Abstract
Aminopeptidase N (APN/CD13) is one of the essential proteins for tumour invasion, angiogenesis and metastasis as it is over-expressed on the surface of different tumour cells. Based on our previous work that L-isoserine dipeptide derivatives were potent APN inhibitors, we designed and synthesized L-isoserine tripeptide derivatives as APN inhibitors. Among these compounds, one compound 16l (IC50 = 2.51 ± 0.2 µM) showed similar inhibitory effect compared with control compound Bestatin (IC50 = 6.25 ± 0.4 µM) and it could be used as novel lead compound for the APN inhibitors development as anticancer agents in the future.
Introduction
Aminopeptidase N (APN; EC 3.4.11.2) is a zinc dependent type II membrane-bond exopeptidaseCitation1,Citation2, which is selectively expressed on the surface of cells such as myeloid progenitors and monocytes, epithelial cells of the intestine and kidney, synaptic membranes in the central nervous system, fibroblasts, endothelial cells, epithelial cellsCitation3–6 and so on. Aminopeptidase N which is expressed on tumour cells can degrade the extra cellular matrix (ECM) and considered as the first step of tumour proliferation and matastasisCitation7. Therefore, APN is a promising target for cancer therapy and APN inhibitors may be of great clinical significance to suppress the invasion, angiogenesis and metastasis of tumour cells.
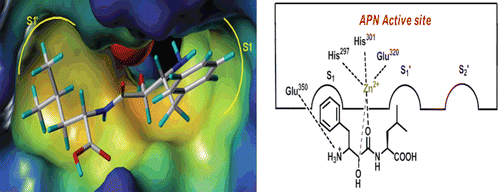
Up to now, a quantity of APN inhibitors such as Bestatin, ProbestinCitation8, AmatatinCitation9, PrebestinCitation8, LapstatinCitation10, AHPA-ValCitation11 have been reported. The first APN inhibitor Bestatin was isolated from a culture filtrate of Streptomyces olivoreticuli in 1976Citation12, and now generally used as the positive control in the assay of small molecule APN inhibitors. The co-crystal complex of Bestatin (AHPA-Leu) and APN from Escherichia coli was reported in 2006, from which we can know that the hydroxyl and carbonyl belong to zinc binding group (ZBG), the phenyl ring in AHPA fragment can insert into the S1 pocket and the amino group in AHPA can interact with Glu 350 ().
Figure 1. The binding character of Bestatin with the active site of APN.
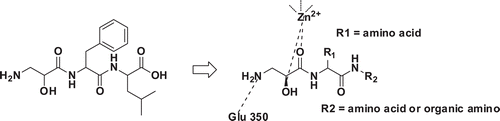
In our previous work, we have designed and synthesized a series of L-isoserine derivatives as APN inhibitors and found that the tripeptide derivative 14b () was the most active compoundCitation13. In order to find better APN inhibitors, we used compound 14b as the leading compound and modified it as follows (): (i) we maintained the L-isoserine scaffold to chelate the zinc ion and the L-phenylalanine group was replaced by different amino acids; (ii) The L-leucine reside was substituted by different amino acids or organic amines in order to closely interact with the hydrophobic pocket of APN.
Figure 2. The strategy for the design of L-isoserine tripeptide derivatives as APN inhibitors.
Material and methods
Chemistry
All the materials were purchased from commercial vendors and used without further purification unless otherwise specified. Solvents were dried over CaCl2 or distilled prior to use and flash chromatography was performed using silica gel (200–300 mesh). All reactions were monitored by thin-layer chromatography on 0.25 mm silica gel plates (60GF-254) and visualize with UV light, or chloride ferric. Proton NMR spectra were determined on a Brucker DRX spectrometer (600 MHz). Measurements were made in DMSO-d6 solutions. ESI-MS were determined on an API 4000 spectrometer. Melting points were conducted on electrothermal melting point apparatus (uncorrected). The final compound was purified by reversed-phase chromatography to give the desired compound.
General procedure for the synthesis of 2a–2e and 4a–4f
The title compounds amino acid methyl ester 2a–2e were prepared from natural amino acids according to methods described in the literaturesCitation14. With the same materials, the title compounds Boc-protected amino acids 4a–4f were prepared.
General procedure for the synthesis of 8a–8n
A 150 mL solution of compound 2a (1.51 g, 8.3 mmol) in dried DCM was gently cooled to 0°C in ice bath. To the solution, TEA (1.72 mL, 24.75 mmol) was added dropwise. After compound 2a was dissolved, HOBt (1.22 g, 9.0 mmol), DMAP (0.18 g, 1.5 mmol), 4a (2 g, 7.5 mmol) and TEA (1.72 mL, 24.75 mmol) were added. The mixture was stirred for 15 min when EDCI (2.87 g, 15 mmol) was added in batches. The reaction mixture was stirred in ice bath for 2 h and then changed to room temperature for 12 h. The solvent was concentrated under vacuum to leave residue. The residue was redissolved with EtOAc (100 mL). The solution was washed with 1 mol/L citric acid solution, saturated NaHCO3, water and saturated sodium chloride solution, dried over MgSO4 and evaporated in vacuo. The residue was purified by flash column chromatography (PE/EtOAc, 3:1(V/V)) to give the desired compound 6a as a white solid (2.6 g), yield: 89.6%, mp. 95–98°C. 1H NMR (DMSO-d6) δ 7.929 (d, J = 7.8 Hz, 1H), 7.292-7.214 (m, 5H), 5.942 (d, 1H), 4.927-4.915 (m, J = 5.5 Hz, 1H), 4.220-4.208 (m, 1H), 3.678 (s, 3H), 3.452-3.438 (m, 1H), 3.212-3.189 (m, 1H), 2.538-3.529 (m, 1H), 1.570-1.552 (m, 2H), 1.428 (s, 9H), 1.121-1.110 (d, 3H), 1.101-0.898 (m, 3H); ESI-MS m/z: [M+H]+ 393.5.
To the dried compound 6a (2.6 g, 6.63 mmol), once in a while, a solution of EtOAc (15 mL) saturated with dry HCl gas was added. The reaction was stirred at room temperature for 4 h before being concentrated in vacuo. The residue was recrystalized with MeOH and ether to give 8a (1.74 g) as a white crystal, yield: 79.96%, 1H NMR (DMSO-d6) δ 8.034 (d, J = 7.8 Hz, 1H), 8.012 (s, 3H), 7.289-7.211 (m, 5H), 4.917-4.905 (m, 1H), 4.210-4.188 (m, 1H), 3.669 (s, 3H), 3.442-3.429 (m, 1H), 3.202-3.172 (m, 1H), 2.542-3.529 (m, 1H), 1.564-1.532 (m, 2H), 1.121-1.110 (d, 3H) 1.101-0.898 (m, 3H); ESI-MS m/z: [M+H]+ 329.2.
General procedure for the synthesis of 9a–9c
Compound 5a (0.62 g, 4.5 mmol) was dissolved in 100 mL of dried DCM. The solution was gently cooled to 0°C in ice bath. Then HOBt (0.73 g, 5.4 mmol), DMAP (0.1 g, 0.9 mmol) and compound 4a (1.2 g, 4.5 mmol) were added. The mixture was stirred for 15 min when EDCI (1.72 g, 9 mmol) was added in batches. The reaction mixture was stirred in 0°C for 2 h and then changed to room temperature for 12 h. The solvent was concentrated under vacuum to leave residue. The residue was redissolved with EtOAc (100 mL). The solution was washed with 1 mol/L citric acid solution, saturated NaHCO3, water and saturated sodium chloride solution, dried over MgSO4 and evaporate in vacuo. The residue was purified by flash column chromatography (PE/EtOAc, 8:1(V/V)) to give the desired compound 7a as a white solid (1.6 g), yield: 92.5%. 1H NMR (DMSO-d6) δ 8.403 (m, J = 7.8 Hz, 1H), 8.032 (dd, 1H), 7.401-7.271 (m, 5H), 7.251(m, 2H), 6.871 (m, 2H), 4.442-4.429 (m, 1H), 4.202-4.172 (m, 1H), 3.843 (s, 3H), 3.212-3.188 (m, 2H), 1.381 (s, 9H); ESI-MS m/z: [M+H]+ 385.2.
To the dried compound 7a (1.2 g, 3.75 mmol) was added once in a while a solution of EtOAc (50 mL) saturated with dry HCl gas. The reaction was stirred at room temperature for 4 h before being concentrated in vacuo. The residue was recrystalized with MeOH and ether to give 9a as a white crystal (1.2 g), yield: 90.2%, 1H NMR (DMSO-d6) δ 8.543 (m, J = 7.8 Hz, 1H), 8.031 (m, 1H), 7.412-7.271 (m, 5H), 7.254 (d, 2H), 6.881 (d, 2H), 4.420-4.329 (m, 2H), 3.950 (m, 1H), 3.843 (s, 3H), 4.402-3.172 (m, 1H), 3.402-3.172 (m, 1H); ESI-MS m/z: [M+H]+ 321.1.
General procedure for the synthesis of Boc-isoserine 11
The starting material L-isoserine is a white solid, (mp. 199–201°C, [a]25D = −32.5 (c 1, H2O)), was purchased from Shanghai Nuotai Chemical Co., Ltd, China. Compound 11 was synthesized following the general procedure as described above (preparation of 4a). A colourless solid, yield: 92%, mp. 85–88°C; [a]25D = +6.7(c 1, MeOH)), 1H NMR (DMSO-d6) δ 7.934 (d, J = 7.8 Hz, 1H), 6.540 (s, 1H), 4.674–4.651 (m, 1H), 3.457 (dd, J = 13.80 Hz, J= 8.4 Hz, 1H), 3.214 (dd, J = 13.80 Hz, J = 8.4 Hz, 1H), 1.435(s, 9H); ESI-MS m/z: [M+H]+ 206.1.
General procedure for the synthesis of 16a–16n
Compound 12a was synthesized from N-protected L-isoserine (11) and L-phenylalanyl-L-isoleucine methyl ester hydrochloride (8a) following the general procedure as described above (preparation of 6a), yield: 68%.
The solution of compound 12a (1.4 g, 2.97 mmol) in MeOH at 0°C was added dropwise a solution of 1 mol/L LiOH (6 mL). The reaction was warmed to room temperature and stirred for 5 h. The solvent was concentrated under vacuum to remove the MeOH. The remaining mixture was added water (5 mL) and acidified to pH 1–2 by a 1 mol/L citric acid solution. The precipitate was collected, washed with water (10 mL) and dried overnight to give the crude product 14a (0.68 g), yield: 50.5%. Compound 16a was obtained through the deprotection of compound 14a in the saturated HCl/EtOAc solution as the preparation of 8a. The crude compound 16a was purified by reversed-phase chromatography to give the desired compound 16a as a white solid, yield: 50%. mp. 200–201°C, 1H NMR (DMSO-d6) δ 12.560 (s, J = 7.8 Hz, 1H), 8.184 (d, J = 7.8 Hz, 1H), 8.117 (s, 2H), 7.776 (d, J = 7.8 Hz, 1H), 7.265-7.176 (m, 5H), 6.518 (s, 1H), 4.468-4.461 (m, 1H), 4.013-3.939 (m, 1H), 4.011-3.929 (m, 1H), 3.047 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.984 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.889-2.872 (m, 1H), 2.765-2.674 (m, 1H), 1.169-1.235 (m, 3H),0.877-0.865 (m, 3H), 0.852-0.802 (d, 3H); ESI-MS m/z: [M+H]+ 366.3.
The compounds 16b–16n were synthesized following the general procedure as described above (preparation of 16a).
General procedure for the synthesis of L-isoserine-L-phenylalanine-L-valine (16b)
A white solid, yield: 53.6%, mp. 190–191°C, 1H NMR (DMSO-d6) δ 12.674 (s, J = 7.8 Hz, 1H), 8.278 (d, J = 7.8 Hz, 1H), 8.119 (s, 2H), 7.756 (d, J = 7.8 Hz, 1H), 7.275-7.266 (m, 5H), 6.498 (s, 1H), 4.458-4.432 (m, 1H), 4.214-3.839 (m, 1H), 4.021-3.897 (m, 1H), 3.111 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.879 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.876-2.858 (m, 1H), 2.775-2.671 (m, 1H), 2.169 (m, 1H), 0.868-0.846 (m, 3H), 0.849-0.832 (d, 3H); ESI-MS m/z: [M+H]+ 352.4.
General procedure for the synthesis of L-isoserine-L-phenylglycine-L-valine (16c)
A white solid, yield: 50.0%, mp. 139–140°C, 1H NMR (DMSO-d6) δ 12.554 (s, J = 7.8 Hz, 1H), 8.372 (d, J = 7.8 Hz, 1H), 8.329 (s, 2H), 8.303 (d, J = 7.8 Hz, 1H), 7.269-7.256 (m, 5H), 6.501 (s, 1H), 4.468-4.432 (m, 1H), 4.209-3.832 (m, 1H), 4.121-3.997 (m, 1H), 3.231 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.979 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.254 (m, 1H), 0.875-0.856 (m, 3H), 0.852-0.8421 (d, 3H); ESI-MS m/z: [M+H]+ 338.4.
General procedure for the synthesis of L-isoserine-L-valine-L-phenylalanine (16d)
A white solid, yield: 52.1%, mp. 210–212°C, 1H NMR (DMSO-d6) δ 12.598 (s, J = 7.8 Hz, 1H), 8.267 (d, J = 7.8 Hz, 1H), 8.219 (s, 2H), 7.743 (d, J = 7.8 Hz, 1H), 7.310 -7.275 (m, 5H), 6.520 (s, 1H), 4.449-4.429 (m, 1H), 4.219-3.939 (m, 1H), 4.111 -3.893 (m, 1H), 3.099 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.902 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.869-2.849 (m, 1H), 2.768-2.688 (m, 1H), 2.321 (m, 1H), 0.897-0.886 (m, 3H), 0.859-0.842 (d, 3H); ESI-MS m/z: [M+H]+ 352.2.
General procedure for the synthesis of L-isoserine-L-tyrosinase-L-valine (16e)
A pink solid, yield: 45.6%, mp. 100–104°C, 1H NMR (DMSO-d6) δ 12.589 (s, J = 7.8 Hz, 1H), 8.310 (d, J = 7.8 Hz, 1H), 8.219 (s, 2H), 8.012 (s, 1H), 7.749 (d, J = 7.8 Hz, 1H), 6.95 (s, 2H), 6.68(s, 2H), 6.500 (s, 1H), 4.449-4.438 (m, 1H), 4.219 -3.798 (m, 1H), 4.051-3.997 (m, 1H), 3.091 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.881 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.881-2.862 (m, 1H), 2.769-2.723 (m, 1H), 2.312 (m, 1H), 0.871-0.856 (m, 3H), 0.839-0.822 (d, 3H); ESI-MS m/z: [M+H]+ 368.2.
General procedure for the synthesis of L-isoserine-L-valine-L-valine (16f)
A white solid, yield: 55.0%, mp. 181–182°C, 1H NMR (DMSO-d6) δ12.600 (s, J = 7.8 Hz, 1H), 8.391 (d, J = 7.8 Hz, 1H), 8.331 (s, 2H), 8.298 (d, J = 7.8 Hz, 1H), 6.499 (s, 1H), 4.471 -4.443 (m, 1H), 4.211-3.732 (m, 1H), 4.119-3.987 (m, 1H), 3.239 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 3.011 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.286 (m, 1H), 2.262 (m, 1H), 0.885-0.867 (m, 3H), 0.880-0.846 (m, 3H), 0.862-0.821(d, 3H), 0.858-0.842(d, 3H); ESI-MS m/z: [M+H]+ 304.4.
General procedure for the synthesis of L-isoserine-L-phenylglycine-L-phenylalanine (16g)
A white solid, yield: 48.1%, mp. 196–198°C, 1H NMR (DMSO-d6) δ 12.598 (s, J = 7.8 Hz, 1H), 8.267 (d, J = 7.8 Hz, 1H), 8.219 (s, 2H), 7.743 (d, J = 7.8 Hz, 1H), 7.310 -7.275 (m, 5H), 7.026-7.014 (m, 5H), 6.520 (s, 1H), 4.449-4.429 (m, 1H), 4.219-3.939 (m, 1H), 4.111 -3.893 (m, 1H), 3.099 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.902 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.869-2.849 (m, 1H), 2.768-2.688 (m, 1H); ESI-MS m/z: [M+H]+ 386.2.
General procedure for the synthesis of L-isoserine-L-valine- L-leucyl (16h)
A white solid, yield: 55.0%, mp. 182–183°C, 1H NMR (DMSO-d6) δ 12.588 (s, J = 7.8 Hz, 1H), 8.389 (d, J = 7.8 Hz, 1H), 8.312 (s, 2H), 8.310 (d, J = 7.8 Hz, 1H), 6.510 (s, 1H), 4.469 -4.439 (m, 1H), 4.199 -3.832 (m, 1H), 4.219-4.001 (m, 1H), 3.301 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 3.021 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.290 (m, 1H), 2.259 (m, 1H), 1.499 -1.435 (m, 3H), 0.895-0.877 (m, 3H), 0.889-0.856 (m, 3H), 0.872-0.831(d, 3H), 0.862-0.851 (d, 3H); ESI-MS m/z: [M+H]+ 318.4.
General procedure for the synthesis of L-isoserine-L-leucyl -L-valine (16i)
A white solid, yield: 53.0%, mp. 181–182°C, 1H NMR (DMSO-d6) δ 12.579 (s, J = 7.8 Hz, 1H), 8.401 (d, J = 7.8 Hz, 1H), 8.308 (s, 2H), 8.309 (d, J = 7.8 Hz, 1H), 6.499 (s, 1H), 4.471 -4.439 (m, 1H), 4.211 -3.932 (m, 1H), 4.221-4.101 (m, 1H), 3.321 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 3.121 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.311 (m, 1H), 2.246 (m, 1H), 1.502 -1.445 (m, 3H), 0.898-0.887 (m, 3H), 0.891-0.876 (m, 3H), 0.868-0.842 (d, 3H), 0.871-0.841 (d, 3H); ESI-MS m/z: [M+H]+ 318.4.
General procedure for the synthesis of L-isoserine-L-chloramphenicol-L-leucyl (16j)
A white solid, yield: 53.0%, mp. 180°C, 1H NMR (DMSO-d6) δ 12.579 (s, J = 7.8 Hz, 1H), 8.168-8.153 (d, J = 7.8 Hz, 2H), 7.868-7.671 (d, 2H), 5.254-5.249 (s, 1H), 4.485-4.480 (s, 1H), 4.053 -4.036 (m, 1H), 3.978-3.960 (m, 1H), 2.908-2.881 (m, 1H), 2.760-2.501 (m, 1H), 1.507-1.496 (d, 2H), 1.429-1.400 (m, 1H), 0.823-0.797 (m, 6H); ESI-MS m/z: [M+H]+ 442.2.
General procedure for the synthesis of L-isoserine-L-phenylglycine-L-leucyl (16k)
A white solid, yield: 49.8%, mp. 205–206°C, 1H NMR (DMSO-d6) δ 12.549 (s, J = 7.8 Hz, 1H), 8.391 (d, J = 7.8 Hz, 1H), 8.298 (s, 2H), 8.313 (d, J = 7.8 Hz, 1H), 7.278-7.250 (m, 5H), 6.511 (s, 1H), 4.471-4.442 (m, 1H), 4.213-3.932 (m, 1H), 4.221-3.997 (m, 1H), 3.242 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 3.012(dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 1.522-1.442 (m, 3H), 0.881-0.876 (m, 3H), 0.852-0.841(d, 3H); ESI-MS m/z: [M+H]+ 352.4.
General procedure for the synthesis of L-isoserine-L-tyrosinase-L-leucyl (16l)
A pink solid, yield: 40.6%, mp. 104–106°C, 1H NMR (DMSO-d6) δ 12.599 (s, J = 7.8 Hz, 1H), 8.289 (d, J = 7.8 Hz, 1H), 8.223 (s, 2H), 8.011 (s, 1H), 7.759 (d, J = 7.8 Hz, 1H), 6.951 (s, 2H), 6.682(s, 2H), 6.495 (s, 1H), 4.455-4.439 (m, 1H), 4.229 -3.804 (m, 1H), 4.151-3.989 (m, 1H), 3.191 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.981 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.981-2.872 (m, 1H), 2.779-2.743 (m, 1H), 1.502-1.446 (m, 3H), 0.869-0.8660 (m, 3H), 0.840-0.812 (d, 3H); ESI-MS m/z: [M+H]+ 382.5.
General procedure for the synthesis of L-isoserine-L-eucyl-L-tyrosinase (16m)
A pink solid, yield: 42.6%, mp. 129–131°C, 1H NMR (DMSO-d6) δ 12.612 (s, J = 7.8 Hz, 1H), 8.314 (d, J = 7.8 Hz, 1H), 8.123 (s, 2H), 8.023 (s, 1H), 7.859 (d, J = 7.8 Hz, 1H), 7.002 (s, 2H), 6.692(s, 2H), 6.512 (s, 1H), 4.465-4.449 (m, 1H), 4.234 -3.814 (m, 1H), 4.161-3.978 (m, 1H), 3.291 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 3.002(dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.981-2.792 (m, 1H), 2.783-2.763 (m, 1H), 1.522-1.486 (m, 3H), 0.872-0.856 (m, 3H), 0.845-0.822 (d, 3H); ESI-MS m/z: [M+H]+ 382.5.
General procedure for the synthesis of L-isoserine-L-phenylalanine-L-phenylalanine (16n)
A white solid, yield: 45.1%, mp. 184–185°C, 1H NMR (DMSO-d6) δ12.588 (s, J = 7.8 Hz, 1H), 8.277 (d, J = 7.8 Hz, 1H), 8.223 (s, 2H), 7.750 (d, J = 7.8 Hz, 1H), 7.311 -7.285 (m, 5H), 7.036-7.024 (m, 5H), 6.519 (s, 1H), 4.454-4.432 (m, 1H), 4.221-3.949 (m, 1H), 4.121 -3.993 (m, 1H), 3.199 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.913 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.879-2.855 (m, 1H), 2.778-2.698 (m, 1H), 2.832-1.794 (m, 1H), 2.509-2.434 (m, 2H); ESI-MS m/z: [M+H]+ 410.2.
General procedure for the synthesis of 15a–15c
Compound 15a was synthesized firstly through the condensation reaction of N-protected L-isoserine(11) and L-phenylalanyl-4-methoxybenzylamine following the general procedure as described above (preparation of 6a), and then deprotecting the Boc group in the saturated HCl/EtOAc solution as the preparation of 8a, yield: 76.1%. mp. 180–182°C, 1H NMR (DMSO-d6) δ 8.597 (d, J = 7.8 Hz, 1H), 7.929-7.917 (s, 2H), 7.907 (d, J = 7.8 Hz, 1H), 7.246-7.111 (m, 5H), 6.875-6.863 (s, 2H), 6.520 (s, 1H), 6.483-6.476 (s, 2H), 4.462 (m, 2H), 4.468-4.461 (m, 1H), 4.013-3.939 (m, 1H), 4.245-4.170 (m, 2H), 3.733 (s, 3H), 3.047 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.984 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.812-2.798 (m, 1H), 2.510-2.439 (m, 1H); ESI-MS m/z: [M+H]+ 372.3.
General procedure for the synthesis of L-isoserine-L-phenylglycine-methoxybenzylamine (15b)
A white solid, yield: 56.1%, mp. 148–150°C, 1H NMR (DMSO-d6) δ8.597 (d, J = 7.8 Hz, 1H), 7.929-7.917 (s, 2H), 7.907 (d, J = 7.8 Hz, 1H), 7.246-7.111 (m, 5H), 6.875-6.863 (s, 2H), 6.520 (s, 1H), 6.483-6.476 (s, 2H), 4.462 (m, 2H), 4.468-4.461 (m, 1H), 4.013-3.939 (m, 1H), 4.245-4.170 (m, 2H), 3.733 (s, 3H), 3.047 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.984 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H); ESI-MS m/z: [M+H]+ 368.3.
General procedure for the synthesis of L-isoserine-L-phenylglycine-2-furfurylamine (15c)
A white solid, yield: 48.1%, mp. 150–152°C, 1H NMR (DMSO-d6) δ 8.892 (d, J = 7.8 Hz, 1H), 7.929-7.917 (s, 2H), 7.907 (d, J = 7.8 Hz, 1H), 7.65 (d, 1H), 7.246-7.111 (m, 5H), 6.46 (d, 1H), 6.26 (d, 1H), 6.875-6.863 (s, 2H), 6.520 (s, 1H), 4.462 (m, 2H), 4.468-4.461 (m, 1H), 4.013-3.939 (m, 1H), 3.047 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.984 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.812-2.798 (m, 1H), 2.510-2.439 (m, 1H); ESI-MS m/z: [M+H]+ 332.4.
In vitro APN inhibition assay
IC50 values against APN were determined as previously described and by using L-Leu-p-nitroanilide as a substrate and microsomal aminopeptidase from Porcine Kidney microsomes (Sigma) in 50 mM PBS (pH 7.2) or suspension of A549, ES-2, HL-60 in PBS (2 × 105/well) as the enzyme. The hydrolysis of the substrate was monitored by following the change in the absorbance measured at 405 nm with a plate reader (Varioskan, Thermo, USA). All the solutions of the inhibitors were prepared in the assay buffer, and the pH was adjusted to 7.5 by the addition of 0.1 M HCl or 0.1 M NaOH. All the inhibitors were preincubated with APN at 37°C. The assay mixture, which contained the inhibitor solution (concentration dependent on the inhibitor), the enzyme solution (4 mg/mL final concentration) or the cells suspension, and the assay buffer, was adjusted to 200 µL.
Antiproliferative activity assay
A549 cells, ES-2 cells (high APN expression) and MDA-MB-231 and K562 (low APN expression) were cultured in RPMI-1640 medium containing 10% FBS in situ at 37°C in 5% CO2 humidified incubator. Cell proliferation was determined by MTT assay. In brief, cells were seeded in a 96-well plate (5 × 103/well for adherent cells and 1 × 104/well for suspension cells) and cultured for 4 h, followed by the addition of different concentrate of inhibitors in medium. After another 44 h, 0.5% MTT (5 mg/mL) solution was added to form the formazan product. At last, DMSO was used to solve the formazan and concentrations were monitored by detecting OD values at 570 nm through a plate reader (Varioskan, Thermo, USA).
Results and discussion
Chemistry
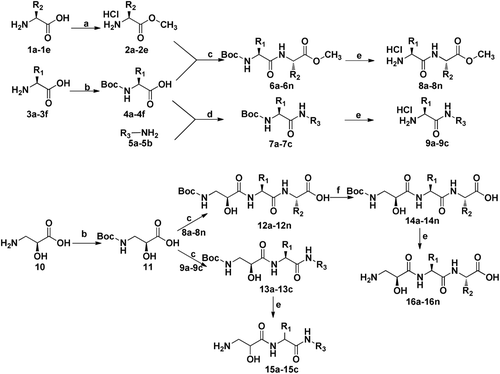
The target compounds were synthesized efficiently following the procedures as shown in . Amino acids 1a–1e were used as the starting materials to yield their methyl esters 2a–2e, while amino acids 3a–3f were protected by (Boc)2O to obtain 4a–4g. Organic amides 5a–5b were used without any protection. N-Boc-protected amino acids 4a–4f were coupled with intermediates 2a–2e and 5a–5b by classical EDCI/HOBt method, and then converted into 8a–8o and 9a–9c by deprotecting the Boc group. Using the same procedure, Boc-protected L-isoserine 11 was coupled with intermediate 8a–8n and 9a–9c to yield tripeptide 12a–12n and 13a–13c containing protecting group. Finally, target compounds 16a–16n were obtained by first reacting with based to deprotect methy group and then deprotecting Boc group while compounds 15a–15c were get merely cleaving Boc group.
Scheme 1. Reagents and conditions:(a) MeOH, concentrated HCl; (b) 1 M NaOH, MeOH, (Boc)2O; (c) HOBT/EDCI/DMAP/Et3N, anhydrous CH2Cl2;(d) HOBT/EDCI/DMAP, anhydrous CH2Cl2; (e) concentrated HCl/MeOH; (f) MeOH, 1 M LiOH.
In vitro APN inhibition assay
All the target compounds were evaluated for their potential inhibitory activities against APN and the results were listed in and . According to the data listed in , inhibitory effect could be detected among all the compounds except compound 16j. Compound 16l (IC50 = 2.51 ± 0.2 µM) is the most potent inhibitor in all target compounds, which displayed inhibitory activity a little better than that of Bestatin(IC50 = 6.25 ± 0.4 µM), suggesting that the tyrosine group may have something to do with hydrogen bond which would be helpful to increase the interaction with APN. On the other hand, the introduction of phenol group in the corresponding R2 site did not improved the activity appreciably as compound 16m did not show outstanding inhibitory result (IC50 = 17.5 ± 2.1 µM). It is possibly indicated that no hydrogen bond interaction is essential in this section. In addition, the phenyl group in R1 position would be much better than benzyl group to increase the activity as the phenylalanine derivatives (16a, 16b, 16n) show generally less inhibitory activity than the phenylqlycine analogues (16c, 16g, 16k). Series 2 compounds were designed and synthesized in order to determine the different influence of natural amino acids with arylamine. From the result of the enzymatic assay, we could see these compounds performed similar inhibitory activities with series 1 compounds. For instance, compounds 15a (IC50 = 38 ± 3.6 µM) and 15b (IC50 = 24.4 ± 2.8 µM) show similar activity with compound 16a (IC50 = 30.4 ± 2.1 µM) and 16b (IC50 = 94 ± 4.6 µM). In our case, despite the inhibitory activities of series 2 compounds maintained, the water-solubility reduced obviously.
Table 1. The structures and inhibitory activities of series 1 compounds and Bestatin against APN.
Table 2. The structures and inhibitory activity of the series 2 compounds against APN.
In addition, the enzymatic inhibitory activity of all the compounds against APN was also determined with A549 (Human lung adenocarcinoma epithelial cell line) cells high-expressing APN. Results were shown in and . From the results, we could learn that the inhibitory activity of these compounds on the cellar level was consistent with the assays on the enzymatic level in general. However, the previous proved compound 16l (IC50 = 2.51 ± 0.2 µM) which was expected to perform the strongest effect against APN showed much weak activity against A549 cells while compound 16b (IC50 = 94 ± 4.6 µM) and 16d (30.2 ± 1.9 µM) showed better inhibitory activity on A549 cells. It was possibly due to the different binding characteristics of APNs among species. Moreover, compounds 16e, 16g, 16h, 16i, 16j, 16k, 16l and Bestatin were evaluated for their activity against ES-2 (Ovarian Clear Cell Adenocarcinoma Cell Line) cells and the inhibitory activity of compounds 16h, 16i, 16l, and Bestatin was also preformed on HL-60 (human promyelocytic leukemia cell line) cells. The results are shown in , from which we could tell that none of these compounds exhibited better activity than the control Bestatin and compound 16j, which performed no activity in the enzymatic level, showed the weakest effect on ES-2 cells still.
Antiproliferative activity assay
On the other hand, all the compounds were detected for their potential effects on proliferation on four tumour cell lines (A549 cells, MDA-MB-231 cells, HL-60 cells and K562 cells) with Bestatin as the control via MTT assay. The results are shown in . It can be seen that some of the compounds showed similar even better anti-proliferation effect than Bestatin on different cell lines. However, compound 16l does not exhibit outstanding anti-proliferation activity like the enzymatic assay. Besides, compound 16e, 16g, 16h, 16i, 16k and 16l which showed good activity in the APN inhibitory assay almost did not perform prominent anti-proliferation activity on the high-expressing APN cells (A549 cells and HL-60 cells) than the low-expressing APN cells (MDA-MB-231 cells and K562 cells), possibly implying a different mechanism from the interaction with APN in the respect of anti- proliferation activity of these compounds.
Table 3. Antiproliferative activities of the L-isoserine derivatives.
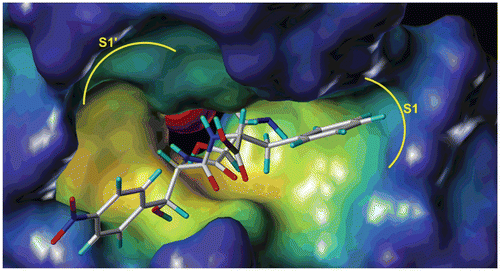
In order to investigate the interaction mode of L-isoserine derivatives with APN, the most active and inactive compound 16l and 16j was sketched and docked into the active site of APN (PDB code: 2DQM) using Surflex-Dock module of Sybyl 8.1. The result suggested that the binding mode of compound 16l was similar with Bestatin and obvious gave the main reason for the distinct activity of both compounds. The zinc ion of APN was coordinating with the L-isoserine part of both compounds ( and ). In addition, the phenyl moiety of phenylalanine residue and tyrosine residue could plunge into the S1 pocket of APN ( and ). Besides, the leucine part of compound 16l could deeply insert into the S1′pocket of APN (, while the chloramphenicol amine residue of compound 16j was out of the S1′ pocket (). Furthermore, compound 16l also could form hydrogen bonds with Arg293, His297, and Try381 at the distance of 9.70, 2.95, and 4.17Å which significantly contributed to improve the binding afïnities (.
Figure 3. The docking result of compound 16l with APN showed by LIGPLOT. (a) The docking result of 16l with APN (16l is showed in capped sticks mode). (b) The docking result of compound 16l is shown by LIGPLOT.
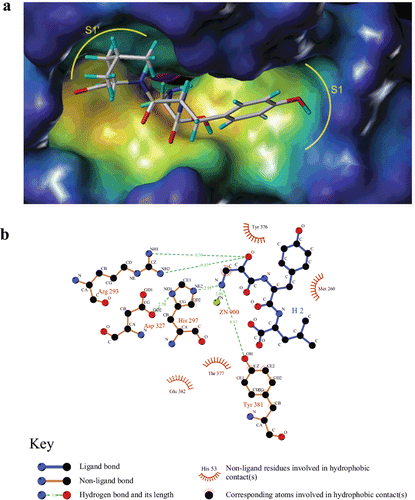
Figure 4. The docking result of 16j with APN (16j is showed in capped sticks mode).
Conclusion
In summary, we designed and synthesized two series of novel L-isoserine tripeptide derivatives as APN inhibitors. The preliminary results showed that compounds 16h, 16i, 16g, 16l and 16n were the most potent compounds and had similar inhibitory activity with Bestatin. The most effective compound 16l (2.51 ± 0.2 µM), exhibited a little better enzymatic inhibitory activity against APN than control compound Bestatin (6.25 ± 0.4 µM).The SAR study of compound 16l Clearly explain the excellent inhibitory activity. Compound 16l could be used as a lead compound for further development of small molecular peptidomimetic APN inhibitors as new anticancer agents.
Declaration of interest
This work was supported by National High Technology Research and Development Program of China (863 project; Grant No. 2007AA02Z314), National Natural Foundation Research Grant (Grant No. 90713041) and The National Natural Science Foundation of China (Grant No. 21172134).
Related Research Data
Reference
- Fukasawa K, Fujii H, Saitoh Y, Koizumi K, Aozuka Y, Sekine K et al. Aminopeptidase N (APN/CD13) is selectively expressed in vascular endothelial cells and plays multiple roles in angiogenesis. Cancer Lett 2006;243:135–143.
- Su L, Fang H, Yang K, Xu Y, Xu W. Design, synthesis and biological evaluation of novel L-lysine ureido derivatives as aminopeptidase N inhibitors. Bioorg Med Chem 2011;19:900–906.
- Dixon J, Kaklamanis L, Turley H, Hickson ID, Leek RD, Harris AL et al. Expression of aminopeptidase-n (CD 13) in normal tissues and malignant neoplasms of epithelial and lymphoid origin. J Clin Pathol 1994;47:43–47.
- Piela-Smith TH, Korn JH. Aminopeptidase N: a constitutive cell-surface protein on human dermal fibroblasts. Cell Immunol 1995;162:42–48.
- Raynaud F, Bauvois B, Gerbaud P, Evain-Brion D. Characterization of specific proteases associated with the surface of human skin fibroblasts, and their modulation in pathology. J Cell Physiol 1992;151:378–385.
- Zhang X, Xu W. Aminopeptidase N (APN/CD13) as a target for anti-cancer agent design. Curr Med Chem 2008;15:2850–2865.
- Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res 2000;60:722–727.
- Aoyagi T, Yoshida S, Nakamura Y, Shigihara Y, Hamada M, Takeuchi T. Probestin, a new inhibitor of aminopeptidase M, produced by Streptomyces azureus MH663-2F6. I. Taxonomy, production, isolation, physico-chemical properties and biological activities. J Antibiot 1990;43:143–148.
- Rich DH, Moon BJ, Harbeson S. Inhibition of aminopeptidases by amastatin and bestatin derivatives. Effect of inhibitor structure on slow-binding processes. J Med Chem 1984;27:417–422.
- Repic Lampret B, Kidric J, Kralj B, Vitale L, Pokorny M, Renko M. Lapstatin, a new aminopeptidase inhibitor produced by Streptomyces rimosus, inhibits autogenous aminopeptidases. Arch Microbiol 1999;171:397–404.
- Chung MC, Lee HJ, Chun HK, Lee CH, Kim SI, Kho YH. Bestatin analogue from Streptomyces neyagawaensis SL-387. Biosci Biotechnol Biochem 1996;60:898–900.
- Umezawa H, Aoyagi T, Suda H, Hamada M, Takeuchi T. Bestatin, an inhibitor of aminopeptidase B, produced by actinomycetes. J Antibiot 1976;29:97–99.
- Yang K, Fen J, Fang H, Zhang L, Gong J, Xu W. Synthesis of a novel series of L-isoserine derivatives as aminopeptidase N inhibitors. J Enzyme Inhib Med Chem 2012;27:302–310.
- Sunggak K, Jae IL, Youn CK. A simple and mild esterification method for carboxylic acids using mixed carboxylic-carbonic anhydrides. J Org Chem 1985;50:560–565.