Abstract
Not only are β-amyloid peptides and senile plaque deposits characteristics in Alzheimer’s disease but there is growing evidence to suggest that oxidative stress also plays a role with a decrease in levels of brain superoxide dismutase (SOD), an enzyme that catalyses the dismutation of superoxide radicals into molecular oxygen and hydrogen peroxide. We show through kinetic and fluorescence analysis that β-amyloid peptides, in the glycine zipper region [Aβ29–33 and Aβ25–37] of Aβ1–40 interact with, and inhibit, SOD directly. The enzyme was purified 15.7-fold from bovine brain by DEAE-Sepharose ion exchange chromatography in a yield of 68.8% and specific activity of 3.66 U.mg−1. The subunit structure of the enzyme was monomeric with a molecular mass of 13 kDa, as estimated by SDS-PAGE. Inhibitor constants (Ki) and dissociation constants (Kd) were calculated as 14.44, 13.16 and 11.72 µM and 9.38, 15.7 and 12.13 for Aβ25–37, Aβ29–33 and Aβ1–40, respectively; the number of binding sites on the enzyme for the peptides was 1.
Introduction
Alzheimer’s disease (AD) is a progressive brain disease that slowly destroys the cognitive function like thinking, memory and reasoning. It is the most common source of dementia which was once considered a rare disorder but now due to the growing number of people with AD, attention has been drawn to finding not only its cause but its treatmentCitation1. AD brains are characterised by intracellular neurofibrillary tangles containing hyper-phosphorylated Tau proteins, senile plaques and cerebrovascular amyloid depositsCitation2.
The β-amyloid peptide fragments [Aβ1–40/42], which have proven to be toxic to neuronal cellsCitation3 are released into the interneuronal space from an amyloid precursor protein (APP) by a series of α-, β- and γ-secretase enzymesCitation4,Citation5. Consequently, these Aβ-peptides aggregate together as oligomers and have been shown to react with neighbouring neurons as well as the synapseCitation2. In essence the oligomers that are not cleared from the brain clump together eventually into fibrils that are insoluble and these further combine to form plaques characteristic of ADCitation6. A prominent feature of Aβ1–40/42 is an amino acid motif GxxxGxxxGxxxG known as the glycine zipper that encompasses residues 25–37 () and which has been attributed to the pathogenicity of the amyloid peptidesCitation7.
Figure 1. Amino acid sequence of Aβ-peptide (1–42). The highlighted region shows the glycine zipper.

Though the deposition of aggregated β-amyloid (Aβ) senile plaques in the human brain is a classic observation in the neuropathology of Alzheimer’s diseaseCitation8–10 an understanding of the mechanism of their formation, however, remains elusive. The astrocytes in the diseased brain not only function to store arginine but are surrounded by the insoluble amyloid plaquesCitation11 and in earlier work from our laboratories it was shown that arginine-metabolising enzymes, in particular neuronal nitricoxide synthase (nNOSCitation12,Citation13) and peptidyl arginine deiminase (PADCitation14) are not only inhibited by β-amyloid peptides but are instrumental as catalysts toward fibrillogenesis. The pathogenesis of Alzheimer’s disease, however, can also be manifested by considering both oxidative and nitrosative stressCitation15. Whether these levels of stress precede, contribute directly or are as a result of AD pathogenesis is not fully understoodCitation15. Both of these enzymes, nNOS and PAD, produce nitric oxide (NO) which apart from being an important signaling molecule in the brain, can be regarded as a ‘janus’ molecule in cell death or cell survival. NO is responsible for the increase of oxidative/nitrosative stress by in vivo formation of peroxynitrite as well as in the modulation of different intracellular pathways associated with Alzheimer’s disease. Large amounts of NO are neurotoxic whereas smaller concentrations are neuroprotectiveCitation16.
The brain has a relatively poor defence against the overproduction and damage by oxygen free radicalsCitation17 and makes use of antioxidants such as superoxide dismutase (SOD), glutathione peroxidase and catalase with the first mentioned purported to be the first line of defence against reactive oxygen species (ROSCitation18). Copper Zinc SOD (Cu-Zn SOD) [E.C. 1.15.1.1] catalyzes the dismutation of superoxide to molecular oxygen and hydrogen peroxideCitation19, is homodimericCitation20 and is the main type found in the brain.
Notwithstanding the importance of oxidative/nitrosative stress within the context of the pathogenicity of Alzheimer’s disease the interaction of Aβ peptides with SOD has not yet been fully investigated and consequently it follows that a therapeutic tool in the etiology and pathogenesis of Alzheimer’s disease may be to investigate SOD and its intimate association with amyloid peptides.
We now report that, not only does Aβ1–40 interact with the enzyme but the three consecutive glycine zipper motifs between residues (25–37) and the single zipper Aβ29–33 does so as well, either individually or in tandem. A BLAST search of the comparison of the sequences between human and bovine SOD have shown a 90% homologyCitation19 and consequently we have purified SOD from bovine brain and used it as the model enzyme.
Methods
Materials
Bovine brain, donated by Rosedale Abattoir (Grahamstown, South Africa), was obtained fresh from animals within 15 min of slaughter and was stored on ice until required. Since the animals had been slaughtered for regular domestic consumption there was no experimental transgression of any moral or ethical animal rights as stipulated by the South African authorities. SOD assay kit was obtained from Sigma Aldrich (South Africa and was used according to their commercial bulletin [Sigma Information Bulletin, 19160]. All other reagents were obtained from Merck (South Africa) and dissolved in milli-Q water. Ultra-violet and fluorometric analyses were carried out on PowerWave microplate spectrophotometer and spectrofluorometer (Bio-Tek Instruments) with 96 well plates, operated at 1 nm bandwidth using the KC Junior software program.
SOD assay
SOD activity of the purified samples was determined by the SOD assay kit using (2-(4-iodophenyl)-3-(4nitrophenyl)-5-(2,4-disulphenyl)-2H-tetrazolium, monosodium salt) [WST] which is a colorimetric assay that monitors the rate of inhibition of WST to a water soluble formazan dye. Purified SOD from bovine brain (20 µl) was incubated (37°C, 20 min) with WST working solution (200 µl) and distilled water (20 µl) according to the Sigma Information Bulletin, 19160 after which SOD activity was determined from the absorbance at 450 nm. One unit (U) of enzyme was defined as that amount of enzyme that will inhibit the rate of WST reduction by 50%.
Protein concentration
The protein concentration for all experiments was routinely determined according to the method of BradfordCitation21. The assay was performed in triplicate in a 96-well microplate. Enzyme extract (5 μl) was incubated (22°C, 10 min) with Bradford reagent (245 μl), the absorbance measured at 595 nm and the concentration determined from a bovine serum albumen standard curve.
Purification of SOD
Fresh bovine brain (374 g) was homogenized by sonication (10 W, 30 sec intervals, 4 min) in Hepes buffer (50 mM, pH 7.6, 600 ml) that contained ethylenediaminetetraacetic acid (1.0 mM), NADPH (1.0 mM), dithiothreitol (0.5 mM) and phenylmethylsulphonylfluoride (0.43 mM). The cell debris was removed by centrifugation (10,000g, 4°C, 30 min) and the crude cell-free extract assayed for SOD activity and protein concentration then stored as 20 ml aliquots at −70°C until required.
Solid ammonium sulphate was slowly added to a stirred solution of crude cell-free extract (20 ml) to give 55% saturation. The solution was allowed to stir (60 min) then centrifuged (10,000g, 15 min, 4°C). The precipitate was resuspended in potassium phosphate buffer (67 mM, pH 7.8) and solid ammonium sulphate added until 70% saturation of ammonium sulphate was reached. The solution was allowed to stir (60 min), centrifuged (10,000g, 15 min, 4°C), the precipitate resuspended in potassium phosphate buffer (67 mM, pH 7.8) followed by dialysis against distilled water. SOD activity and protein content was determined as described above.
A diethylaminoethyl (DEAE)-Sepharose column (1.5 × 30 cm) was equilibrated with potassium phosphate buffer (67 mM, pH 7.8). Enzyme solution (10.0 ml), obtained from the ammonium sulphate precipitation, was applied to the column and the column washed with the same buffer until A280 was at the baseline. The adsorbed proteins were then eluted with NaCl (0–1 M) in the same buffer at a flow rate of 0.5 ml.min−1 and fractions (3.0 ml) collected, assayed for SOD activity and protein content and active fractions pooled and dialysed against distilled water.
SDS-PAGE of SOD
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to check the purity of fractions and determine the molecular mass. Samples from each purification step (20 µl) and a standard molecular weight marker, were electrophoresed at 200 V on a discontinuous gel with 13% resolving gel and 8% stacking gel. After electrophoresis the gel was stained with Coomassie brilliant blue R-250 then destained in methanol:acetic acid:water (1:1:8 v/v/v).
Interaction of β-amyloid peptides with SOD
Kinetic analysis
Purified SOD from bovine brain (20 µl) was incubated (37°C, 60 min) with Aβ25–37, Aβ29–33 or Aβ1–40 (20 µl, 5 µM) followed by incubation (37°, 20 min) with WST working solution (200 µl) according to the Sigma Information Bulletin, 19160 after which SOD activity was determined from the absorbance at 450 nm.
Fluorimetric analysis
Spectrofluorimetry was used to analyse the interaction of amyloid peptides [Aβ25–37, Aβ29–33 and Aβ1–40] with SOD. The excitation wavelength was fixed at 295 nm, the wavelength at which tryptophan absorbs, and the emission wavelength was at 482 nm. The change in fluorescence of the solution was monitored as increasing concentrations of amyloid peptides (0–40 µM) were added to a reaction mixture of SOD (5.0 µl) in Tris-HCl buffer (pH 8.0, 50 mM) in a final volume of 300 µl.
Statistical analyses
All analyses were carried out in triplicate and values reported as the means with standard deviation p < 0.05 versus controls. Where necessary analysis of variance was conducted using Statistica for Windows, version 8 (Statsoft Inc.) and Microsoft Excel 2007.
Results and discussion
Purification of SOD from bovine brain
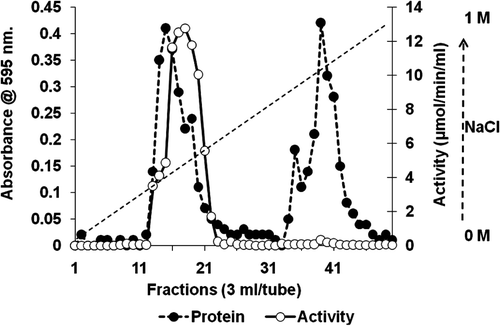
Bovine brain was subjected to 70% ammonium sulphate precipitation and this was followed by anion exchange chromatography using DEAE-Sepharose. SOD was eluted from the matrix by a linear gradient of NaCl (0–1 M). The active enzyme was eluted in fractions 14–22 () in a yield of 68.76%, with a purification fold of 15.71 and a specific activity of 3.66 µmol.min−1.mg−1 ().
Table 1. Purification of superoxide dismutase from bovine brain.
Figure 2. Chromatography profile for purification of SOD on DEAE- Sepharose (1.5 × 30 cm) ion exchange. SOD solution (10 ml) in potassium phosphate buffer (67 mM, pH 7.8) was applied to the column and adsorbed proteins eluted with 0–1 M NaCl gradient at a flow rate of 0.5 ml.min−1. Fractions (3.0 ml) were collected, assayed for protein and SOD activity and active fractions pooled and dialysed against distilled water.
SDS-PAGE of SOD
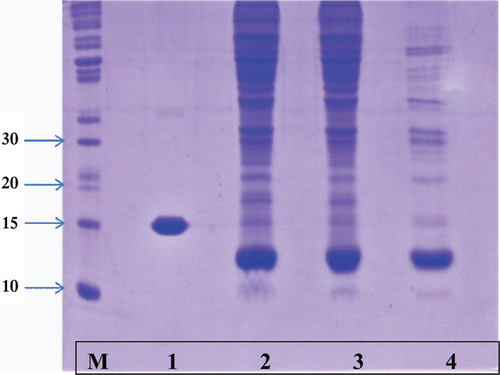
Molecular weight of purified SOD as determined by SDS-PAGE () showed a single band of 13 kDa while a band of 16 kDa represented a standard commercial SOD sample that had been isolated from liver. Cu-Zn SOD from bovine brain has been reported to be a homodimer of 13 kDaCitation20 while that from liver and/or blood erythrocytes to be in the region of 16.5 kDa. It should be mentioned that the presence of the other enzyme isoform (Mn SOD) may be a possibility. Literature reportsCitation22, however, that Mn SOD was a homotetramer with subunit molecular mass of 22 kDa. There was no evidence of any band at 22 kDa from the fractions after ion-exchange chromatography (, lane 4) as only the Cu/Zn SOD isoform (13–16 kDa) was seen.
Figure 3. SDS Polyacrylamide gel electrophoresis of purified SOD fractions. Samples from each purification step (20 µl) and a standard molecular weight marker, were electrophoresed at 200 V on a discontinuous gel with 13% resolving gel and 8% stacking gel. After electrophoresis the gel was stained with Coomassie brilliant blue R-250 then destained in methanol:acetic acid:water (1:1:8 v/v/v). M: molecular weight marker, 1: commercial SOD, 2: Crude homogenate, 3: ammonium sulphate fraction and 4: DEAE-Sepharose ion exchange.
Interaction of β-amyloid peptides with SOD
Kinetic analysis
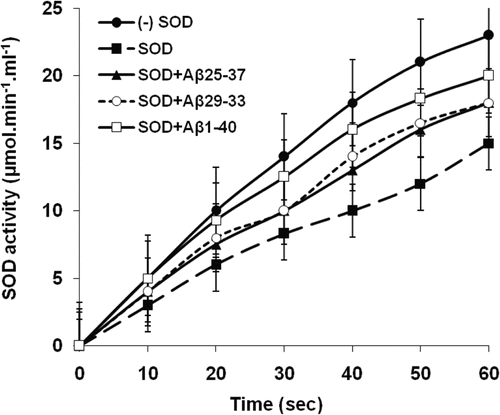
The effect of β-amyloid peptides (Aβ) on SOD was tested after incubation of the purified SOD with Aβ25–37, Aβ29–33 and Aβ1−40 (). The results are expressed as the rate of WST reduction in units of µmol.min−1.ml−1 with samples with no SOD as 23 µmol.min−1.ml−1 representing 100% reduction. The results indicated that without any Aβ-peptide [only SOD present] the reduction of WST was 35% which represented a SOD activity of 15 µmol.min−1.ml−1. In the presence of Aβ25–37, Aβ29–33 and Aβ1−40 the reduction of WST changed to 70%, 73% and 78%, respectively which interpreted as 35%, 38% and 43% inhibition of SOD by Aβ25–37, Aβ29–33 and Aβ1−40 (SOD activity of 9.8 µmol.min−1.ml−1; 9.3 µmol.min−1.ml−1 and 8.6 µmol.min−1.ml−1), respectively (). An increase in the percentage inhibition implied that the Aβ-peptides had interacted with the SOD such that an increase in WST reduction was noted. The inhibitor constant (Ki) calculated from Equation 1, indicated the binding affinity of enzyme for inhibitor ().
Table 2. Binding parameters for the interaction of Aβ-peptides with SOD determined by kinetic and fluorimetric analysis. All analyses were carried out in triplicate and values reported as the means with standard deviation p < 0.05 versus controls.
Figure 4. Kinetic analysis for the interaction of Aβ-peptides with purified SOD. Purified SOD from bovine brain (20 µl) was incubated (37°C, 60 min) with Aβ25–37, Aβ29–33 or Aβ1–40 (20 µl, 5 µM) followed by incubation (37°, 20 min) with WST working solution (200 µl) according to the Sigma Information Bulletin, 19160 after which SOD activity was determined from the absorbance at 450 nm. All analyses were carried out in triplicate and values reported as the means with standard deviation p < 0.05 versus controls.
where Aβ = concentration of amyloid peptide; Vm and Vmapp = maximum enzyme velocity in the absence and presence of respective amyloid peptides. Aβ1−40 inhibited SOD slightly greater (Ki = 11.72 µM) compared to Aβ25–37 and Aβ29–33 with Ki values of 14.44 and 13.16 μM, respectively (). There was no significant difference between Ki values of the three amyloid peptides which, in our opinion, reflected that the regions of Aβ1−40 represented as the glycine zippers (Aβ25–37 and Aβ29–33) were responsible for the actual inhibition of SOD. In view of the fact that the pentapeptide ‘glycine zipper’ Aβ29–33 had a slightly lower Ki value than the multiple zipper region within Aβ25–37 it was tempting to speculate that this individual pentapeptide was crucial for the inhibition of SOD by the amyloid peptide. In an earlier study it was reported that Aβ peptides produced in AD caused a modification in SOD that led to its inactivation in mice expressing AβCitation23 but no kinetic analysis was forthcoming. Furthermore it was suggested that Aβ peptides sequested transition metals from enzymes such as SOD and since Cu2+ is fundamental to the function of SOD and is crucial for the redox reaction involving the dismutation of superoxide radical to molecular oxygen and hydrogen peroxideCitation22 it could explain a mechanism for the inhibition of SODCitation24.
Fluorimetric analysis
Since tryptophan fluorescence is a well established principle to study the micro-environment for ligand binding to biological macromolecules fluorimetric analysis was used to identify the actual binding pocket for amyloid peptides in the SOD. The titration of SOD with the various amyloid peptides showed typical hyperbolic relationships (). The concentration of binding sites (n) on the purified enzyme, that were available for each of the amyloid peptides, and the dissociation constants (Kd) were estimated, respectively from the slopes of the linear regressions and the intercept on the log [(F-Fo)/(Fmax-F)] axis () [Equation 2].
Where Fo and F are respective fluorescence values in the absence and presence of Aβ-peptides; Fmax is the fluorescence of the enzyme after saturation with amyloid peptide and [Aβ] the concentration of the peptide. Since the concentration of SOD was 2 µM, there was only one binding sites available for each Aβ-peptide and respective values for Kd were 9.38 [Aβ25–37], 15.7 [Aβ29–33] and 12.13 [Aβ1–40] (). Typical saturation binding curves are also represented (, insets) supporting the finding that the binding sites on SOD for Aβ were independent and indistinguishable. Once again there was no significant difference between the Kd values of the three amyloid peptides which reflected that the glycine zippers regions, Aβ25–37 and Aβ29–33, within the Aβ1−40 peptide were responsible for the actual binding to SOD. Furthermore the association of the pentapeptide Aβ29–33 with SOD was the strongest (Kd = 15.7) supporting our initial finding that this pentapeptide was critical in the binding and interaction of amyloid peptides with SOD.
Figure 5. Fluorimetric analysis for the binding and interaction of amyloid peptides with SOD. Increasing concentrations of amyloid peptides (0–40 µM) were added to a reaction mixture of SOD (5.0 µl) in Tris-HCl buffer (pH 8.0, 50 mM) in a final volume of 300 µl. Excitation wavelength was at 295 nm and the emission wavelength was at 482 nm. All analyses were carried out in triplicate and values reported as the means with standard deviation p < 0.05 versus controls.
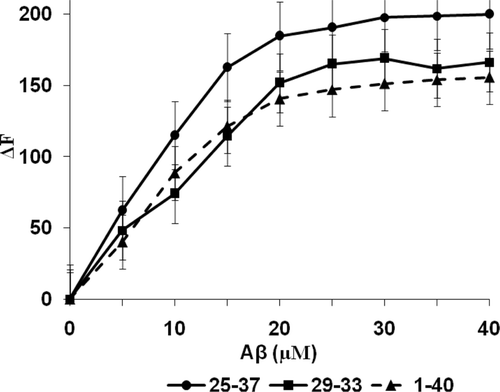
Figure 6. Modified Hill plots for the interaction of [A] [Aβ25–37(•), [B] Aβ29–33(▪), [C] Aβ1–40(▴)] with purified SOD (5 µl) in Tris-HCl buffer (pH 8.0, 50 mM). Inset: Saturation binding curves. All analyses were carried out in triplicate and values reported as the means with standard deviation p < 0.05 versus controls.
![Figure 6. Modified Hill plots for the interaction of [A] [Aβ25–37(•), [B] Aβ29–33(▪), [C] Aβ1–40(▴)] with purified SOD (5 µl) in Tris-HCl buffer (pH 8.0, 50 mM). Inset: Saturation binding curves. All analyses were carried out in triplicate and values reported as the means with standard deviation p < 0.05 versus controls.](/cms/asset/e50b3f1a-c84f-471b-a279-637a3960ad34/ienz_a_680063_f0006_b.gif)
In view of the fact that Aβ1−40 was, not only found within senile plaques but in the implication of oxidative stress and that it may act as a trigger in the formation of ROS, it was tempting to consider that the three consecutive glycine zipper motifs [Gly25–Ser26–Asn27–Lys28–Gly29–Ala30–Ile31–Ile32–Gly33–Leu34–Met35–Val36–Gly37] were critical in the initial interaction of the amyloid peptide with SOD. When it came to the interaction of these Aβ-peptides with SOD it was significant to understand that all three peptides mirrored each other with similar kinetic and fluorimetric analyses.
An in depth molecular modelling and docking study of the interaction and binding of all of the amyloid peptide fragments to SOD is currently on going and will be reported elsewhere.
From and it was established that the binding of the amyloid peptides caused a change in tryptophan fluorescence of purified SOD. The only intrinsic fluorophores present in the sequence Aβ1–40 were three phenyl rings (Phe4, Phe19 and Phe20) and one tyrosine (Tyr10). Since phenylalanine has a very low fluorescent quantum yield the possibility of any internal quenching by these amino acids when Aβ1–40 was bound to SOD was negligible. Nevertheless when Aβ1–40 interacted with SOD it showed a slightly lower value of ΔFmax when compared to Aβ25–37 and Aβ29–33 and this, in all three cases, must be due to fluorescence quenching by Tyr10.
The extent of interaction of the amyloid peptides with SOD is reflected in the dissociation constants for the SOD-Aβ complex which is largely determined by the size, structure and configuration of the peptide. The capability of the peptide to bind non-covalently at, or close to, the active site could also influence these values. Although no evidence of active site co-operativity was obvious in the present study the polarity of the active region precluded any binding of the hydrophobic amyloid peptides to this region. This meant that these peptides were bound at a site that was remote to the active site. The binding of the amyloid peptides with SOD was also influenced by solvating properties of the active catalytic region and since the peptides themselves were unable to change the micro-environment of the binding locus it was assumed that they induced conformational changes within the SOD molecule leading to the exposure of new groups near the active site.
Conclusion
Detection of fluorimetric changes in a protein upon binding with ligands was one of the simplest and most direct methods to study ligand induced conformational changes. In the present investigation the change in fluorescence of the enzyme upon binding with Aβ25–37, Aβ29–33 or Aβ1–40 was used to study SOD-Aβ interactions. Our investigations provided substantial evidence indicating that the binding sites on SOD were independent and indistinguishable. The saturation binding curves (, inset) from the binding of the amyloid peptides to SOD supports the absence of any subunit interaction.
The interaction of, not only Aβ1–40 but the three consecutive glycine zipper motifs between residues (25–37) and the single zipper Aβ29–33, with SOD, either individually or in tandem has now been investigated. There were no significant differences between Ki and Kd values of the three amyloid peptides that reflected that the regions of Aβ1–40 represented as the glycine zippers (Aβ25–37 and Aβ29–33) were responsible for the actual binding of the amyloid peptides to SOD as well as inhibition of SOD. In view of the fact that the pentapeptide ‘glycine zipper’ Aβ29–33 had a higher affinity (Kd = 15.7) with slightly lower Ki value than the multiple zipper region within Aβ25–37 it was tempting to speculate that this individual pentapeptide was crucial for the binding and inhibition of SOD by the amyloid peptide.
These findings may explain the enhanced production of ROS in Alzheimers disease and may serve as a therapeutic tool in the etiology and pathogenesis of the disease.
Related Research Data
References
- Hardy J. A hundred years of Alzheimer’s disease research. Neuron 2006;52:3–13.
- Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer’s disease. J Biol Chem 1996;271:18295–18298.
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans 2002;30:552–557.
- Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc Natl Acad Sci USA 2005;102:14278–14283.
- Gralle M, Ferreira ST. Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog Neurobiol 2007;82:11–32.
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging 2005;26:1235–1244.
- Jarrett JT, Lansbury PT Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993;73:1055–1058.
- Soto C, Brañes MC, Alvarez J, Inestrosa NC. Structural determinants of the Alzheimer’s amyloid beta-peptide. J Neurochem 1994;63:1191–1198.
- Findeis MA. The role of amyloid beta peptide 42 in Alzheimer’s disease. Pharmacol Ther 2007;116:266–286.
- Soto C, Castano E, Frangione B, Inestrosa NC. The α-helical to β-strand transition in the N-terminal fragment of the amyloid β-peptide modulates amyloid formation. J Biol Chem 1995;266:4025–4028.
- Poulos TL, Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Zhong WE, Jia Q, Wang PG. The novel binding mode of N-Alkyl-N’-hydroxyguanidine to neuronal nitric oxide synthase provides mechanistic insights into NO biosynthesis. Biochemistry 2002;41:3868–13875.
- Padayachee ER, Whiteley CG. Spectrofluorimetric analysis of the interaction of amyloid peptides with neuronal nitric oxide synthase: implications in Alzheimer’s disease. Biochim Biophys Acta 2011;1810:1136–1140.
- Padayachee E, Ngqwala NP, Whiteley CG. Association of β-amyloid peptide fragments with neuronal nitric oxide synthase: Implications in the etiology of Alzheimers disease. Enz Inhib Med Chem 2011 doi: 10.3109/14756366.2011.590805
- Mohlake P, Whiteley CG. Arginine metabolising enzymes as therapeutic tools for Alzheimer’s disease: peptidyl arginine deiminase catalyses fibrillogenesis of beta-amyloid peptides. Mol Neurobiol 2010;41:149–158.
- Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN et al. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res 2007;1148:243–248.
- Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 2007;8:766–775.
- Celsi F, Ferri A, Casciati A, D’Ambrosi N, Rotilio G, Costa A et al. Overexpression of superoxide dismutase 1 protects against beta-amyloid peptide toxicity: effect of estrogen and copper chelators. Neurochem Int 2004;44:25–33.
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem 2005;93:953–962.
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969;244:6049–6055.
- Carloni P, Blichl PE, Parrinello M. Electronic structure of the Cu-Zn superoxide dismutase active site and its interactions with the substrate. J Phys Chem 1995;99:1338–1348.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254.
- Barra D, Schinina ME, Simmaco M, Bannister JV, Bannister WH, Rotilio G et al. The primary structure of human liver manganese superoxide dismutase. J Biol Chem 1984;259:12595–12601.
- Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK et al. Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am J Pathol 2006;168:1608–1618.
- Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 1998;158:47–52.