Abstract
Cross metathesis (CM) of 9-butenylpurines with 4-butenyloxycoumarin in the presence of Grubbs 2nd generation catalyst under MW irradiation resulted to conjugated compounds containing homo-N-nucleosides and coumarins. Analogous derivatives received by the CM reaction of 9-butenyl-6-piperidinylpurine with 6- or 7-butenyloxycoumarins, allyloxycoumarins or coumarinyl acrylate. These compounds were tested in vitro for their antioxidant activity and they present significant scavenging activity. The presence of a pentenyloxy moiety, the attachment position on coumarin ring as well as a purine homo-N-nucleoside group are considered as important structural features.
Introduction
Nucleosides and modified nucleosides represent classes of compounds that possess very interesting biological activitiesCitation1–9 especially antiviral, anticancer, antimetabolic. Such activities appears also in acyclic nucleosidesCitation4,Citation5 bearing an alkyl chain in the place of sugar moiety. The acyclovir (I) is an antiviral drug; the adefovir (II) is in use for the treatment of the Hepatitis B virus (HBV) infections; the tenofovir (III) is the most important component in the anti-HIV drug cocktailsCitation1. The AP23464 (IV) and (V) are potent inhibitors of Src and Abl tyrosine kinases, which regulate angiogenesisCitation10. The hydroxamic acids VI have been synthesized as inhibitors of adenylyl cyclaseCitation11 whereas fluorinated pyranonucleoside analogues of N4-benzoylcytosine and N6-benzoyladenine have been tested for antioxidant activityCitation12. Compounds like VII show cardiostimulant and antiarrhythmic activitiesCitation13.
Coumarin derivatives, naturally occurring or synthetic, exhibit a broad range of biological activitiesCitation14–19 including anticoagulant, antibiotic, antifungal, antipsoriasis, cytotoxic, anti-HIV, anti-inflammatory, antioxidant, antithrombin etc. The geiparvarin (VIII) and their derivatives exhibit antitumorial activityCitation20,Citation21 and they also appeared as MAO-B inhibitorsCitation22. The auraptene (IX) is a chemopreventative agent against different kinds of cancer and an antiiflammatory agentCitation23. In terms of structural characteristics all the above mentioned compounds include a linker–substituent ().
Figure 1 Modified nucleosides and coumarin derivatives with important biological activities.
In continuation to our previous studies on coumarin derivativesCitation16,Citation24–29 and on modified nucleosidesCitation30,Citation31 and taking under consideration the linker’s presence and its contribution to the biological activity of molecules I–IX, we decided to synthesize a new series of conjugated molecules as modified nucleosides, combining the coumarin, the purine and a spacer moieties. The new compounds will be studied as free radical scavengers and lipid peroxidation inhibitors. Free radicals and the consequent lipid peroxidation could trigger coagulation and inflammation. Evidence points toward extensive cross-talk between coagulation and inflammation. It has been proved that inflammation plays an important role in post-thrombolytic complications. In patients with acute myocardial infarctionCitation32, inflammation is induced by thrombolytic therapy. Thrombin, a proteolytic enzyme can elicit many inflammatory responses in microvascular endothelium. Its multiple role in thrombosis makes thrombin an important target for the therapeutic agents designed for thrombus preventionCitation33.
For our syntheses we will apply the powerfull olefin metathesisCitation34–37, utilizing its application the cross-metathesis (CMCitation36,Citation38–40). The olefin metathesis reaction has been emerged the last decade in the nucleoside chemistryCitation41, while with the CM have been synthesized especially a few acyclic nucleosidesCitation42,Citation43 and dinucleosidesCitation44–47. Hetero-dinucleosides have been prepared in one case by CM under MW irradiation in shorter reaction times and better selectivity of heterodimer over the homodimerCitation48. Generally dramatic improvement of yields and reaction times have been reported in the MW-assisted CM reactionsCitation49–51. For those reasons we studied the new reactions under MW irradiation. The studied reactions and the obtained products are depicted in , . The derived compounds are examined as possible lipoxygenase (LOX) inhibitors, as potential inhibitors of lipid peroxidation and thrombin, as well as hydroxyl radical scavengers. The results will be discussed in terms of the structural characteristics of the compounds and the role of the combined moieties will be delineated.
Materials and methods
Melting points were determined on a Kofler hot-stage apparatus and are uncorrected. IR spectra were obtained with a Perkin-Elmer 1310 spectrophotometer as KBr pellets or Nujol mulls. NMR spectra were recorded on a Bruker AM 300 (300 MHz and 75 MHz for 1H and 13C, respectively) using CDCl3 as solvent and TMS as an internal standard. J values are reported in Hz. Mass spectra were determined on a LCMS-2010 EV Instrument (Shimadzu) under Electrospray Ionization (ESI) conditions. Microanalyses were performed on a Perkin-Elmer 2400-II Element analyzer. Silica gel No 60, Merck A.G. was used for column chromatography. All the chemicals used were of analytical grade and commercially available. 1, 1-Diphenyl-2-picrylhydrazyl (DPPH), nordihydroguairetic acid (NDGA), Trolox, Soybean Lipoxygenase, linoleic acid sodium salt, 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH), thrombin, tosyl-Gly-Pro-Arg-pNA are purchased from the Aldrich Chemical Co. & Sigma, Milwaukee, WI, USA. UV-Vis spectra were obtained on a Perkin-Elmer Lambda 20 beam spectrophotometer and on a Hitachi U-2001 spectrophotometer. The MW experiments were performed in a Biotage (Initiator 2.0) scientific MW oven.
Synthesis
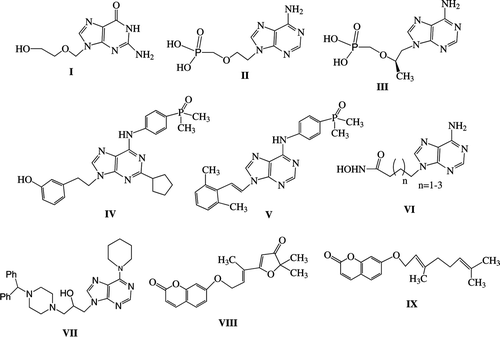
Compounds 4aCitation52, 8Citation53 and 10Citation54 were prepared according to the literature.
General procedure for the butenylation of purines. 6-Piperidinylpurine (1aCitation29) (0.53 g, 2.61 mmol), 4-bromo-1-butene (0.705 g, 0.536 mL, 5.22 mmol) and anhydrous K2CO3 (0.72 g, 5.22 mmol) were suspended in 10 mL dry DMF and the mixture was refluxed under N2 for 15 h. The resulting precipitate was filtered while hot and washed with CH2Cl2. The filtrate was evaporated and subjected to column chromatography [silica gel, hexane /EtOAc (1:1)] to give, from the faster moving band, compound 1a (0.47 g, 70% yield).
9-(But-3-en-1-yl)-6-(piperidin-1-yl)-9H-purine (2a). Oil, IR (Neat): 3080, 2920, 2840, 1575, 1545, 1470 cm−1; 1H-NMR (CDCl3) δ 1.63–1.80 (m, 6H), 2.62 (q, 2H, J = 6.9 Hz), 4.23 (t, 6H, J = 6.9 Hz), 5.05 (d, 1H, J = 17.3 Hz), 5.06 (d, 1H, J = 11.1 Hz), 5.70–5.86 (m, 1H), 7.70 (s, 1H), 8.34 (s, 1H); 13C-NMR (CDCl3) δ 24.9, 26.2, 34.1, 43.1, 46.4, 118.3, 120.0, 133.8, 138.0, 150.9, 152.5, 154.1; MS (ESI): 258 [M+H]+.; Anal. Calcd for C14H19N5: C, 65.34; H, 7.44; N, 27.22. Found: C, 65.23; H, 7.54; N, 27.08.
9-(But-3-en-1-yl)-6-(pyrrolidin-1-yl)-9H-purine (2b). [70% Yield, prepared from 6-pyrrolidinylpurineCitation33], m.p. 56–58°C (EtOH), IR (Nujol): 3060, 1580 cm−1; 1H-NMR (CDCl3) δ 1.97–2.12 (m, 4H), 2.63 (q, 2H, J = 6.9 Hz), 3.70–4.20 (m, 4H), 4.24 (t, 2H, J = 6.9 Hz), 5.06 (dd, 1H, J1 = 1.5 Hz, J2 = 17.4 Hz), 5.07 (dd, 1H, J1 = 1.5 Hz, J2 = 10.8 Hz), 5.72–5.87 (m, 1H), 7.69 (s, 1H), 8.37 (s, 1H); 13C-NMR (CDCl3) δ 25.5, 34.1, 43.1, 48.1, 118.2, 120.3, 133.7, 138.7, 150.2, 152.8, 153.2; MS (ESI): 244 [M+H]+.; Anal. Calcd for C13H17N5: C, 64.17; H, 7.04; N, 28.78. Found: C, 64.42; H, 6.79; N, 28.67.
9-(But-3-en-1-yl)-6-chloro-9H-purine (2c). (75% Yield, prepared from 6-chloropurine), oil, IR (Neat): 3058, 2910, 2835, 1580, 1547, 1490 cm−1; 1H-NMR (CDCl3) δ 2.71(q, 2H, J = 6.9 Hz), 4.42 (t, 2H, J = 6.9 Hz), 5.04 (dd, 1H, J1 = 1.2 Hz, J2 = 17.1 Hz), 5.08 (dd, 1H, J1 = 1.2 Hz, J2 = 10.2 Hz), 5.72–5.87 (m, 1H), 8.18 (s, 1H), 8.75 (s, 1H); 13C-NMR (CDCl3) δ 33.4, 43.5, 118.5, 131.2, 132.7, 145.0, 150.4, 151.4, 151.5; MS (ESI): 209/211 [M+H]+.; Anal. Calcd for C9H9N5Cl: C, 51.81; H, 4.36; N, 26.85. Found: C, 51.76; H, 4.14; N, 26.98.
General procedure for the butenylation of coumarins. 6-Hydroxy-4-methylcoumarin (3b) (0.25 g, 1.42 mmol), 4-bromo-1-butene (0.192 g, 0.146 ml, 1.42 mmol) and anhydrous K2CO3 (0.35 g, 2.53 mmol) were added in 10 ml dry acetone and the mixture was refluxed for 20 h. More 4-bromo-1-butene (96 mg, 0.073 ml, 0.71 mmol, totally 2.13 mmol) and anh. K2CO3 (0.175g, 1.27mmol, totally 3.8 mmol) were added and the reflux continued for 28 h. The resulting precipitate was filtered while hot and washed with acetone. The filtrate was evaporated and subjected to column chromatography [silica gel, hexane /EtOAc (3:1)] to give, from the faster moving band, compound 4b (0.229 g, 70% yield).
6-(But-3-en-1-yloxy)-4-methyl-2H-chromen-2-one (4b). M.p. 74–76°C (EtOAc/ hexane), IR (Nujol): 3070, 1705, 1575 cm−1; 1H-NMR (CDCl3) δ 2.40 (s, 3H), 2.57 (q, 2H, J = 6.7 Hz), 4.06 (t, 2H, J = 6.7 Hz), 5.14 (dd, 1H, J1 = 1.6 Hz, J2 = 12.9 Hz), 5.19 (dd, 1H, J1 = 1.6 Hz, J2 = 17.2 Hz), 5.84–5.99 (m, 1H), 6.31 (s, 1H), 7.03 (d, 1H, J = 2.9 Hz), 7.10 (dd, 1H, J1 = 2.9 Hz, J2 = 8.9 Hz), 7.26 (d, 1H, J = 8.9 Hz); 13C-NMR (CDCl3) δ 18.6, 33.5, 68.1, 108.7, 115.4, 117.2, 117.8, 119.1, 120.4, 134.1, 147.9, 151.8, 155.3, 160.5; MS (ESI): 253 [M+Na]+., 231 [M+H]+.; Anal. Calcd for C14H14O3: C, 73.03; H, 6.13. Found: C, 73.08; H, 5.86.
7-(But-3-en-1-yloxy)-4-methyl-2H-chromen-2-one (4c). (74% Yield, prepared from 7-hydroxy-4-methylcoumarin), m.p. 68–69°C (EtOAc/hexane), IR (Nujol): 3070, 1710, 1595 cm−1; 1H-NMR (CDCl3) δ 2.36 (s, 3H), 2.56 (q, 2H, J = 6.6 Hz), 4.04 (t, 2H, J = 6.6 Hz), 5.13 (dd, 1H, J1 = 1.5 Hz, J2 = 10.2 Hz), 5.18 (dd, 1H, J1 = 1.5 Hz, J2 = 17.4 Hz), 5.83–5.97 (m, 1H), 6.06 (s, 1H), 6.71 (d, 1H, J = 2.4 Hz), 6.82 (dd, 1H, J1 = 2.4 Hz, J2 = 8.7 Hz), 7.44 (d, 1H, J = 8.7 Hz); 13C-NMR (CDCl3) δ 18.5, 33.2, 67.7, 101.4, 111.8, 112.5, 113.5, 117.3, 125.4, 133.8, 152.4, 155.2, 161.0, 161.9; MS (ESI): 253 [M+Na]+., 231 [M+H]+.; Anal. Calcd for C14H14O3: C, 73.03; H, 6.13. Found: C, 73.27; H, 6.19.
Synthesis of 4-methyl-2-oxo-2H-chromen-6-yl acrylate (12). 6-Hydroxy-4-methyl-2H-chromen-2-one (3b) (0.5 g, 2.84 mmol) was dissolved in 20 ml dry acetone. Acryloyl chloride (0.257 g, 0.15 ml, 2.84 mmol) and anhydrous K2CO3 (0.392 g, 2.84 mmol) were added and the mixture was refluxed for 15 h. The resulting precipitate was filtered while hot and washed with acetone. The filtrate was evaporated and subjected to column chromatography [silica gel, hexane /EtOAc (3:1)] to give, from the faster moving band, compound 12 (0.57 g, 87% yield), m.p. 137–139°C (hexane/EtOAc); IR (Nujol): 3060, 1720, 1700, 1580 cm−1; 1H-NMR (CDCl3) δ 2.04 (s, 3H), 6.08 (dd, 1H, J1 = 1.2 Hz, J2 = 10.5 Hz), 6.34 (s, 1H), 6.35 (dd, 1H, J1 = 10.5 Hz, J2 = 17.4 Hz), 6.65 (dd, 1H, J1 = 1.2 Hz, J2 = 17.4 Hz), 7.30 (dd, 1H, J1 = 2.4 Hz, J2 = 9.0 Hz), 7.37 (d, 1H, J = 9.0 Hz), 7.39 (d, 1H, J = 2.4 Hz); 13C-NMR (CDCl3) δ 18.6, 115.8, 117.2, 118.1, 120.6, 125.2, 127.5, 133.2, 146.6, 151.1, 151.6, 160.3, 164.4; MS (ESI): 253 [M+Na]+., 231 [M+H]+.; Anal. Calcd for C13H10O4: C, 67.82; H, 4.38. Found: C, 67.77; H, 4.24.
General procedure for the CM reactions. Dry CH2Cl2 (2 ml) was placed in a tube (10 ml) for the Biotage Initiator 2.0, MW oven, equipped with a Teflon-coated stirring bar; the compounds 4aCitation52 (22 mg, 0.1 mmol) and 2a (77 mg, 0.3 mmol) were added followed by the catalyst 5 (8.5 mg, 10 mol%), argon was flushed in the tube and the mixture was irradiated at 100°C for 2 h (checked by TLC every 30 min). Another portion of 5 (3.5 mg, 4 mol%, totally 12 mg, 14 mol%) was added and the solution was irradiated for further 1 h. After cooling and evaporation of the solvent the residue was separated by column chromatography [silica gel No 60, hexane/EtOAc (3:1)] to give after the elution of unreacted starting materials the compound 6a (38 mg, 86% yield).
4-{[(3E)-6-(6-piperidin-1-yl-9H-purin-9-yl)-hex-3-en-1-yl]oxy}-2H-chromen-2-one (6a). M.p. 91–92°C (EtOAc/hexane), IR (Nujol): 3060, 1700, 1610, 1570, 980 cm−1; 1H-NMR (CDCl3) δ 1.64–1.80 (m, 6H), 2.53 (q, 2H, J = 6.3 Hz), 2.63 (q, 2H, J = 6.9 Hz), 4.03 (t, 2H, J = 6.3 Hz), 4.22 (t, 6H, J = 6.9 Hz), 5.46–5.67 (m, 2H), 5.62 (s, 1H), 7.23–7.34 (m, 2H), 7.54 (t, 1H, J = 8.7 Hz), 7.66 (s, 1H), 7.75 (d, 1H, J = 8.1 Hz), 8.32 (s, 1H); 13C-NMR (CDCl3) δ 24.8, 26.1, 31.7, 33.0, 43.2, 46.3, 68.4, 90.5, 115.7, 116.7, 119.9, 122.9, 123.8, 128.6, 128.9, 132.3, 137.8, 150.8, 152.4, 153.4, 153.9, 162.8, 165.4; MS (ESI): 468 [M+Na]+., 446 [M+H]+.; Anal. Calcd for C25H27N5O3: C, 67.40; H, 6.11; N, 15.72. Found: C, 67.59; H, 6.24; N, 15.83.
4-{[(3E)-6-(6-pyrrolidin-1-yl-9H-purin-9-yl)-hex-3-en-1-yl]oxy}-2H-chromen-2-one (6b). (89% Yield), m.p. 137–139°C (EtOAc/hexane), IR (Nujol): 3050, 1705, 1620, 1580, 965 cm−1; 1H-NMR (CDCl3) δ 1.92–2.10 (m, 4H), 2.54 (q, 2H, J = 6.4 Hz), 2.64 (q, 2H, J = 6.9 Hz), 3.67–4.18 (m, 4H), 4.01 (t, 2H, J = 6.4 Hz), 4.23 (t, 2H, J = 6.9 Hz), 5.46–5.68 (m, 2H), 5.60 (s, 1H), 7.22–7.34 (m, 2H), 7.53 (t, 1H, J = 7.3 Hz), 7.66 (s, 1H), 7.72 (d, 1H, J = 8.0 Hz), 8.34 (s, 1H); 13C-NMR (CDCl3) δ 27.0, 31.7, 33.1, 43.2, 48.3, 68.5, 90.5, 115.8, 116.8, 120.4, 122.9, 123.8, 128.7, 128.9, 132.3, 138.7, 150.2, 151.7, 152.7, 153.6, 162.8, 165.4; MS (ESI): 454 [M+Na]+., 432 [M+H]+.; Anal. Calcd for C24H25N5O3: C, 66.81; H, 5.84; N, 16.23. Found: C, 66.72; H, 5.92; N, 16.18.
4-{[(3E)-6-(6-chloro-9H-purin-9-yl)-hex-3-en-1-yl]oxy}-2H-chromen-2-one (6c). (76% Yield), m.p. 105–106°C (EtOAc/hexane), IR (Nujol): 3080, 1714, 1645, 1582, 960 cm−1; 1H-NMR (CDCl3) δ 2.56 (q, 2H, J = 6.0 Hz), 2.70 (q, 2H, J = 6.6 Hz), 4.07 (t, 2H, J = 6.0 Hz), 4.37 (t, 2H, J = 6.6 Hz), 5.48-5.72 (m, 2H), 5.64 (s, 1H), 7.28 (t, 1H, J = 7.8 Hz), 7.31 (d, 1H, J = 7.8 Hz), 7.55 (t, 1H, J = 7.8 Hz), 7.73 (d, 1H, J = 7.8 Hz), 8.12 (s, 1H), 8.74 (s, 1H); 13C-NMR (CDCl3) δ 31.6, 32.9, 44.0, 68.2, 90.6, 115.6, 116.8, 122.8, 123.8, 128.1, 129.5, 131.6, 132.4, 145.1, 151.1, 151.9, 152.0, 153.3, 162.7, 165.3; MS (ESI): 419/421 [M+Na]+.; 397/399 [M+H]+.; Anal. Calcd for C20H17N4O3Cl: C, 60.53; H, 4.52; N, 14.12. Found: C, 60.65; H, 4.45; N, 13.98.
9,9′-(3E)-Hex-3-ene-1,6-diylbis(6-chloro-9H-purine (7)). (51% Yield), m.p. 140–141°C (DCM), IR (Nujol): 3080, 1580, 1540, 983 cm−1; 1H-NMR (CDCl3) δ 2.58 (q, 2H, J = 6.6 Hz), 4.25 (t, 2H, J = 6.6 Hz), 5.38–5.43 (m, 1H), 8.03 (s, 1H), 8.74 (s, 1H); 13C-NMR (CDCl3) δ 32.8, 43.9, 129.2, 131.7, 144.9, 151.3, 151.8, 152.0; MS (ESI): 411/413/415 [M+Na]+.; 389/391/393 [M+H]+.; Anal. Calcd for C16H14Cl2N8: C, 49.37; H, 3.63; N, 28.79. Found: C, 49.21; H, 3.69; N, 28.52.
4-Methyl-6-{[(3E)-6-(6-piperidin-1-yl-9H-purin-9-yl)-hex-3-en-1-yl]oxy}-2H-chromen-2-one (6d). (63% Yield), m.p. 109–111°C (EtOAc/hexane), IR (Nujol): 3030, 1710, 1583, 990 cm−1; 1H-NMR (CDCl3) δ 1.62–1.78 (m, 6H), 2.41 (s, 3H), 2.46 (q, 2H, J = 6.6 Hz), 2.61 (q, 2H, J = 6.6 Hz), 3.92 (t, 2H, J = 6.6 Hz), 4.22 (t, 6H, J = 6.6 Hz), 5.48–5.65 (m, 2H), 6.29 (s, 1H), 7.00 (d, 1H, J = 2.7 Hz), 7.06 (dd, 1H, J1 = 2.7 Hz, J2 = 9.0 Hz), 7.25 (d, 1H, J = 9.0 Hz), 7.69 (s, 1H), 8.33 (s, 1H); 13C-NMR (CDCl3) δ 18.7, 24.5, 25.8, 32.0, 32.7, 42.9, 45.9, 67.7, 108.4, 115.1, 117.4, 118.8, 119.5, 120.1, 128.0, 129.2, 137.9, 147.7, 150.6, 151.5, 151.9, 153.6, 155.0, 160.2; MS (ESI): 482 [M+Na]+.; Anal. Calcd for C26H29N5O3: C, 67.95; H, 6.36; N, 15.24. Found: C, 68.17; H, 6.50; N, 15.12.
4-Methyl-7-{[(3E)-6-(6-piperidin-1-yl-9H-purin-9-yl)-hex-3-en-1-yl]oxy}-2H-chromen-2-one (6e). (74% Yield), m.p. 96–97°C (EtOAc/hexane), IR (Nujol): 3020, 1705, 1605, 1570, 980 cm−1; 1H-NMR (CDCl3) δ 1.64–1.78 (m, 6H), 2.39 (s, 3H), 2.47 (q, 2H, J = 6.6 Hz), 2.61 (q, 2H, J = 6.6 Hz), 3.94 (t, 2H, J = 6.6 Hz), 4.22 (t, 6H, J = 6.6 Hz), 5.45–5.63 (m, 2H), 6.13 (s, 1H), 6.78 (d, 1H, J = 2.4 Hz), 6.82 (dd, 1H, J1 = 2.4 Hz, J2 = 9.0 Hz), 7.48 (d, 1H, J = 9.0 Hz), 7.68 (s, 1H), 8.33 (s, 1H); 13C-NMR (CDCl3) δ 18.6, 24.8, 26.1, 32.1, 33.1, 43.4, 46.4, 67.7, 101.5, 112.0, 112.6, 113.6, 119.8, 125.5, 127.4, 128.3, 129.3, 138.0, 150.7, 152.4, 153.7, 155.3, 161.2, 161.9; MS (ESI): 482 [M+Na]+., 460 [M+H]+.; Anal. Calcd for C26H29N5O3: C, 67.95; H, 6.36; N, 15.24. Found: C, 68.06; H, 6.48; N, 15.41.
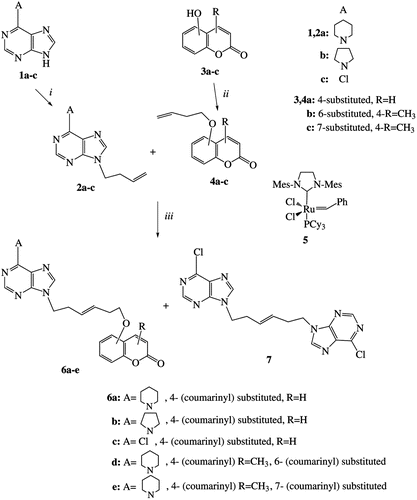
4-{[(2E)-5-(6-piperidin-1-yl-9H-purin-9-yl)-pent-2-en-1-yl]oxy}-2H-chromen-2-one (9). (63 % Yield), m.p. 83–85°C (EtOAc/hexane), IR (Nujol): 3020, 1700, 1615, 1575, 975 cm−1; 1H-NMR (CDCl3) δ 1.67–1.80 (m, 6H), 2.74 (q, 2H, J = 7.2 Hz), 4.20–4.37 (m, 6H), 4.58 (d, 2H, J = 5.1 Hz), 5.62 (s, 1H), 5.78 (dt, 1H, J1 = 5.1 Hz, J2 = 15.4 Hz), 5.92 (dt, 1H, J1 = 7.2 Hz, J2 = 15.4 Hz), 7.21–7.37 (m, 2H), 7.55 (t, 1H, J = 8.4 Hz), 7.71 (s, 1H), 7.80 (d, 1H, J = 8.4 Hz), 8.35 (s, 1H); 13C-NMR (CDCl3) δ 22.7, 26.2, 31.9, 42.9, 46.6, 69.1, 90.9, 115.7, 116.8, 119.8, 123.1, 123.9, 126.4, 131.9, 132.4, 138.1, 144.2, 153.5, 155.4, 160.4, 162.8, 165.1; MS (ESI): 454 [M+Na]+., 432 [M+H]+.; Anal. Calcd for C24H25N5O3: C, 66.81; H, 5.84; N, 16.23. Found: C, 66.72; H, 5.73; N, 16.45.
4-Methyl-6-{[(2E)-6-(6-piperidin-1-yl-9H-purin-9-yl)-pent-2-en-1-yl]oxy}-2H-chromen-2-one (11). (58% Yield), m.p. 81–82°C (EtOAc/hexane), IR (Nujol): 3010, 1703, 1620, 1565, 970 cm−1; 1H-NMR (CDCl3) δ 1.65–1.83 (m, 6H), 2.39 (s, 3H), 2.70 (q, 2H, J = 6.6 Hz), 4.26 (t, 6H, J = 6.6 Hz), 4.47 (d, 2H, J = 5.1 Hz), 5.73 (dt, 1H, J1 = 5.1 Hz, J2 = 15.6 Hz), 5.84 (dt, 1H, J1 = 6.6 Hz, J2 = 15.6 Hz), 6.29 (s, 1H), 6.99 (d, 1H, J = 2.7 Hz), 7.05 (dd, 1H, J1 = 2.7 Hz, J2 = 9.0 Hz), 7.24 (d, 1H, J = 9.0 Hz), 7.67 (s, 1H), 8.33 (s, 1H); 13C-NMR (CDCl3) δ 18.6, 24.9, 26.2, 31.9, 43.2, 46.1, 68.8, 109.2, 115.6, 117.1, 118.0, 119.2, 120.0, 128.7, 137.9, 139.9, 147.9, 148.1, 150.2, 151.3, 152.0, 155.6, 160.1; MS (ESI): 468 [M+Na]+.,460 [M+H]+.; Anal. Calcd for C25H27N5O3: C, 67.40; H, 6.11; N, 15.72. Found: C, 67.65; H, 6.26; N, 15.67.
4-Methyl-2-oxo-2H-chromen-6-yl(2E)-5-(6-piperidin-1-yl-9H-purin-9-yl)pent-2-enoate (13). (34% Yield), m.p. 89–91°C (EtOAc/hexane), IR (Nujol): 3070, 1702, 1630, 1560, 975 cm−1; 1H-NMR (CDCl3) δ 1.63–1.85 (m, 6H), 2.41 (s, 3H), 2.92 (q, 2H, J = 6.9 Hz), 4.18–4.32 (m, 4H), 4.38 (t, 2H, J = 6.9 Hz), 6.05 (d, 1H, J = 15.6 Hz), 6.32 (s, 1H), 7.15 (dt, 1H, J1 = 6.9 Hz, J2 = 15.6 Hz), 7.23–7.37 (m, 3H), 7.71 (s, 1H), 8.35 (s, 1H); 13C-NMR (CDCl3) δ 18.6, 22.7, 26.1, 31.9, 42.2, 46.4, 115.8, 117.1, 118.1, 119.9, 120.5, 122.8, 123.2, 125.2, 137.6, 146.7, 147.9, 151.0, 151.7, 157.9, 160.4, 161.7, 164.0; MS (ESI): 460 [M+H]+.; Anal. Calcd for C25H25N5O4: C, 65.35; H, 5.48; N, 15.24. Found: C, 66.31; H, 5.53; N, 15.09.
Biological assay
In vitro experiments
In the in vitro assays each experiment was performed at least in triplicate and the standard deviation of absorbance was less than 10% of the mean.
Determination of the reducing activity of the stable radical 1,1-diphenyl-picrylhydrazyl (DPPH)30
To a solution of DPPH in absolute ethanol (final concentration 0.05 mM) the tested compounds (stock solutions in DMSO) was added (final concentration 0.05 and 0.1 mM). Ethanol was used as control solution. After 20 and 60 min at room temperature the absorbance was recorded at 517 nm () and compared with the reference compound NDGA.
Table 1. Interaction-reducing activity with DPPH (RA%); competition with DMSO for hydroxyl radical (HO˙ %); inhibition of lipid peroxidation (AAPH%); in vitro inhibition of soybean lipoxygenase (LOX); in vitro inhibition of thrombin (% Thr).
Competition of the tested compounds with DMSO for hydroxyl radicals28
The hydroxyl radicals generated by the Fe3+/ascorbic acid system, were detected according to Nash, by the determination of formaldehyde produced from the oxidation of DMSO. The reaction mixture contained EDTA (0.1 mM), Fe3+ (167 μM), DMSO (33 mM) in phosphate buffer (50 mM, pH 7.4), the tested compounds at 0.1 mM and ascorbic acid (10 mM). After 30 min of incubation (37°C) the reaction was stopped with CCl3COOH (17 % w/v) (). Trolox was used as a standard drug.
Inhibition of linoleic acid lipid peroxidation30
Production of conjugated diene hydroperoxide by oxidation of linoleic acid in an aqueous dispersion is monitored at 234 nm. AAPH is used as a free radical initiator. This assay can be used to follow oxidative changes and to understand the contribution of each tested compound.
Azo compounds generating free radicals through spontaneous thermal decomposition are useful for in vitro studies of free radical production. The water-soluble azo compound AAPH has been extensively used as a clean and controllable source of thermally produced alkylperoxyl free radicals. Ten microlitre of the 16 mM linoleic acid sodium salt solution was added to the UV cuvette containing 0.93 mL of 0.05 M phosphate buffer, pH 7.4 prethermostated at 37°C. The oxidation reaction was initiated at 37°C under air by the addition of 50 μL of 40 mM AAPH solution. Oxidation was carried out in the presence of aliquots (10 μL) in the assay without antioxidant; lipid oxidation was measured in the presence of the same level of DMSO. The rate of oxidation at 37°C was monitored by recording the increase in absorption at 234 nm caused by conjugated diene hydroperoxides. Trolox was used as a reference compound ().
Soybean lipoxygenase inhibition study in vitro28–30
In vitro study was evaluated as reported previously. The tested compounds dissolved in ethanol were incubated at room temperature with sodium linoleate (0.1 mM) and 0.2 ml of enzyme solution (1/9 × 104 w/v in saline). The conversion of sodium linoleate to 13-hydroperoxylinoleic acid at 234 nm was recorded and compared with the appropriate standard inhibitor (). NDGA was used as a reference.
Inhibition of thrombin30
Tosyl-Gly-Pro-Arg-pNA was used as a substrate for thrombin at 1 mM final concentration. Compounds were dissolved at a final concentration of 0.1 mΜ in a Tris-buffer (0.05 M Tris, 0.154 M NaCl, ethanol 5%, pH 8.0). Three minutes after the addition of bovine thrombin (2.5 unit/mg), the reaction was ended by adding 0.1 ml acetic acid 50%. The absorption of the released p-nitroaniline was measured at 405 nm. NAPAP was used as a standard.
Determination of lipophilicity as clog P
Lipophilicity was theoretically calculated as clog P values in n-octanol-buffer by CLOGP Programme of Biobyte CorpCitation67.
Results and discussion
Synthesis
The butenylation of 6-piperidinylpurine (1a)Citation29 with excess of 4-bromo-1-butene in DMF in the presence of K2CO3, in analogy to allylationCitation55, led after refluxing for 9 h to 9-(but-3-en-1-yl)-6-(piperidin-1-yl)-9H-purine 2a (70%). The CM reaction of 4-(but-3-en-1-yloxy)-2H-chromen-2-one (4a)Citation52 with three equivalents of compound 2a seems to increase the ratio of the desired cross metathesis product to the possible homodimerCitation46. The reaction is performed in CH2Cl2 in the presence of the Grubbs 2nd generation catalyst 5 (14 mol%, added in two portions) under MW irradiation at 100°C over 3 h() and resulted only to the product 6a in 86% yield, while no homodimerization products detected. From the 1H-NMR of 6a the ratio E/Z is 3.2/1 with the E-isomer to give in the 13C-NMR absorptions for the CH2–CH=CH–CH2 at 33.0 and 31.7 ppm, while in the Z-isomer appear, as expectedCitation47,Citation56, at lower values at 29.7 and 27.9 ppm. When we studied the above reaction with 5% of the catalyst 5 under the same conditions the yield for 6a was only 29%.
The CM reaction of 9-but-3-en-1-yl-6-(pyrrolidin-1-yl)-9H-purine (2b) (3 equivalents) with alkene 4a in the presence of the catalyst 5 (14 mol% in two portions) under microwaves for 3 h gave the product 6b (89% yield) (E/Z ratio 3/1 from the 1H-NMR spectra). The same experiment with 11 mol% of the catalyst 5, loaded at once led to 79% yield of the product 6b. The analogous reaction of alkenes 2c and 4a with the catalyst 5 (14 mol% in two portions) under MW led to heteroalkene 6c (76%) [E/Z ratio 3.1/1, from the 1H-NMR spectrum] followed in this case by the purine homodimer 7 (51%) [E/Z ratio 3/1, from the 1H-NMR spectrum]. As we can see from the above reactions the CM of the Type ICitation57 alkenes 2a–c (3 equiv.) and 4a proceeds under MW irradiation in high yield by using enough amount of the catalyst 5 (14 mol % loaded in two portions) ().
Scheme 1. Reagents and conditions: (i) 4-bromo-1-butene, anhydrous K2CO3, dry DMF, reflux under Ar2, 15 h; (ii) 4-bromo-1-butene, anhydrous K2CO3, dry acetone, reflux, 20 h; (iii) CH2Cl2, 5 (14 mol% in two portions, the second after 2 h), MW, 100°C, 3 h.
The products 6d (63%) and 6e (74%) [E/Z ratio 2.8/1 and ratio 3/1, respectively, from the 1H-NMR spectra] are isolated from the CM reactions () of purine derivative 2a with the butenyloxycoumarins 4b,c (prepared in analogy to 4a) in the presence of catalyst 5 (14 mol% in two portions). No CM products are received from the efforts for reaction of coumarin derivative 4c with the 6-chloro-9-vinylpurine in the presence of the catalyst 5 or the Hoveyda-Grubbs 2nd generation catalyst under different ratios of the starting materials and under reflux or MW irradiation ().
Scheme 2. Reagents and conditions: (i) CH2Cl2, 3 (14 mol% in two portions, the second after 2 h), MW, 100°C, 3 h.
The CM reaction of derivative 2a with the 4-allyloxycoumarin (8)Citation53 by treatment with the catalyst 5 (14 mol% in two portions) under MW irradiation resulted to the product 9 (63%). This product seems to be the E-isomer since in the 13C-NMR spectrum the shift is only at 69.1 ppm, but nothing at 65.1 ppm as expected for the possible Z-isomerCitation45. In the 1H-NMR spectrum there are also two peaks at 5.78 ppm (dt, 1H, J1 = 5.1 Hz, J2 = 15.4 Hz) and 5.92 ppm (dt, 1H, J1 = 7.2 Hz, J2 = 15.4 Hz) for the olefinic protons revealing that this is the E-isomer. The reaction of coumarin 8 with the 9-allyl-6-chloropurine in the presence of catalyst 5 under reflux gave no CM products.
The analogous reaction between the purine derivative 2a and the 6-allyloxy-4-methylcoumarin (10)Citation54 with 5 (14 mol% in two portions) gave the heteroproduct 11 (58%). This product is the E-isomer since in the 1H-NMR spectrum there are two peaks at 5.73 ppm (dt, 1H, J1 = 5.1 Hz, J2 = 15.6 Hz) and 5.84 ppm (dt, 1H, J1 = 6.6 Hz, J2 = 15.6 Hz) for the olefinic protons and in the 13C-NMR spectra45 there is a shift at 68.8 ppm. In this case no Z-isomer was isolated from the column chromatography. By using a higher loading for the catalyst (16 mol%) but at once, the product 11 has been received in lower yield (52%). The reaction of allyloxycoumarin 10 with the 9-allyl-6-chloropurine (10:1 equivalents), in the presence of the catalyst 5 under reflux resulted to unchanged starting materials.
The CM reaction of derivative 2a and acrylate 12 [prepared from the reaction of 4-methyl-6-hydroxycoumarin (3b) with acryloyl chloride] with the catalyst 5 (14 mol%, in two portions) led to the E-isomer 13 (50%) (J = 15.6 Hz for the coupling of olefinic protons). No Z-isomer was detected in the fractions of column chromatography. When we used 17 mol% of the catalyst 5 (added at once) the isolated yield of the product 13 was only 34%. The reaction of acrylate 12 with the 9-allyl-6-chloropurine in the presence of Hoveyda-Grubbs 2nd generation catalyst or the catalyst 5 under reflux or MW irradiation didn’t resulted to CM products. These reactions gave only the 6-chloro-9-(prop-1-enyl)purine (isomerization product of the purine derivative).
The yields for the above CM reactions of allyloxycoumarins are lower than the analogous reactions with butenyloxycoumarins as expected due to the possible complexation of allylic oxygen to the Ru metal in the intermediate steps. The analogous complexations also of vinylic or allylic nitrogen from purines to the Ru metal, in the intermediate steps, are possibly responsible for no reaction of 9-vinyl- or 9-allylpurine derivatives.
Biological studies
In the present investigation, the compounds 6a–e, 9, 11 and 13 were studied with regard to their antioxidant ability as well as to their ability to inhibit soybean LOX. Compounds with antioxidant properties could be expected to offer protection in thrombosis and inflammation and to lead to potentially effective drugs. Taking into account the multifactorial character of oxidative stress and inflammation, we decided to evaluate the in vitro antioxidant activity of the synthesized molecules using three different antioxidant assays: (a) interaction with the stable free radical DPPH, (b) competition of the tested compounds with dimethyl sulfoxide (DMSO) for hydroxyl radicals and (c) interaction with the water-soluble azo compound AAPH. In view of the differences among the test systems available, the results of a single assay can give only a suggestion on the protective potential of tested compounds.
The DPPH test is very useful in the micromolar range demanding minutes to hours for both lipophilic and hydrophilic samples. In cases where the structure of the electron donor is not known, this method can afford data on the reduction potential of the sample, and hence can be helpful in comparing the reduction potential of unknown materials. The interaction of the examined compounds with the stable free radical DPPH was studied by the use of DPPHCitation58 at 0.05 mM and 0.1 mM after 20 and 60 min (). This interaction indicates their reducing ability in an iron-free system. The compounds did not show any interaction at 0.05 mM. The results showed that this interaction is time dependent but low compared to the reference compound NDGA. This can be attributed to the absence of any easily oxidizable functionalities like the ones (two catechol subunits) present in NDGA which is used as a reference compound. Compounds 9, 13, 11 and 6a present the higher interaction values at 60 min (31–63%). Among the derivatives of subgroup 6a–c, no significant changes are observed. The attachment of hexenyloxy group at position 6-, namely compound 6d, resulted in complete loss of activity. The presence of the ester group in compound 13 improves significantly the reducing ability, whereas in compound 11 the absence of this group leads to complete loss of antioxidant activity. The low interaction may be also due to the fact that they are large molecules and they can’t reach the radical site or due to steric reasons. Lipophilicity is not well correlated with the results. However, comparing the reducing abilities of 6a (clog P = 3.98, 31%) to 9 (clog P = 3.65, 63%) and of 6d (clog P = 4.34, no) to 11 (clog P = 4.01, 33%), it seems that lower lipophilicity (due to smaller aliphatic chain), is correlated with higher antioxidant ability.
It is consistent that rates of reactive oxygen species (ROS) production are increased in most diseasesCitation59. Cytotoxicity of O2-. and H2O2 in living organisms is mainly due to their transformation into HO., reactive radical metal complexes and 1O2. During the inflammatory process, phagocytes generate the superoxide anion radical at the inflamed site and this is connected to other oxidizing species as HO. Hydroxyl radicals are among the most reactive oxygen species and are considered to be responsible for some of the tissue damage occurring in inflammation.
The competition of compounds with DMSO for ·OH radicalsCitation58, generated by the Fe3+/ascorbic acid system, expressed as the inhibition of formaldehyde production, was used for the evaluation of their hydroxyl radical scavenging activity. Derivatives 6a, 6d, 9 and 13 highly compete with DMSO (33 mM) at 0.1 mM in comparison to Trolox (). Perusal of the percentage results shows that the 4-substituted hybrids 6a and 9 as well as the 6-substituted 6d and 13 are more potent. The magnitude of the ring (6- or 5-membered) 6a and 6b seems to influence the competition as well as the attachment position of the hexenyloxy group on the aromatic ring of the coumarin. Thus, the 6-substituted derivative compound 6d is a potent scavenger (100%, ), whereas in the corresponding 7-substituted the competition is dramatically vanished. Small differences are observed between the hexenyloxy- and pentylenyloxy analogues (6a and 9). The presence of the ester group in compound 13 improves significantly the competition, whereas in compound 11 the absence of this group leads to complete loss of hydroxyl radical scavenging activity. Lipophilicity is not correlated with the results. Antioxidants of hydrophilic or lipophilic character are both needed to act as radical scavengers in the aqueous phase or as chain-breaking antioxidants in biological membranes.
In our studies, AAPH was used as a free radical initiator to follow oxidative changes of linoleic acid to conjugated diene hydroperoxide. Azo compounds generating free radicals through spontaneous thermal decomposition are useful for free radical production studies in vitroCitation60. The water-soluble azo compound AAPH has been extensively used as a clean and controllable source of thermally produced C-centered radicals. The activity of the radicals produced by the action of AAPH shows a similarity to cellular activities such as lipid peroxidationCitation61. Effective hydrogen atom transfer (HAT) agents are compounds with high hydrogen atom donating abilityCitation62. These include: (i) compounds with low heteroatom-H bond dissociation energies and/or (ii) compounds from which hydrogen abstraction leads to sterically hindered radicals and (iii) compounds from which abstraction of hydrogen leads to C-centered radicals stabilized by resonance.
All compounds caused inhibition of lipid peroxidation (LPO), with the exception of compounds 6a and 13. The two most powerful and almost equipotent inhibitors of LPO (compounds 6d and 11) include the same structural characteristics. The only difference is the pentenyl/hexenyloxy chain in 11/6d compounds. Within the 6 subgroup, complete loss is observed when A is piperidinyl residue. No significant differences are observed between compounds 6b and 6c. The 6-substituted derivative, namely compound 6d is more potent (73%) than the corresponding 7-substituted, 6e (34%).
Our compounds were further evaluated for inhibition of soybean lipoxygenase LOX by the UV absorbance based enzyme assayCitation63. While one may not extrapolate the quantitative results of this assay to the inhibition of mammalian 5-LOX, it has been shown that inhibition of plant LOX activity by NSAIDs is qualitatively similar to their inhibition of the rat mast cell LOX and may be used as a simple qualitative screen for such activity. Many flavonoids and other phenolics as well as coumarin derivatives inhibit soybean lipoxygenase. This inhibition is related to their ability to reduce from the iron species in the active site to the catalytically inactive ferrous formCitation64. Lipoxygenases oxidize certain fatty acids at specific positions to hydroperoxides that are the precursors of leukotrienes, which contain a conjugated triene structure. Most of the LOX inhibitors are antioxidants or excellent ligands for Fe3+ or free radical scavengers, since lipoxygenation occurs via a carbon-centered radical. It is known that soybean lipoxygenase, is inhibited by NSAIDs in a qualitatively similar way to that of the rat mast cell lipoxygenase and may be used in a reliable screen for such activity. NDGA a known inhibitor of soybean LOX has been used as a reference compound. Perusal of percentage inhibition values () shows that the percentage inhibition values of all compounds range from 32 to 47, with the exception of 6a and 13 which are very poor inhibitors. There is no evidence for any special structural characteristic that influences the anti-LOX activity within this dataset. For all the compounds the LOX% inhibition values are in agreement with these on lipid peroxidation. The inhibitory activity of the compounds which do not contain an easily oxidizable moiety might be attributed mainly to their ability to attach to the active site of the enzyme and to a lesser degree to their electron donating ability. Although lipophilicity is referredCitation65,Citation66 as an important physicochemical property for LOX inhibitors, all the above tested derivatives do not follow this concept.
We evaluated the ability of some potent representative structures (6d, 9 and 11) to inhibit thrombin. Compound 9 shows 11% inhibition, while 6d and 11 do not exhibit any activity under the reported experimental conditions.
The above research will be used for the design and the synthesis in the near future of more potent compounds.
Conclusion
In conclusion, we have synthesized acyclic purine N-nucleosides modified with coumarin skeleton by the CM reaction under MW irradiation in moderate to high yields without receiving the possible purine homodimers, by the use of three equivalents excess of purine alkene derivative and the catalyst (14 mol%) added in two portions. Preliminary biological evaluation of compounds containing a linker in a coumarin modified homo-N-nucleoside resulted to significant antioxidant activity mostly for 6d, 9 and 13. Compounds 9 and 13 interact to the free stable radical DPPH and scavenge potently HO˙ radicals. Our studies showed that the presence of a pentenyloxy moiety, the attachment position on coumarin ring as well as a purine homo-N-nucleoside group are important structural features for the antioxidant and anti-LPO activity. This provides an impetus for designing new interesting molecules for the development of a new pharmacophore for disease-modifying agents useful in the treatment of a variety of inflammatory and coronary artery diseases.
Acknowledgements
We would like to thank Dr. A. Leo and C. Hansch†, as well as to Biobyte Corp. 201 West 4th Str., Suite 204, Claremont CA California, 91711, USA for free access to the C-QSAR program.
Declaration of Interest
The authors report no conflicts of interest.
Related Research Data
References
- De Clercq E. Highlights in the discovery of antiviral drugs: a personal retrospective. J Med Chem 2010;53:1438–1450.
- Mehellou Y, De Clercq E. Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go? J Med Chem 2010;53:521–538.
- De Clercq E. New approaches toward anti-HIV chemotherapy. J Med Chem 2005;48:1297–1313.
- De Clercq E. Acyclic nucleoside phosphonates: past, present and future. Bridging chemistry to HIV, HBV, HCV, HPV, adeno-, herpes-, and poxvirus infections: the phosphonate bridge. Biochem Pharmacol 2007;73:911–922.
- De Clercq E. The acyclic nucleoside phosphonates from inception to clinical use: historical perspective. Antiviral Res 2007;75:1–13.
- Herdewijn P (2008). In Modified Nucleosides in Biochemistry, Biotechnology and Medicine. Weinheim, Germany: Wiley-VCH.
- Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev 2009;109:2880–2893.
- Mieczkowski A, Roy V, Agrofoglio LA. Preparation of cyclonucleosides. Chem Rev 2010;110:1828–1856.
- Matsuda A, Sasaki T. Antitumor activity of sugar-modified cytosine nucleosides. Cancer Sci 2004;95:105–111.
- Wang Y, Shakespeare WC, Huang WS, Sundaramoorthi R, Lentini S, Das S et al. Novel N9-arenethenyl purines as potent dual Src/Abl tyrosine kinase inhibitors. Bioorg Med Chem Lett 2008;18:4907–4912.
- Levy D, Marlowe C, Kane-Maguire K, Bao M, Cherbavaz D, Tomlinson J et al. Hydroxamate based inhibitors of adenylyl cyclase. Part 1: the effect of acyclic linkers on P-site binding. Bioorg Med Chem Lett 2002;12:3085–3088.
- Spanou C, Manta S, Komiotis D, Dervishi A, Kouretas D. Antioxidant activity of a series of fluorinated pyrano-nucleoside analogues of N4-benzoyl cytosine and N6-benzoyl adenine. Int J Mol Sci 2007; 8:625–704.
- Οtt H.(1989) US Patent 4.849.423.
- O’Kennedy R, Thornes RD. (1997) “Coumarins: biology, applications and mode of action”. Chichester: J. Wiley & Sons.
- Murray DH, Mendez J, Brown SA. (1982) “The natural coumarins: occurrence, chemistry and biochemistry”. N. York: J. Wiley & Sons.
- Fylaktakidou KC, Hadjipavlou-Litina DJ, Litinas KE, Nicolaides DN. Natural and synthetic coumarin derivatives with anti-inflammatory/ antioxidant activities. Curr Pharm Des 2004;10:3813–3833.
- Borges F, Roleira F, Milhazes N, Santana L, Uriarte E. Simple coumarins and analogues in medicinal chemistry: occurrence, synthesis and biological activity. Curr Med Chem 2005;12:887–916.
- Lacy A, O’Kennedy R. Studies on coumarins and coumarin-related compounds to determine their therapeutic role in the treatment of cancer. Curr Pharm Des 2004;10:3797–3811.
- Kostova I. Synthetic and natural coumarins as antioxidants. Mini Rev Med Chem 2006;6:365–374.
- Chimichi S, Boccalini M, Cravotto G, Rosati O. A new convenient route to enantiopure 2-coumarinyloxypropanals: application to the synthesis of optically active geiparvarin analogues. Tetrahedron Lett 2006; 47:2405–2408.
- Viola G, Vedaldi D, dall’Acqua F, Basso G, Disarò S, Spinelli M et al. Synthesis, cytotoxicity, and apoptosis induction in human tumor cells by geiparvarin analogues. Chem Biodivers 2004;1:1265–1280.
- Carotti A, Carrieri A, Chimichi S, Boccalini M, Cosimelli B, Gnerre C et al. Natural and synthetic geiparvarins are strong and selective MAO-B inhibitors. Synthesis and SAR studies. Bioorg Med Chem Lett 2002;12:3551–3555.
- Curini M, Cravotto G, Epifano F, Giannone G. Chemistry and biological activity of natural and synthetic prenyloxycoumarins. Curr Med Chem 2006;13:199–222.
- Nicolaides DN, Gautam DR, Litinas KE, Hadjipavlou-Litina DJ, Fylaktakidou KC. Synthesis and evaluation of the antioxidant and antiinflammatory activities of some benzo[l]khellactone derivatives and analogues. Eur J Med Chem 2004;39: 323–332.
- Galariniotou E, Fragos V, Makri A, Litinas KE, Nicolaides DN. Synthesis of novel pyridocoumarins and benzo-fused 6-azacoumarins. Tetrahedron 2007; 63:8298–8304.
- Baldoumi V, Gautam DR, Litinas KE, Nicolaides DN. Convenient synthesis of linear pyrano[3,2-g]-, [2,3-g]- and angular pyrano[3,2-f]coumarins from 4[(1,1-dimethyl-2-propynyl)oxy]phenol. Tetrahedron 2006; 62:8016–8020.
- Gautam DR, Protopapas J, Fylaktakidou KC, Litinas KE, Nicolaides DN, Tsoleridis K. Unexpected one-pot synthesis of new polyclic coumarin[4,3-c]pyridine derivatives via a tandem hetero Diels-Alder and 1,3-dipolar cycloaddition reaction. Tetrahedron Lett 2009; 50:448–451.
- Kontogiorgis C, Litinas KE, Makri A, Nicolaides DN, Vronteli A, Hadjipavlou-Litina DJ et al. Synthesis and biological evaluation of novel angular fused Pyrrolocoumarins. J Enzyme Inhib Med Chem 2008;23:43–49.
- Symeonidis T, Chamilos M, Hadjipavlou-Litina DJ, Kallitsakis M, Litinas KE. Synthesis of hydroxycoumarins and hydroxybenzo[f]- or [h]coumarins as lipid peroxidation inhibitors. Bioorg Med Chem Lett 2009;19:1139–1142.
- Thalassitis A, Hadjipavlou-Litina DJ, Litinas KE, Miltiadou P. Synthesis of modified homo-N-nucleosides from the reactions of mesityl nitrile oxide with 9-allylpurines and their influence on lipid peroxidation and thrombin inhibition. Bioorg Med Chem Lett 2009;19:6433–6436.
- Litinas KE, Thalassitis A. Synthesis of fused dihydropyrido[e]purines via ring closing metathesis. Tetrahedron Lett 2010; 51:6451–6453.
- Merlini PA, Cugno M, Rossi ML, Agricola P, Repetto A, Fetiveau R et al. Activation of the contact system and inflammation after thrombolytic therapy in patients with acute myocardial infarction. Am J Cardiol 2004;93:822–825.
- Maraganore JM. Thrombin, thrombin inhibitors, and the arterial thrombotic process. Thromb Haemost 1993;70:208–211.
- Vougioukalakis GC, Grubbs RH. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem Rev 2010;110:1746–1787.
- Grubbs RH. Olefin metathesis. Tetrahedron 2004; 60:7117–7140
- Schrock RR, Hoveyda AH. Molybdenum and tungsten imido alkylidene complexes as efficient olefin-metathesis catalysts. Angew Chem Int Ed Engl 2003;42:4592–4633.
- Grubbs, RH. (2003) In Handbook of Metathesis. Weinheim, Germany: Wiley-VCH.
- Hoveyda AH, Gillingham DG, Van Veldhuizen JJ, Kataoka O, Garber SB, Kingsbury JS et al. Ru complexes bearing bidentate carbenes: from innocent curiosity to uniquely effective catalysts for olefin metathesis. Org Biomol Chem 2004;2:8–23.
- Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. A general model for selectivity in olefin cross metathesis. J Am Chem Soc 2003;125:11360–11370.
- Connon SJ, Blechert S. Recent developments in olefin cross-metathesis. Angew Chem Int Ed Engl 2003;42:1900–1923.
- Amblard F, Nolan SP, Agrofoglio LA. Metathesis strategy in nucleoside chemistry. Tetrahedron 2005; 61:7067–7080.
- Amblard F, Nolan SP, Gillaizeau I, Agrofoglio LA. A new route to acyclic nucleosides via palladium-mediated allylic alkylation and cross-metathesis. Tetrahedron Lett 2003; 44:9177–9180
- Amblard F, Nolan SP, Schinazi RF, Agrofoglio LA. Efficient synthesis of various acycloalkenyl derivatives of pyrimidine using cross-metathesis and Pd(0) methodologies. Tetrahedron 2005; 61:537–544.
- Zhong S, Mondon M, Pilasrd S, Len C. Synthesis of novel dinucleosides via tandem cross-metathesis and ring-closing metathesis. Tetrahedron Lett 2006; 47:6221–6224
- Roy V, Zerrouki R, Krausz P. New dinucleoside analogues via cross-coupling metathesis. Nucleosides Nucleotides Nucleic Acids 2005;24:289–301.
- Murata S, Ichikawa S, Matsuda A. Synthesis of galactose-linked uridine derivatives with simple linkers as potential galactosyltransferase inhibitors. Tetrahedron 2005; 61: 5837–5842
- Batoux N, Benhaddou-Zerrouki R, Bressolier P, Granet R, Laumont G, Aubertin AM, Krausz P. Nucleoside homodimerisation by cross metathesis. Tetrahedron Lett 2001; 42:1491–1493.
- Colombeau L, Zerrouki R, Krausz P, Champavier Y. Dinucleoside analog synthesis via microwave activated cross coupling metathesis. Lett Org Chem 2005; 2:613–615.
- Coquerel Y, Rodriguez J. Microwave-assisted olefin metathesis. Eur J Org Chem 2008; 1125–1132
- Gebauer J, Arseniyadis S, Cossy J. Total synthesis of cystothiazole A by microwave-assisted olefin cross-metathesis. Eur J Org Chem 2008; 2701–2704
- Fustero S, Jiménez D, Sanchez-Roselló M, del Pozo C. Microwave-assisted tandem cross metathesis intramolecular Aza-Michael reaction: an easy entry to cyclic beta-amino carbonyl derivatives. J Am Chem Soc 2007;129:6700–6701.
- Jackson S, Nieduzak T, Rebello S, Liang G, Chiang Y, Merrill J. Coumarins as iNOS inhibitors. 2005; US Patent 20050054681 A1.
- Avetisyan AA, Alvandzhyan AG. Syntheses on the basis of 2H-chromen-2-one and 2H-chromen-2-thione. Russ J Org Chem (Engl Transl) 2006; 42:1063–1067.
- Kaufman KD, Keana JFW, Kelly RC, McBride DW, Slomp G. Synthetic furocoumarins. VI analogs of psoralene derived from hydroquinone. J Org Chem 1962; 27:2567–2572.
- Gundersen LL. Synthesis of purinecarbonitriles by Pd(0)-catalysed coupling of halopurines with Zinc cyanide. Acta Chem Scand 1996; 50:58–63.
- Breitmaier E, Voelter W (1987). Carbon-13 NMR spectroscopy. High resolutions methods and applications in organic chemistry and biochemistry. New York: VCH, p. 192.
- Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. A general model for selectivity in olefin cross metathesis. J Am Chem Soc 2003; 125:11360–11370.
- Pontiki E, Hadjipavlou-Litina D. Synthesis and pharmacochemical evaluation of novel aryl-acetic acid inhibitors of lipoxygenase, antioxidants, and anti-inflammatory agents. Bioorg Med Chem 2007;15:5819–5827.
- Halliwell B Gutterridge JMC (1989) Editors, In Free Radicals in Biology and Medicine Oxford, UK Clarendon
- Huang D, Ou B, Prior RL. The chemistry behind antioxidant capacity assays. J Agric Food Chem 2005;53:1841–1856.
- Górnicki A, Gutsze A. In vivo and in vitro influence of etretinate on erythrocyte membrane fluidity. Eur J Pharmacol 2001;423: 127–134.
- Liégeois C, Lermusieau G, Collin S. Measuring antioxidant efficiency of wort, malt, and hops against the 2,2′-azobis(2-amidinopropane) dihydrochloride-induced oxidation of an aqueous dispersion of linoleic acid. J Agric Food Chem 2000;48:1129–1134.
- Taraporewala IB, Kauffman JM. Synthesis and structure-activity relationships of anti-inflammatory 9,10-dihydro-9-oxo-2-acridine-alkanoic acids and 4-(2-carboxyphenyl)aminobenzenealkanoic acids. J Pharm Sci 1990;79:173–178.
- Müller K. 5-Lipoxygenase and 12-lipoxygenase: attractive targets for the development of novel antipsoriatic drugs. Arch Pharm (Weinheim) 1994;327:3–19.
- Pontiki E Hadjipavlou-Litina D. Review in QSARs on LOX inhibitors.Mini Rev Med Chem 2003;3:487–499
- Pontiki E, Hadjipavlou-Litina D. Lipoxygenase inhibitors: a comparative QSAR study review and evaluation of new QSARs. Med Res Rev 2008;28:39–117.
- Biobyte Corp. C-QSAR Database 201 West 4th Str., Suite 204, Claremont CA, California 91711, USA.