benzoic acids containing fluorine, iodine and sulfur. J Am Chem Soc 1924;46:1645–1653.
- NCCLS. Reference method for broth dilution antifungal susceptibility testing of yeasts; Approved Standard M27-A2. National Committee on Clinical Laboratory Standards, 2002.
- NCCLS. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; Approved Standard, 7th Edn. M7-A7, National Committee on Clinical Laboratory Standards, 2006.
- NCCLS. Susceptibility testing of mycobacteria, nocardia, and other aerobic actinomycetes; Tentative Standard—Approved Standard, M24-A. National Committee on Clinical Laboratory Standards, 2003.
- Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J Clin Microbiol 1998;36:362–366.
- NCCLS. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard, M38-A. National Committee on Clinical Laboratory Standards, 2002.
- Valter RE, Flitch W. Ring-Chain Tautomerism. New York and London: Plenum Press, 1985.
- Zhang J, Blazecka PG, Berven H, Belmont D. Metal-mediated allylation of mucohalic acids: facile formation of γ-allylic α,β-unsaturated γ-butyrolactones. Tetrahedron Lett 2003;44:5579–5582.
- DeTar DF, Relyea DI. Intramolecular Reactions. II. The mechanism of the cyclization of diazotized 2-aminobenzophenones. J Am Chem Soc 1954;76:1680–1685.
- Momin RA, Nair MG. Mosquitocidal, nematicidal, and antifungal compounds from Apium graveolens L. seeds. J Agric Food Chem 2001;49:142–145.
- Sekiya K, Tezuka Y, Tanaka K, Prasain JK, Namba T, Katayama K et al. Distribution, metabolism and excretion of butylidenephthalide of Ligustici chuanxiong rhizoma in hairless mouse after dermal application. J Ethnopharmacol 2000;71:401–409.
- Zhang H, Zhang S, Liu L, Luo G, Duan W, Wang W. Synthesis of chiral 3-substituted phthalides by a sequential organocatalytic enantioselective aldol-lactonization reaction. Three-step synthesis of (S)-(-)-3-butylphthalide. J Org Chem 2010;75:368–374.
- Höller U, Gloer JB, Wicklow DT. Biologically active polyketide metabolites from an undetermined fungicolous hyphomycete resembling Cladosporium. J Nat Prod 2002;65:876–882.
- Brady SF, Wagenaar MM, Singh MP, Janso JE, Clardy J. The cytosporones, new octaketide antibiotics isolated from an endophytic fungus. Org Lett 2000;2:4043–4046.
- Yoganathan K, Rossant C, Ng S, Huang Y, Butler MS, Buss AD. 10-Methoxydihydrofuscin, fuscinarin, and fuscin, novel antagonists of the human CCR5 receptor from Oidiodendron griseum. J Nat Prod 2003;66:1116–1117.